

Abstract
We have found that human organs such as colon, lung, and muscle, as well as their derived tumors, share nearly all mitochondrial hotspot point mutations. Seventeen hotspots, primarily G → A and A → G transitions, have been identified in the mitochondrial sequence of base pairs 10,030–10,130. Mutant fractions increase with the number of cell generations in a human B cell line, TK6, indicating that they are heritable changes. The mitochondrial point mutation rate appears to be more than two orders of magnitude higher than the nuclear point mutation rate in TK6 cells and in human tissues. The similarity of the hotspot sets in vivo and in vitro leads us to conclude that human mitochondrial point mutations in the sequence studied are primarily spontaneous in origin and arise either from DNA replication error or reactions of DNA with endogenous metabolites. The predominance of transition mutations and the high number of hotspots in this short sequence resembles spectra produced by DNA polymerases in vitro.
In 1958, Seymour Benzer and Ernest Freese demonstrated that the distribution of point mutations with regard to kind and position depended on the mutagen used (1). Since then, the specificity of mutational spectra for many mutagens has been extended to bacteria and yeast and rodent and human cells (ref. 2 and references therein and refs. 3 and 4). In particular it has been recognized that spontaneous mutation also is characterized by a reproducible set of predominant point mutations (1, 5).
All reports of mutational spectra have involved sequences in genes for which selective conditions could be devised, and none has included mitochondrial sequences. Our recent development of means to measure point mutations in certain DNA sequences at mutant fractions as low as 10−5 without reference to phenotypic changes gave us the opportunity to examine mitochondrial point mutational spectra (6). Our first and most important question was whether mitochondrial mutations accumulated during a human lifetime were different from those that arise in human cell cultures in the absence of added xenobiotic chemicals. If they were different, it would perhaps point to a particular environmental cause of mitochondrial mutation. If not, it would suggest focusing on spontaneous mechanisms of mutation as a high priority in understanding the reasons for mitochondrial mutagenesis.
We recognized that the most significant source of mutations in mtDNA would probably not be the same as for nuclear DNA. Nevertheless, mitochondrial mutagenesis itself has been implicated in several important areas of human health. Accumulation of mitochondrial mutations may be a molecular clock for organ degeneration in the aging process (7, 8). Also, human diseases including Leber hereditary optic neuropathy, myoclonic epilepsy and ragged-red fiber disease, inherited adult-onset diabetes, deafness, and many others arise as a result of inherited mitochondrial mutations (8).
MATERIALS AND METHODS
Cells and Tissue Samples.
The human B cell line TK6 (9) was recloned and maintained in exponential growth phase by daily dilution of a 600-ml culture at 106 cells/ml for more than 400 generations. Surgical discard samples of colon and muscle were provided by S. Singer of the Brigham and Women’s Hospital, Boston. Lung cell samples were brush biopsies obtained via bronchoscopy of volunteers at the University of Rochester Medical Center, New York, under the supervision of M. Frampton and M. Utell. Lung cells also were obtained from organ donors through the University of Rochester Medical Center and Brigham and Women’s Hospital.
Mutational Spectrometry.
Mutational spectrometry was performed as described (6). Our approach was based on the seminal work of Fischer and Lerman (10) who introduced the use of denaturing gradient gel electrophoresis as a means to separate single base change mutants lying within short (100-bp) contiguous isomelting DNA domains. The mitochondrial sequence of base pairs 10,030–10,130 is such a domain. In brief, DNA was isolated by ethanol precipitation after proteinase and RNase digestion of cells or macerated tissue. After restriction digestion with RsaI and DdeI to liberate the desired fragment (base pairs 10,009–10,231), the mutant homoduplexes were separated from wild-type DNA by constant denaturant gel electrophoresis (11). Fractions containing enriched mutants were amplified with Pfu DNA polymerase (Stratagene) and subsequently further enriched by constant denaturant capillary electrophoresis (CDCE) (12). The enriched mutant fractions were amplified with fluorescein-labeled PCR primers, and individual peaks present in the mutational spectra were observed and measured by using laser-induced fluorescence and high resolution CDCE conditions (13). Purified mutants that would not interfere with visualization of the spectrum were added to the samples before restriction digestion at precise fractions of the total copy number to serve as internal standards. Thus, we were able to estimate the mutant fraction that each peak represented in the initial samples as the ratio of the area under that peak to the area under the internal standard. Initially, all peaks were isolated and individually sequenced. When it became clear that all samples were expressing the same set of hotspots, we used comigration with tetramethylrhodamine-labeled authentic mutant standards to identify peaks. The identity of many peaks also was confirmed by capillary hybridization (6).
