

Abstract
Aging in vivo and cell division in vitro are associated with telomere shortening. Several lines of evidence suggest that telomere length may be a good predictor of the long term replicative capacity of cells. To investigate the natural fate of chromosome telomeres of hematopoietic stem cells in vivo, we measured the telomere length of peripheral blood granulocytes from 11 fully engrafted bone marrow transplant recipients and from their respective donors. In 10 of 11 donor–recipient pairs, the telomere length was significantly reduced in the recipient and the extent of reduction correlated inversely with the number of nucleated cells infused. These data provide internally controlled in vivo evidence that, concomitantly with their proliferation, hematopoietic stem cells lose telomere length; it is possible that, as a result, their proliferative potential is reduced. These findings must be taken into account when developing new protocols in which few stem cells are used for bone marrow transplantation or for gene therapy.
Telomeres, the physical ends of eukaryotic chromosomes, are essential for chromosome structure and stability (1, 2). Telomeres are characterized by specific proteins (3, 4) and by the presence of a large number of highly conserved, tandemly repeated short DNA sequences (5) that, in humans and in other vertebrates, have the structure TTAGGG (6). The length of telomeres is remarkably variable because of high variability in the number of TTAGGG repeats (7). Telomeres become shorter during the aging of humans (8, 9), and during the aging of primary fibroblasts cultured in vitro, as telomere sequences are lost with each round of cell division (10). Indeed, telomere length has been regarded as a “mitotic clock” (11), i.e., as a good predictor of the remaining replicative capacity of human fibroblasts (12). Telomeres are shorter in adult bone marrow cells than in fetal liver or in cord blood cells, and telomeres become shorter in the progeny of cultured primitive hematopoietic progenitor cells (13, 14), suggesting that a similar trend applies also to hematopoietic cells.
Sensitive assays for telomerase (15), a ribonucleoprotein that synthesizes telomeric DNA (16–18), have revealed low but detectable activity in human hematopoietic cells (19, 20), in highly enriched human hematopoietic progenitors (21, 22), and in purified murine hematopoietic progenitors (23). An increase in telomerase activity was detected in very early hematopoietic progenitors when they were “stimulated” with growth factors (14, 21, 22). The observations carried out in peripheral blood cells as a function of age (8, 9, 24) and in cultures of hematopoietic cells (13, 14) suggest that this telomerase activity is not sufficient to prevent telomere shortening. However, it is not known whether this also is true of bona fide stem cells because these are not available in pure form. Therefore, telomere dynamics of stem cells can be only studied in vivo, and, because of considerable individual variability in telomere length (24), in principle it would have to be assessed on individual subjects by using a longitudinal approach; given the low rate of stem cell division, it would take years to complete such a study.
We therefore resorted to analyzing a situation in which stem cell proliferation is accelerated, namely the reconstitution of hematopoiesis after bone marrow transplantation (BMT). Indeed, repopulation of the bone marrow of a myelo-ablated recipient is the most definitive assay for stem cells. In addition, after allogeneic BMT, we have an opportunity to analyze the donor’s telomeres in the recipient’s blood.
Based on the use of a sub-telomeric probe specific for the long arm of chromosome 7 (25), we measured telomere length in the peripheral blood granulocytes from the donor and from the recipient at the same time. Each pairwise comparison was thereby internally controlled because the donor telomeres were analyzed in each recipient after engraftment. In nearly all cases, the telomere length was reduced significantly in the recipient, suggesting that the proliferative potential of stem cells may be reduced after BMT.
MATERIALS AND METHODS
Patients.