To test the reproducibility of the method, we compared the set of ratios of peak areas to internal standard peak areas in replicate samples. DNA isolated from a single lung sample was divided into two parts, and both samples were subjected to mutational spectra procedures. The resulting homoduplex spectra are shown in Fig. 1 A and B. The reproducibility is excellent; the areas under the peaks representing each of the mutants show little variability.
Figure 1.
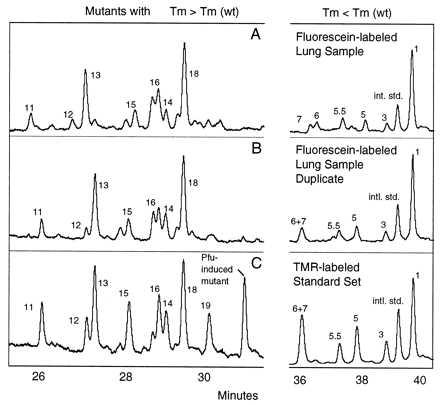
Reproducibility of the procedure and identification of mutants. Restriction-digested DNA containing the equivalent of ≈3 × 108 copies of the mitochondrial target sequence was divided into two samples, and each was doped at 10−4 with two internal standards. A higher melting temperature mutant originally isolated from human tissue, mutant 13 (T → C, base pair 10,072), and a constructed mutant with a lower melting temperature than wild type (G → A, base pair 10,040) were used as internal standards. The samples were subjected to the mutational spectrometry procedure as described in Materials and Methods. (A and B) The results of CDCE separations of mutant homoduplexes isolated from duplicate DNA samples. The x axis reflects the time since the beginning of the run at which the peak reaches the detector and the y axis marks the relative intensity of fluorescence. (Left) Mutants with melting temperatures higher than wild type. (Right) Mutants with melting temperatures lower than wild type. The fluorescein-labeled samples were separated at the same time and in the same capillary as the tetramethylrhodamine (TMR)-labeled standard set containing all previously identified peaks. (C) TMR-labeled standard set separated simultaneously with the sample in B. Peaks in the samples were identified based on comigration with TMR-labeled standards and are numbered accordingly.
Mutant peaks numbered 1, 2, 3… have homoduplex melting temperatures below that of the wild-type homoduplex and thus move more slowly than the wild type through the separation zone in CDCE. These are mostly G → A transitions. Mutant peaks 11, 11.3, 11.5, 12, 13… have higher melting temperatures than the wild type and thus move more rapidly through the CDCE separation zone. These consist mainly of A → G transitions.
Tests for Errors.
Of the peaks in the set of 17 we report, only one (peak 6) was found to arise from the PCR process itself (14). When constant denaturant gel electrophoresis-purified wild-type material devoid of mutants was mixed with rat cells as an overall negative control for the procedure, no prominent signals were observed. Furthermore, for most of the mutants in selected samples, we checked both DNA strands independently to discover whether our signals could have arisen from mismatch intermediates in DNA replication or endogenous DNA adducts that were converted to mutations during PCR. This analysis revealed that neither mismatch intermediates nor adducts contributed significantly (>10%) to the signals observed from human tissues with the exception of one peak (peak 3). Finally, we tested whether the mutant fractions increased with cell divisions in TK6 cells. Were our signals to arise from endogenous DNA damage or from unresolved mismatch intermediates, the observed mutant fractions would be expected to remain constant in growing cell cultures. Such an argument would not necessarily apply to observations in aging humans because nonreplicating cells could accumulate damage without the dilution of cell division. The monotonic increase in mutant fraction with cell division number for the G → A hotspot mutation at base pair 10,068 (peak 1) is shown in Fig. 5B.
Figure 5.
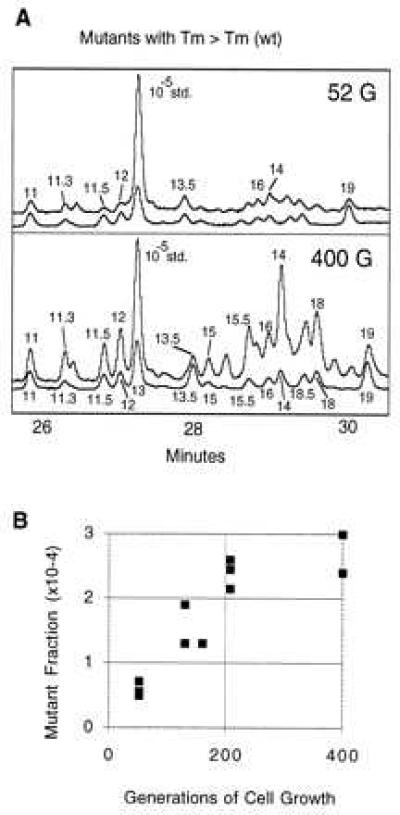
Mutational spectra of cultured human cells. (A) TK6 cells grown for 52 and 400 generations were sampled. DNA was isolated from an aliquot of cells and subjected to mutational spectrometry. CDCE runs of the mutational spectra observed are shown. The axes are as in Fig. 1. Only the mutants with melting temperatures higher than wild type are shown. (B) Increase in mutant fraction of mutant 1 (G → A, base pair 10,068) with generations of human cells grown in culture. TK6 cells were grown in spinner cultures and sampled at the number of generations designated on the horizontal axis. Mutant fraction is plotted on the y axis. Individual points represent separate mutant fraction determinations from independent cell aliquots taken from a single culture.
RESULTS
Normal Human Tissues.
Fig. 2 shows the set of mutant peaks observed on the final CDCE run when mutational spectra analysis was applied to four samples of normal tissues: colon, muscle, and lung epithelium (two samples). Each peak that has been identified is designated by number. The specific mutations designated by numbers in Fig. 2 are shown in Fig. 3, which provides the kind of mutation and the position of the 17 mutational hotspots in the context of the local DNA sequence. Also shown are the average mutant fractions for all peaks in many of the lung epithelium samples analyzed to date.
Figure 2.
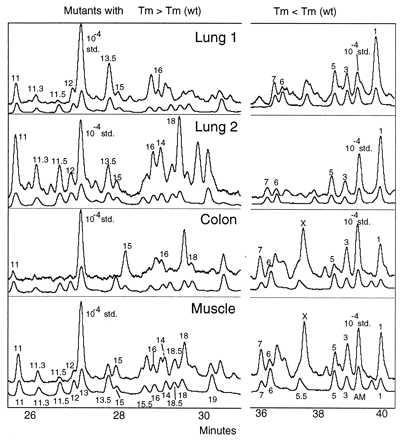
Mutational spectra of lung, colon, and muscle samples. DNA was isolated from ≈1 g of epithelial cells from a human colon, 1 g of human muscle tissue, and two samples of 106 human bronchial epithelial cells each. DNA was restriction digested and mixed with internal standards, and the mutational spectra were determined. Depicted are separations of fluorescein-labeled samples along with the coseparated tetramethylrhodamine-labeled authentic standards (lower of each pair of curves) for samples of two lungs, colon, and muscle. The x and y axes and labels are as in Fig. 1. “X” marks a peak that has not yet been sequenced.
Figure 3.