Eleven recipient–donor pairs were studied at 4–83 months (median: 23 months) after BMT. At the time of transplantation, five patients were in complete remission from acute leukemia, and six had chronic phase Philadelphia-positive chronic myelogenous leukemia. All patients had received a conventional conditioning regimen based on busulfan and cyclophosphamide. All patients were in complete remission at the time of the study. For a quantitative assessment of long term engraftment, we used cytogenetics and serological tests. In cytogenetic analysis, at least 25 metaphases were counted, and we accepted for analysis only patients in whom all metaphases were of donor type. Serological analysis was carried out by a gel test technique, and no red cells of the recipient’s blood group were found. Based on the results of artificial mixing experiments, this technique would easily detect 2.5% red cells of a different blood group. By these criteria, full engraftment was proven in 10 of 11 patients (A-J). In patient K, we had no reliable marker of engraftment, but the patient is in long lasting hematological complete remission, and he remains Philadelphia-negative (Table 1).
Table 1.
Patients investigated
| Don/Rec pair | Diagnosis | Age* Don/Rec | Sex Don/Rec | Blood group* Don/Rec | Infused cells (×10−8/Kg) | Recipient data at the time of the study
|
|||
|---|---|---|---|---|---|---|---|---|---|
| Months from BMT | Blood group | Cytogenetics | CR | ||||||
| A | AML | 38/32 | F/M | B,+/B,+ | 1.8 | 4 | B,+ | 46 XX | Yes |
| B | CML | 39/32 | M/M | O,+/O,− | 1.9 | 5 | O,+ | 46 XY Ph- | Yes |
| C | ALL | 22/21 | M/F | O,+/O,+ | 3.3 | 8 | O,+ | 46 XY | Yes |
| D | CML | 51/38 | M/F | A,+/A,+ | 2.6 | 9 | A,+ | 46 XY Ph- | Yes |
| E | AML | 33/37 | F/M | A,+/A,+ | 2.5 | 20 | A,+ | 46 XX | Yes |
| F | AML | 42/44 | F/F | A,+/A,− | 2.8 | 23 | A,+ | 46 XX | Yes |
| G | AML | 8/12 | F/F | O,+/O,− | 2.6 | 33 | O,+ | 46 XX | Yes |
| H | CML | 28/37 | M/M | O,+/A,+ | 2.7 | 37 | O,+ | 46 XY Ph- | Yes |
| I | CML | 16/20 | F/M | O,+/A,+ | 3.3 | 38 | O,+ | 46 XX Ph- | Yes |
| J | CML | 33/37 | F/F | A,+/A,− | 1.9 | 42 | A,+ | 46 XX Ph- | Yes |
| K | CML | 24/29 | M/M | B,+/B,+ | 2.1 | 83 | B,+ | 46 XY Ph- | Yes |
Don, bone marrow donor; Rec, bone marrow recipient; CR, complete remission; AML, acute myelogenous leukemia; ALL, acute lymphoblastic leukemia; CML, chronic myelogenous leukemia; ∗, at time of transplantation; +, Rh(D) blood group positive; −, Rh(D) blood group negative; Ph, absence of Philadelphia chromosome.
Bold: data informative of allogeneic engraftment.
Cell Separation and DNA Extraction.
Samples of peripheral blood were obtained after informed consent and at the same time from each BMT pair. Neutrophils were isolated by Ficoll/Paque (Pharmacia LKB) density gradient centrifugation and recovered from the top of the red blood cells pellet. High molecular weight DNA was extracted by a standard method (26).
Telomere Restriction Fragment Length Measurement by Using a (TTAGGG)3 Probe.
The method of telomere restriction fragment length measurement by using a (TTAGGG)3 probe is a slight variation of that described by Harley et al. (10). In brief, DNA was digested to completion with restriction enzymes MspI and RsaI and subjected to electrophoresis on a 0.6% agarose gel. DNA was transferred onto a nylon membrane (Hybond N, Amersham) after HCl depurination and hybridized in 6× SSC at 50°C with an end-labeled (TTAGGG)3 probe. The membrane was washed in 3× SSC at 42°C, and telomere signals were visualized by autoradiography.
Measurement of Telomere Length of a Single Chromosome End.