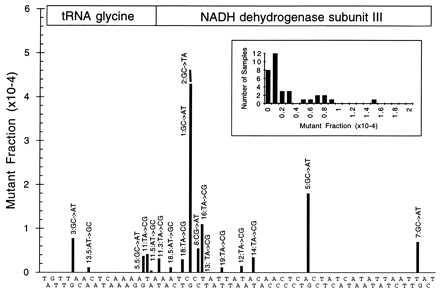
Position, type of mutation, and frequency of hotspot mutations. The type of mutation and its position for each of the mutants sequenced so far are shown. The target sequence is printed along the x axis to provide information on sequence context (the sequence is staggered for clarity). The target sequence starts with T(10,031) and ends with T(10,129). The positions of the two genes encoded by this sequence, tRNAgly and NADH dehydrogenase subunit III, are designated. The height of each peak reflects the average mutant fraction of this peak in most of the lung samples analyzed to date. Note that mutants 1 and 2 are different substitutions at the same base pair. Also note that mutant 13 (T → C, base pair 10,072) was used as an internal standard to measure the size of other peaks. We estimate the average mutant fraction for mutant 13 in tissues to be 2 × 10−5 based on four samples. Error bars are not shown in this figure because the data are not normally distributed (see Discussion). Instead, the Insert demonstrates the distribution of mutant fractions among bronchial epithelial samples for a representative mutant, p11.3.
As shown in Fig. 3, the sequence studied comprises fragments of both the tRNAgly and the NADH dehydrogenase subunit III genes. No differences in the number or kind of mutations present in the two genes were observed. Both contained the A → G and G → A mutations that dominate the spectrum.
The 101 base pairs scanned (base pairs 10,030–10,130) contained 27 GC base pairs; 7 of 17 hotspots identified originated with GC base pairs. Inspection of the hotspots’ nearest neighbors found no significant deviation from the expectation of chance.
Most members of the set of 17 mutants have been observed in each of the samples analyzed to date, but the variation in mutant fractions for individual hotspots among samples is large. As a result of this variation, in some samples, certain mutants displayed low mutant fractions indistinguishable from the background (e.g., p18 in Fig. 2, Lung 1). In other samples, these same or other mutants displayed mutant fractions much higher than the mean for that mutant, which resulted in large peaks (p18 in Fig. 2, Lung 2). A number of other peaks appeared to recur among samples, but these have not yet been isolated and sequenced. Such large variation is not a result of irreproducibility of our procedures. Indeed, when mutational spectra of the same sample were measured in replicate, the results were quite reproducible (compare spectra A and B in Fig. 1, where the DNA isolated from a single lung sample was split into two parts and each was used to measure a mutational spectrum). The basis of the variability between samples is discussed below (see Discussion). Despite quantitative variability, the overwhelming majority of mutants in the spectra of different samples and different tissues fell within the same set of 17 mutations.
Human Tumors.
Fig. 4 shows the set of mutant peaks observed for samples derived from tumors of the colon and muscle. The same set of hotspot mutations as in normal tissues was observed in these tumors. As in the case of normal tissues, replicate assays on the same sample derived by splitting the sample after DNA isolation were essentially identical.
Figure 4.
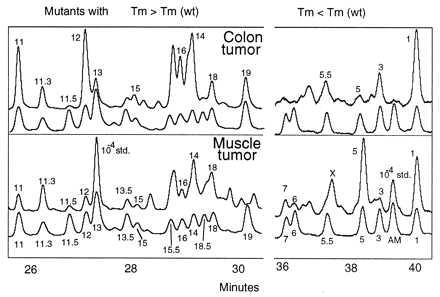
Mutational spectra of colon and muscle tumor samples. The mutational spectra were determined for colon and muscle tumor samples of 1 g and 20 mg, respectively. Shown are the CDCE separations of the mutational spectra for colon and muscle tumor samples as well as the coseparations of the tetramethylrhodamine-labeled set (lower curve of each pair of curves). The axes and labels are as in Fig. 1.
Human Cells in Culture.
Fig. 5A shows the set of mutant peaks for a culture grown from a single cell at 52 and 400 successive generations. Although the mutants observed in tissue samples were present in the cultured cells, the mutant fractions were lower in cells at this number of generations after cloning than in tissues (note change in the size of the internal standard peak area between Figs. 1 and 5A). The exception is peak 1, which was at approximately the same mutant fraction in tissues and cultured cells at 100 generations. The ratio of peak 1 area to that of other peaks was ≈10-fold greater in TK6 cells at 100 generations than in samples derived from human tissues. The relatively larger size of peak 1 made it a good candidate to study the increase in mutant fraction with cell division (Fig. 5B). Peak 1, a G → A transition at base pair 10,068, increased almost linearly relative to an internal control up to 200 generations. The mutant fraction did not increase linearly from 200 to 400 generations. One possible reason is that, by 400 generations of continuous culture, a faster growing subpopulation of cells may have displaced the other cells in the culture, thus making mutant fractions no longer reliable, as has been documented (15). Other peaks also rose monotonically with cell division (Fig. 5A), but the relatively low mutant fractions of these peaks prevented us from obtaining a satisfactory degree of statistical precision (16).
DISCUSSION
Based on these observations and previous tests for experimental bias (6), we conclude that the samples we have investigated contain a remarkably similar set of hotspot point mutations with regard to the position and kinds of mutations in the mitochondrial sequence examined. Our study included healthy tissues, tumor samples, and a human B cell line. There are, however, significant quantitative differences in the hotspot mutant fractions among the tissue samples, even among samples taken from the same organ. We recognize that comparing the mutational spectra for a given colon sample and a given lung sample will show significant differences in the frequencies of specific mutants; however, because we observed differences in mutant frequencies within a single organ that were similarly large, we focused on the observations of the same set of mutants in different organs and in cultured cells to support our conclusion that similar processes are creating these mutants and that they are likely to be spontaneous in nature.
Comparison of Mutation Rate for Mitochondrial and Nuclear Mutations in Vivo and in Vitro.
The spontaneous mutation rate in mtDNA is apparently more than two orders of magnitude higher than that observed in nuclear sequences both in vitro and in vivo. In vivo, the mutant fractions we observed in mtDNA in human tissues can be compared with the mutant fractions observed for selectable genes in nuclear DNA. In humans 50 years of age, the approximate age of the individuals who donated their surgical discard samples for our research, the mutant fractions at the nuclear genes encoding hypoxanthine–guanine phosphoribosyl transferase and HLA in lymphocytes have been reported to be ≈1.5 × 10−5, of which some 85% are point mutations (17, 18). Assuming a target size of ≈1000 bp for these genes, the estimated nuclear mutant fraction/base pair is ≈1 × 10−8. In the 100-bp fragment assayed for mitochondrial mutations, the sum of all peaks yielded a total mutant fraction of ≈3 × 10−4 in tissue samples or ≈3 × 10−6 per base pair. It would thus appear that mitochondrial in vivo mutant fractions were several hundred-fold higher than those reported for nuclear in vivo mutant fractions. Assuming that these mutant fractions reflected constant mutation rates over a lifetime and that mtDNA doubled once per cell division, we conclude that mitochondrial mutation rates were approximately several hundred-fold greater than nuclear gene mutation rates.
In terms of in vitro comparisons, the hprt spontaneous mutation rate in TK6 cells was ≈10−7 mutations/base pair generation (5). Approximately 60% of these did not involve large deletions and were thus presumably point mutants (19). Using an estimated target size of 1000 bp, this rate of 6 × 10−11 point mutations per base pair per cell division is estimated for this nuclear gene. In the mtDNA of TK6 cells, our studies showed an overall point mutation rate of ≈2 × 10−6 per cell division for a 100× bp target or an average mitochondrial point mutation rate of 2 × 10−8 per base pair per cell division. If we assume that mtDNA doubles once per cell division, we estimate that the mitochondrial mutation rate in this sequence is several hundred-fold times the nuclear rate in hprt in vitro as well as in vivo.
What Processes Create the Same Set of Mitochondrial Point Mutations in These Organs, Tumors, and Cultured Cells?
The appearance of the same kinds and positions of hotspot mutations in tissues, such as colon and lung, and human cells grown in the laboratory in the absence of added xenobiotic chemicals (compare Figs. 2, 4, and 5A) suggests to us that the mutations we observed were most likely to be “spontaneous” in origin, that is, not induced by environmental nonnutritional factors. The observed mutations could arise either from DNA replication error or from replicative bypass of DNA adducts created by reaction with endogenous factors, for instance, the elements of the oxygen and NO cascades.