The TelBam8 probe is unique for the subtelomeric region of long arm of chromosome 7 (25). To measure the length of the 7q telomeric fragment (including ≈8 kb of subtelomeric region), genomic DNA was digested to completion with BamHI, loaded on 1% agarose gel, and resolved by using field-inversion gel electrophoresis (FIGE Mapper Electrophoresis System, Bio-Rad) with the following parameters: forward voltage 6 V/cm, reverse voltage 4 V/cm, switch time ramping in a nonlinear mode from 0.1 to 0.4 sec, and run time 26–30 h (27). DNA was nicked by UV irradiation, transferred onto a nylon membrane (ZetaProbe GT, Bio-Rad) under alkaline conditions, and hybridized in 6× SSC at 65°C with the radiolabeled TelBam8 probe. The membrane was washed under high stringency conditions. The length of the BamHI telomeric fragment was calculated by densitometry from the position of the peak of maximum intensity on autoradiograph films. All data represent the average of three determinations.
Statistical Analysis.
Statistical analysis was performed by using Fisher’s exact test, the Wilcoxon rank sum test, and the linear regression, as appropriate and as stated.
RESULTS AND DISCUSSION
Measurement of Telomere Length by Single Chromosome Analysis.
We have used the chromosome 7q subtelomeric probe TelBam8 (25) to measure the telomere length of one end of a single chromosome pair. In this way, we were able to reduce the variation in size of the telomere length that is seen in blots hybridized to a (TTAGGG)n telomeric repeat probe (Fig. 1A), in which the signal must be attributed to the telomeres of all 92 chromatid ends present in each human cell (184 during mitosis and in G2). This approach is similar to that used for studying telomere dynamics in a human cell line engineered to have a plasmid sequence inserted in the 13q telomere (28). By using the same technique on DNA extracted from individual single clones of human lymphoblastoid cell lines recently transformed by Epstein–Barr virus (29), even sharper signals were seen (Fig. 1B). This suggests that another factor contributing to the spread of hybridization signals is the heterogeneity of telomere length in different cells, presumably reflecting different mitotic histories. To establish the accuracy of this technique, 14 individual DNA samples were run in duplicate. The positions of the peaks of the autoradiography signals obtained in these duplicates were highly reproducible. The average difference in migration for the 14 duplicates, converted to base pair, was 81 (absolute value), and the SD was 80. Therefore, we decided that the difference in telomere length measured by our method between any two DNA samples (ΔTEL) can be safely regarded as significant when it is greater than 321 bp (mean + 3 SD of the values observed in the duplicates). An alternative method for estimating the size of individual telomeres by quantitating the fluorescent signals obtained by in situ hybridization has been reported (30, 31). This method has the advantage that, in the cells selected for analysis, all individual telomeres are measured; conversely, the method we have used has the advantage that the values we obtained are representative of some 106 cells.
Figure 1.
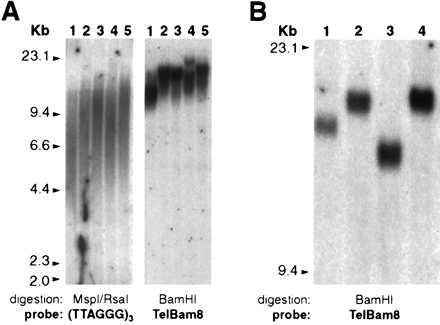
Comparison between the standard technique and the “single chromosome end technique” for the measurement of telomere length. (A) Genomic DNA from peripheral blood granulocytes from five individuals of different age (range: 24–73 years) was analyzed by Southern blotting. (Left) DNA was digested with MspI and RsaI, and the filter was hybridized to a (TTAGGG)3 probe. (Right) DNA was digested with BamHI, and the filter was hybridized to the TelBam8 probe. (B) Four individual clones from a single lymphoblastoid cell line (29) were analyzed as A (Right).
Telomeres Are Shorter After Repopulation of the Bone Marrow in Transplant Recipient.