In terms of distinguishing between replication errors vs. endogenous adducts as the cause of the mutations observed, we noted that a spectrum dominated by A → G and G → A transitions was reminiscent of the spectrum produced by the Klenow fragment of DNA polymerase I from Escherichia coli (20). It is also suggestive of the types of spectrum observed in the lacI gene in E. coli that are defective in mismatch repair (21, 22). The average mutation rate of 3 × 10−8 per base pair per division is, however, some 200 times lower than the error rate of any DNA polymerase studied in pure form. We also note that no frameshift mutations were detected despite the presence of a run of six consecutive adenines and one run of three consecutive guanines. Such a finding would be consistent with reports that the DNA polymerase responsible for replicating human mtDNA, polymerase γ, has been observed in vitro to make frameshifts less frequently than human polymerases α and β (23). However, it is also possible that some frameshift mutations might be missed in our procedure because some frameshift mutants have homoduplex melting temperatures close to the wild type.
On the other hand, it is certainly reasonable to continue to consider hypotheses regarding endogenous chemical reaction products as premutagenic lesions because these would accumulate as a function of time in vivo and in vitro, and our experiments do not differentiate between cell division and time dependence. We favor the replication error hypothesis on the basis of the predominance of transitional hotspots found in high number in this short sequence, which is similar to what we have seen in studies of DNA polymerization in vitro (24, 25) and is different from the point mutations induced by oxidative stressors in a human cell nuclear gene (5). However, the data contained in this paper do not allow us to distinguish between replicative errors and endogenous adducts or both as the causes of mitochondrial point mutations.
What Is the Basis of the Large Variation in Individual Hotspot Mutant Fractions Among Samples from the Same Organ or Tumor?
Shown in the Fig. 3 insert is a histogram of the number of bronchial epithelial cell samples containing specific amounts of a sample mitochondrial mutant, p11.3. This distribution is far from normal in that several samples display mutant fractions significantly above the mean. We interpret this as an indication that mutant-rich cells may be distributed as clusters within a tissue. According to this interpretation, samples containing unusually high mutant fractions may have included a cluster of mutant-rich cells. Such a cluster could be a “turnover unit” in which one stem cell gives rise to descendant transitional cells and terminal cells. All cells in such a unit would inherit mutant mitochondrial copies from the stem cell, so a single mutant-rich stem cell could result in a mutant-rich turnover unit. We plan to explore this further in a separate paper (H.A.C., K.K., Pablo Herrero, and W.G.T., unpublished work). The clustering of mitochondrial mutants within individual muscle fibers has been observed by others both by in situ hybridization to detect deleted copies of mRNA (26) and by histochemical staining for cytochrome c oxidase-deficient organelles (27). Clustering of mitochondrial haplotypes within colonic turnover units recently was observed in mice (28).
Relationship to Previous Reports.
Large deletions of mtDNA have been discovered to show tissue-specific differences in frequency (29, 30). The levels of deleted mtDNA also were observed to increase with age (29–32). In one report, the fraction of mtDNA that was deleted was 5.5 × 10−4 in muscle, 1 × 10−4 in heart, and 0.5 × 10−4 in spleen, and much lower levels or no levels were detectable in liver, kidney, and lung in an 84-year-old man (29). In contrast, we found that the point mutant fractions in the tissues examined, colon, muscle, and lung, were similar.
By using allele-specific PCR, mitochondrial point mutations associated with pathologic conditions also have been found in several undiseased organs. A mutant frequency of 2.4% was reported for an A → G mutation at bp 8344 associated with MERRF (myoclonic epilepsy and ragged-red fiber disease) (33). For an A → G transition at bp 3243 associated with MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), the reported frequencies are 0.1% (34) and from an undetectable level to 0.7% (35). These frequencies are higher than those we observed for mitochondrial mutants in regions of mtDNA that have not been directly implicated in disease.
Studies of the types of mitochondrial point mutations observed as polymorphisms have shown that both G → A and A → G occur more frequently than transversions (36, 37). This is consistent with our observations of predominantly transition mutations.