Hematopoietic re-constitution after BMT places a much greater proliferative demand on stem cells than normal hematopoiesis (32). To evaluate the effect of in vivo proliferation on the telomere length of hematopoietic stem cells, we have measured in parallel the length of telomere in the granulocytes from BMT recipient and donor. In fact, after engraftment, we are comparing the donor’s telomeres in the donor blood with the donor’s telomeres in the recipient’s blood; thus, each pairwise comparison is internally controlled. In analyzing clinical samples, we do not have the option of using recently cloned cell populations. However, we did reduce the heterogeneity of blood leukocytes by collecting granulocytes free of mononuclear cells. By this analysis, we found that the average telomere length is consistently shorter in the BMT recipient than in the respective donor (Figs. 2A and 3A; P = 0.001, Wilcoxon rank sum test for paired samples). The extent of telomere shortening (ΔTEL, Fig. 2B) ranged from 79 to 1446 bp, and it was significantly greater than the values obtained in the 14 controls (see above), which ranged from 13 to 256 bp (Fig. 3B; P < 10−5, Wilcoxon rank sum test for unpaired samples). In 10 of 11 donor–recipient pairs, ΔTEL was above the background threshold we previously set (see above; Fig. 3B; P < 10−5, Fisher’s exact test). In this series, we have found no correlation between ΔTEL and donor age or between ΔTEL and time elapsed since the BMT procedure. In contrast, there was a significant inverse correlation (Fig. 4) between ΔTEL and the number of nucleated bone marrow cells infused (per kilogram of recipient body weight). Although the majority of nucleated cells infused do not contribute to long term hematopoiesis in the recipient, it is reasonable to assume that their total number is roughly proportional to the number of bona fide stem cells. The simplest interpretation of our finding is that the fewer the stem cells transferred to the recipient, the more cell divisions are needed for reconstitution of hematopoiesis. Consequently, a greater consumption of telomeres takes place.
Figure 2.
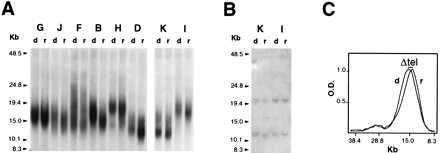
Telomere length in representative cases of BMT donor–recipient pairs. (A) Representative cases of BMT donor–recipient pairs. Southern blot analysis of granulocyte DNA digested with BamHI, resolved by field-inversion gel electrophoresis, and hybridized to the TelBam8 probe. Each pair is indicated by a capital letter (see Table 1); in each pair, d is the donor and r is the recipient. (B) Uniform migration of nontelomeric unique sequence fragments. The filter shown in A (Left) was rehybridized with a PIG-A cDNA probe, yielding the two expected bands of 20.6 Kb and 12 Kb, respectively. (C) Densitometric tracings of the two lanes of a representative case (pair J). The difference in telomere size, ΔTEL, is measured directly as the distance between the modal values of the peaks from donor (d) and recipient (r), respectively.
Figure 3.
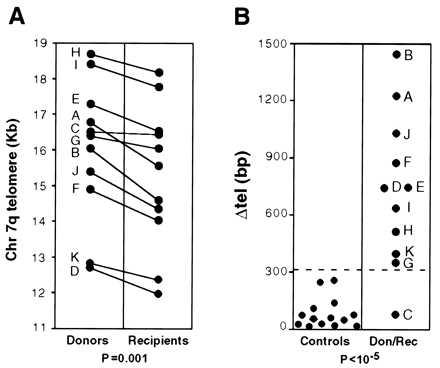
Shortening of telomere (ΔTEL) after BMT. (A) The differences in telomere size between the paired DNA samples in the original donor and in the recipient after BMT are shown by individual straight lines. Each pair is indicated by a capital letter (see Table 1). Each data point represents the average of three determinations. The donor–recipient difference, analyzed by the Wilcoxon rank sum test for paired samples, is highly significant (P = 0.001). (B, Left) A scattergram of the difference in telomere length measured between two aliquots of the same DNA sample that were run side by side; in these controls, the apparent difference in telomere length was 81.5 ± 80 bp (range: 13–256 bp). (Right) The ΔTEL values for each donor–recipient pair (indicated by a capital letter, see Table 1) also are displayed as a scattergram. The broken line across the entire figure (3 SD above the mean of control data) is taken to be the threshold of background of our method of analysis. In 10 of 11 donor–recipient pairs, ΔTEL is above the threshold (P < 10−5, Fisher’s exact test). Don, donor; Rec, recipient.