CONCLUSION
Our data indicate that mitochondrial point mutations in the organs and sequence studied are spontaneous in nature. The three organs sampled as well as derived tumors displayed a spectrum similar to that observed in human cells growing in the laboratory, suggesting a common pathway. The hypothesis that environmental mutagens are important contributors to mitochondrial point mutagenesis no longer seems tenable. Our data did not permit us to distinguish between the pathways of misincorporation error in DNA replication or replicative bypass of DNA reaction products with endogenous chemicals such as the elements of the intracellular oxygen or nitric oxide cascades.
Acknowledgments
We acknowledge the assistance of Dr. S. Singer (Brigham and Women’s Hospital) and Drs. M. Frampton, A. Torres, and M. Utell (University of Rochester) in obtaining human tissue samples. The authors also thank L. Marcelino (Massachusetts Institute of Technology) and Prof. G. Hu (Shanghai Cell Biology Institute) for sharing unpublished results. Our development of the CDCE technology was the result of a long-standing collaboration with Prof. Barry Karger and Dr. Frantisek Foret of the Barnett Institute of Northeastern University. This work was supported by grants from the National Institute for Environmental Health Sciences: Mutagenic Effects of Air-Borne Toxicants (P01-ES07168), Superfund Basic Research (P42-ES04675), and Genetics and Toxicology (PO1-ES03926) and by a grant from the U.S. Department of Energy, Office of Health and Environmental Research (DE-FGO2-86ER60448), Comparative Mutagenesis of Human Cells In Vitro and In Vivo.
ABBREVIATION
- CDCE
constant denaturant capillary electrophoresis
References
- 1.Benzer S, Freese E. Proc Natl Acad Sci USA. 1958;44:112–119. doi: 10.1073/pnas.44.2.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thilly W G. Annu Rev Pharmacol Toxicol. 1990;30:369–385. doi: 10.1146/annurev.pa.30.040190.002101. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong J D, Kunz B A. Proc Natl Acad Sci USA. 1990;87:9005–9009. doi: 10.1073/pnas.87.22.9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roy A, Fuchs R P. Mol Gen Genet. 1994;245:69–77. doi: 10.1007/BF00279752. [DOI] [PubMed] [Google Scholar]
- 5.Oller A R, Thilly W G. J Mol Biol. 1992;228:813–826. doi: 10.1016/0022-2836(92)90866-i. [DOI] [PubMed] [Google Scholar]
- 6.Khrapko K, Coller H, André P, Li X-C, Foret F, Belinky A, Karger B L, Thilly W G. Nucleic Acids Res. 1997;25:685–693. doi: 10.1093/nar/25.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bandy B, Davison A J. Free Radical Biol Med. 1990;8:523–539. doi: 10.1016/0891-5849(90)90152-9. [DOI] [PubMed] [Google Scholar]
- 8.Wallace D C. Proc Natl Acad Sci USA. 1994;91:8739–8746. doi: 10.1073/pnas.91.19.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skopek T R, Liber H L, Penman B W, Thilly W G. Biochem Biophys Res Commun. 1978;84:411–416. doi: 10.1016/0006-291x(78)90185-7. [DOI] [PubMed] [Google Scholar]
- 10.Fischer S G, Lerman L S. Proc Natl Acad Sci USA. 1983;80:1579–1583. doi: 10.1073/pnas.80.6.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hovig E, Smith-Sørensen B, Brøgger A, Børresen A-L. Mutat Res. 1991;262:63–71. doi: 10.1016/0165-7992(91)90108-g. [DOI] [PubMed] [Google Scholar]
- 12.Khrapko K, Hanekamp J S, Thilly W G, Belenkii A, Foret F, Karger B L. Nucleic Acids Res. 1994;22:364–369. doi: 10.1093/nar/22.3.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khrapko K, Coller H, Thilly W G. Electrophoresis. 1996;17:1867–1874. doi: 10.1002/elps.1150171211. [DOI] [PubMed] [Google Scholar]