Figure 4.
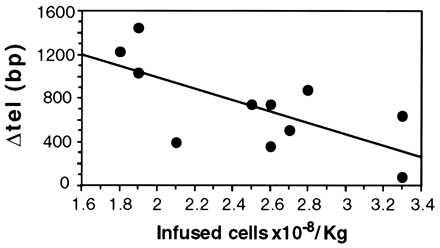
Graft size and telomere shortening (ΔTEL) after BMT. ΔTEL correlates inversely with graft size (number of nucleated bone marrow cells infused per kilogram of recipient body weight) after BMT. The straight line was obtained by linear regression from data representing the average of three determinations. The slope of regression is significantly different from 0 (r = −0.686; P < 0.02).
The donor’s stem cells must presumably undergo a larger number of telomere-shortening rounds in the engrafting recipient than have occurred in the steady-state in the donor. According to current estimates, the loss of telomere length is 40–120 bp per cell division (9, 10, 13). Assuming that shortening of telomeres is indeed proportional to the number of cell divisions, the lowest loss we have measured may correspond to just one additional cell division whereas the highest loss may correspond to ≈12–36 additional cell divisions in the recipient after BMT.
Until now, the loss of telomere length associated with the aging of stem cells was inferred (i) from age-related population studies (8, 9, 24) and (ii) from culture studies on bone marrow progenitor cells (downstream of stem cells) (13, 14). By contrast, this work shows what happens to stem cell telomeres by analyzing them in vivo at two discrete points in time, before and after expansion associated with BMT; the results are in keeping with the previous observations just quoted. Thus, we can state confidently that stem cell proliferation is associated with loss of telomere length. This implies that the telomerase activity, which recently has been observed in hematopoietic progenitors and in their various progeny (14, 19–22), is unable to prevent telomere shortening, either because it is quantitatively insufficient or because it has additional or other functions (19). Telomere length correlates with the number of remaining allowable cell divisions in somatic cells (11, 12), and this may also apply to hematopoietic stem cells (30). Therefore, our findings suggest that the proliferative potential of human stem cells is decreased after hematopoietic reconstitution of myelo-ablated patients. This is reminiscent of the well known progressive dilution in hematopoiesis-reconstituting cells after serial bone marrow transplantation in the mouse (33–37); indeed, this latter phenomenon could be closely related to telomere shortening.
These observations may have significant practical implications. It is possible that, in normal persons, the telomere length of stem cells is sufficient to support hematopoiesis for a longer time than the average human life span, and serial transplants in mice show that the original stem cell pool of mouse has the capacity to sustain hematopoiesis for several multiples of the mouse’s lifespan (32). In keeping with this, late failure of a well established graft after BMT in humans is rare. However, there is an increasing interest in the transplantation of a reduced number of stem cells [e.g., after expansion in vivo (38) or ex vivo (39)]. It will be important to determine whether such procedures significantly increase ΔTEL because telomere length may become limiting for long term hematopoiesis. For similar reasons, the fate of telomeres may be crucial for the outcome of gene therapy protocols in which one or few stem cells are expected to repopulate the bone marrow.