- 14.André P, Kim A, Khrapko K, Thilly W G. Genome Methods. 1997;7:843–852. doi: 10.1101/gr.7.8.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kubitschek H E. Introduction to Research with Continuous Cultures. Englewood Cliffs, NJ: Prentice–Hall; 1970. [Google Scholar]
- 16.Oller A R, Rastogi P, Morgenthaler S, Thilly W G. Mutat Res. 1989;216:83–97. doi: 10.1016/0165-1161(89)90001-0. [DOI] [PubMed] [Google Scholar]
- 17.Grist S A, McCarron M, Kutlaca A, Turner D R, Morley A A. Mutat Res. 1992;266:189–196. doi: 10.1016/0027-5107(92)90186-6. [DOI] [PubMed] [Google Scholar]
- 18.Robinson D R, Goodall K, Albertini R J, O’Neill J P, Finette B, Sala-Trepat M, Moustacchi E, Tates A D, Beare D M, Green M H L, Cole J. Mutat Res. 1994;313:227–247. doi: 10.1016/0165-1161(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 19.Gennett I N, Thilly W G. Mutat Res. 1988;201:149–160. doi: 10.1016/0027-5107(88)90121-2. [DOI] [PubMed] [Google Scholar]
- 20.Keohavong P, Thilly W G. Proc Natl Acad Sci USA. 1989;86:9253–9257. doi: 10.1073/pnas.86.23.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaaper R M, Dunn R L. Proc Natl Acad Sci USA. 1987;84:6220–6224. doi: 10.1073/pnas.84.17.6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leong P M, Hsia H C, Miller J H. J Bacteriol. 1986;168:412–416. doi: 10.1128/jb.168.1.412-416.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunkel T A. J Biol Chem. 1985;260:12866–12874. [PubMed] [Google Scholar]
- 24.Keohavong P, Thilly W G. Proc Natl Acad Sci USA. 1989;86:9253–9257. doi: 10.1073/pnas.86.23.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keohavong P, Ling L, Dias C, Thilly W G. PCR Methods Applications. 1993;2:288–292. doi: 10.1101/gr.2.4.288. [DOI] [PubMed] [Google Scholar]
- 26.Shoubridge E A, Karpati G, Hastings K E M. Cell. 1990;62:43–49. doi: 10.1016/0092-8674(90)90238-a. [DOI] [PubMed] [Google Scholar]
- 27.Müller-Höcker J, Schneiderbanger K, Stefani F H, Kadenbach B. Mutat Res. 1992;275:115–124. doi: 10.1016/0921-8734(92)90016-i. [DOI] [PubMed] [Google Scholar]
- 28.Jenuth J P, Peterson A C, Shoubridge E A. Nat Genet. 1997;16:93–95. doi: 10.1038/ng0597-93. [DOI] [PubMed] [Google Scholar]
- 29.Simonetti S, Chen X, DiMauro S, Schon E A. Biochim Biophys Acta. 1992;1180:113–122. doi: 10.1016/0925-4439(92)90059-v. [DOI] [PubMed] [Google Scholar]
- 30.Yen T-C, Su J-H, King K-L, Wei Y-H. Biochem Biophys Res Commun. 1991;178:124–131. doi: 10.1016/0006-291x(91)91788-e. [DOI] [PubMed] [Google Scholar]
- 31.Cortopassi G A, Arnheim N. Nucleic Acids Res. 1990;18:6927–6933. doi: 10.1093/nar/18.23.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linnane A W, Baumer A, Maxwell R J, Preston H, Zhang C, Marzuki S. Biochem Int. 1990;22:1067–1076. [PubMed] [Google Scholar]
- 33.Muenscher C, Rieger T, Muller-Hocker J, Kadenbach B. FEBS Lett. 1993;317:27–30. doi: 10.1016/0014-5793(93)81484-h. [DOI] [PubMed] [Google Scholar]
- 34.Zhang C, Linnane A W, Nagley P. Biochem Biophys Res Commun. 1993;195:1104–1110. doi: 10.1006/bbrc.1993.2158. [DOI] [PubMed] [Google Scholar]
- 35.Pallotti F, Chen X, Bonilla E, Schon E A. Am J Hum Genet. 1996;59:591–602. [PMC free article] [PubMed] [Google Scholar]
- 36.Aquadro C F, Greenberg B D. Genetics. 1983;103:287–312. doi: 10.1093/genetics/103.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horai S, Hayasaka K. Am J Hum Genet. 1990;46:828–842. [PMC free article] [PubMed] [Google Scholar]