Acknowledgments
We thank W. R. A. Brown (Oxford University, Oxford, England) for the TelBam8 probe, on which we based our method to measure telomere length; M. Bessler and K. Nafa for providing DNA from lymphoblastoid cell clones; C. Selleri for clinical information on patients; L. Luciano for cytogenetic analysis; S. Formisano and R. Romano for blood serological tests; and J. Beckmann, N. Hastie, D. Higgs, A. Karadimitris, P. P. Pandolfi, V. Rosti, A. Rovira, M. Sadelain, D. Schlessinger, G. Tremml, and J. W. Young for their helpful suggestions in reviewing the manuscript. This work was supported in part by New York State Legislative Grant C960170. Preliminary data on part of this work were communicated at the American Society of Hematology Annual Meeting, December 6–9, 1996, in Orlando, FL.
ABBREVIATIONS
- BMT
bone marrow transplantation
- ΔTEL
telomere shortening
References
- 1.Blackburn E H. Nature (London) 1991;350:569–573. doi: 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- 2.Zakian V A. Science. 1995;270:1601–1607. doi: 10.1126/science.270.5242.1601. [DOI] [PubMed] [Google Scholar]
- 3.Chong L, van Steensel B, Broccoli D, Erdjument-Bromage H, Hanish J, Tempst P, de Lange T. Science. 1995;270:1663–1667. doi: 10.1126/science.270.5242.1663. [DOI] [PubMed] [Google Scholar]
- 4.Smith S, de Lange T. Trends Genet. 1997;13:21–26. doi: 10.1016/s0168-9525(96)10052-4. [DOI] [PubMed] [Google Scholar]
- 5.Blackburn E H, Szostak J W. Annu Rev Biochem. 1984;53:163–194. doi: 10.1146/annurev.bi.53.070184.001115. [DOI] [PubMed] [Google Scholar]
- 6.Moyzis R K, Buckingham J M, Cram L S, Dani M, Deaven L L, Jones M D, Meyne J, Ratliff R L, Wu J R. Proc Natl Acad Sci USA. 1988;85:6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Lange T, Shiue L, Myers R M, Cox D R, Naylor S L, Killery A M, Varmus H E. Mol Cell Biol. 1990;10:518–527. doi: 10.1128/mcb.10.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hastie N D, Dempster M, Dunlop M G, Thompson A M, Green D K, Allshire R C. Nature (London) 1990;346:866–868. doi: 10.1038/346866a0. [DOI] [PubMed] [Google Scholar]
- 9.Vaziri H, Schachter F, Uchida I, Wei L, Zhu X, Effros R, Cohen D, Harley C B. Am J Hum Genet. 1993;52:661–667. [PMC free article] [PubMed] [Google Scholar]
- 10.Harley C B, Futcher A B, Greider C W. Nature (London) 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 11.Harley C B. Mutat Res. 1991;256:271–282. doi: 10.1016/0921-8734(91)90018-7. [DOI] [PubMed] [Google Scholar]
- 12.Allsopp R C, Vaziri H, Patterson C, Goldstein S, Younglai E V, Futcher A B, Greider C W, Harley C B. Proc Natl Acad Sci USA. 1992;89:10114–10118. doi: 10.1073/pnas.89.21.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaziri H, Dragowska W, Allsopp R C, Thomas T E, Harley C B, Lansdorp P M. Proc Natl Acad Sci USA. 1994;91:9857–9860. doi: 10.1073/pnas.91.21.9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engelhardt M, Kumar R, Albanell J, Pettengell R, Han W, Moore M A S. Blood. 1997;90:182–193. [PubMed] [Google Scholar]
- 15.Kim N W, Piatyszek M A, Prowse K R, Harley C B, West M D, Ho P L, Coviello G M, Wright W E, Weinrich S L, Shay J W. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 16.Greider C W, Blackburn E H. Cell. 1987;51:887–898. doi: 10.1016/0092-8674(87)90576-9. [DOI] [PubMed] [Google Scholar]
- 17.Morin G B. Cell. 1989;59:521–529. doi: 10.1016/0092-8674(89)90035-4. [DOI] [PubMed] [Google Scholar]
- 18.Feng J, Funk W D, Wang S S, Weinrich S L, Avilion A A, et al. Science. 1995;269:1236–1241. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- 19.Broccoli D, Young J W, de Lange T. Proc Natl Acad Sci USA. 1995;92:9082–9086. doi: 10.1073/pnas.92.20.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Counter C M, Gupta J, Harley C B, Leber B, Bacchetti S. Blood. 1995;85:2315–2320. [PubMed] [Google Scholar]
- 21.Hiyama K, Hirai Y, Kyoizumi S, Akiyama M, Hiyama E, Piatyszek M A, Shay J W, Ishioka S, Yamakido M. J Immunol. 1995;155:3711–3715. [PubMed] [Google Scholar]
- 22.Chiu C P, Dragowska W, Kim N W, Vaziri H, Yui J, Thomas T E, Harley C B, Lansdorp P M. Stem Cells. 1996;14:239–248. doi: 10.1002/stem.140239. [DOI] [PubMed] [Google Scholar]
- 23.Morrison S J, Prowse K R, Ho P, Weissman I L. Immunity. 1996;5:207–216. doi: 10.1016/s1074-7613(00)80316-7. [DOI] [PubMed] [Google Scholar]
- 24.Slagboom P E, Droog S, Boomsma D I. Am J Hum Genet. 1994;55:876–882. [PMC free article] [PubMed] [Google Scholar]
- 25.Brown W R, MacKinnon P J, Villasante A, Spurr N, Buckle V J, Dobson M J. Cell. 1990;63:119–132. doi: 10.1016/0092-8674(90)90293-n. [DOI] [PubMed] [Google Scholar]
- 26.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. pp. 9.14–9.19. [Google Scholar]
- 27.Birren B W, Lai E, Hood L, Simon M I. Anal Biochem. 1989;177:282–286. doi: 10.1016/0003-2697(89)90052-3. [DOI] [PubMed] [Google Scholar]
- 28.Murnane J P, Sabatier L, Marder B A, Morgan W F. EMBO J. 1994;13:4953–4962. doi: 10.1002/j.1460-2075.1994.tb06822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hillmen P, Bessler M, Crawford D H, Luzzatto L. Blood. 1993;81:193–199. [PubMed] [Google Scholar]
- 30.Lansdorp P M, Verwoerd N P, van de Rijke F M, Dragowska V, Little M T, Dirks R W, Raap A K, Tanke H J. Hum Mol Genet. 1996;5:685–691. doi: 10.1093/hmg/5.5.685. [DOI] [PubMed] [Google Scholar]
- 31.Henderson S, Allsopp R, Spector D, Wang S S, Harley C. J Cell Biol. 1996;134:1–12. doi: 10.1083/jcb.134.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrison D E, Astle C M. J Exp Med. 1982;156:1767–1779. doi: 10.1084/jem.156.6.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siminovitch L, Till J E, McCulloch E A. J Cell Comp Physiol. 1964;64:23–31. doi: 10.1002/jcp.1030640104. [DOI] [PubMed] [Google Scholar]
- 34.Ogden D A, Micklem H S. Transplantation. 1976;22:287–293. doi: 10.1097/00007890-197609000-00010. [DOI] [PubMed] [Google Scholar]
- 35.Harrison D E, Astle C M, Delaittre J A. J Exp Med. 1978;147:1526–1531. doi: 10.1084/jem.147.5.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harrison D E, Stone M, Astle C M. J Exp Med. 1990;172:431–437. doi: 10.1084/jem.172.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrison S J, Wandycz A M, Akashi K, Globerson A, Weissman I L. Nat Med. 1996;2:1011–1016. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- 38.Goldman J. Blood. 1995;85:1413–1415. [PubMed] [Google Scholar]
- 39.Emerson S G. Blood. 1996;87:3082–3088. [PubMed] [Google Scholar]