

Abstract
Oligosaccharyl transferase (OT) is a multi-subunit enzyme that catalyzes N-linked glycosylation of nascent polypeptides in the lumen of the endoplasmic reticulum. In the case of Saccharomyces cerevisiae, OT is composed of nine integral membrane protein subunits. Defects in N-linked glycosylation cause a series of disorders known as congenital disorders of glycosylation (CDG). The C-terminal domain of Stt3p subunit has been reported to contain the acceptor protein recognition site and/or catalytic site. We report here the subcloning, overexpression, a robust but novel method of production of pure C-terminal domain of Stt3p at 60∼70 mg/L in E. coli. CD spectra indicate that the C-terminal Stt3p is highly helical and has a stable tertiary structure in SDS micelles. The well dispersed 2D {1H-15N}-HSQC spectrum in SDS micelles indicates that it is feasible to determine the atomic structure by NMR. The effect of the conserved D518E mutation on the conformation of the C-terminal Stt3p is particularly interesting. The comparative analysis of the fluorescence and NMR data of the mutant and the wild-type C-terminal domain of Stt3p revealed that the replacement of the key residue Asp518, which is located within the WWDYG signature motif (residues 516-520), led to a distinct tertiary structure, even though both proteins have similar overall secondary structures. This observation strongly suggests that Asp518, which was previously proposed to primarily function as a catalytic residue, also plays a critical structural role. Moreover, the activity of the protein was confirmed by Saturation Transfer Difference (STD) and NMR titration studies.
Keywords: N-glycosylation, NMR, Oligosaccharyl Transferase, Stt3p, Integral Membrane Protein, Saturation Transfer Difference
N-linked glycosylation, the most ubiquitous protein modification in eukaryotic cells, is catalyzed by oligosaccharyl transferase (OT). OT is a remarkably complex multisubunit enzyme that, in the case of the yeast Saccharomyces cerevisiae, contains nine non-identical integral membrane protein (IMP) subunits: Ost1p, Ost2p, Ost3p, Ost4p, Ost5p, Ost6p, Stt3p, Wbp1p, and Swp1p (1). Among these, Ost3p and Ost6p are homologous, interchangeable subunits, while Stt3p, Wbp1p, Swp1p, Ost1p, and Ost2p are essential for the viability of the cell (2). In the central reaction, OT transfers a preassembled oligosaccharide moiety from a dolichol-linked pyrophosphate (Dol-PP-oligosaccharide) donor onto the side chain of the Asn of the nascent polypeptide chain specified by the N-X-T/S consensus sequence, where X can be any amino acid except proline (1, 3).
N-glycoproteins have been implicated in a multitude of cellular processes including immune response, intracellular targeting, intercellular recognition, protein folding, and protein stability (4, 5). Human genetic defects in the N-linked glycosylation pathway cause 18 inherited human disorders known as Congenital Disorders of Glycosylation (CDG, 6). These conditions affect multiple organs with severe clinical manifestations including mental retardation, developmental delay, hypoglycemia, liver dysfunction, etc. Complete loss of N-linked glycosylation is lethal to all eukaryotic organisms.
Given its extreme importance, numerous efforts have been made in understanding the structure and mechanisms of this enzyme complex over the last few decades. However, very limited success has been achieved with regard to the determination of structures at atomic level. High-resolution structures of only two OT subunits have been reported so far. One is the NMR structure of 36-residue Ost4p, a non-essential yeast OT subunit (7). The other is the crystal structure of C-terminal domain of Stt3p from an archaea, which has very little sequence similarities to the eukaryotic Stt3p (8). As a result, even the most fundamental questions concerning the function of each subunit and the mechanisms of catalysis, still remain unanswered. The major bottleneck in research progress is due primarily to the inherent difficulties associated with the preparation of milligram quantities of IMPs, which are necessary for structure/function studies. Another barrier is finding a suitable detergent that can solubilize the protein during and after purification steps, since structure/function studies of IMPs typically rely upon detergent micelles as faithful mimics of the native lipid bilayer. Moreover, there is no unique detergent that can solubilize every IMP and provide a stable environment for structure/function studies. Thus, finding a suitable detergent for an IMP is still very much a process of trial and error (9).
Stt3p is the only conserved subunit in all three domains of life (10). During the last several years, overwhelming amount of evidences have been obtained indicating the direct involvement of C-terminal domain of Stt3p in the catalytic process of glycosylation in eukaryotes (11-13), the bacterium Campylobacter jejuni (14), and the archaea Pyrococcus furiosus (8). The most direct evidence demonstrating Stt3p as the catalytic subunit of the eukaryotic OT has come from the finding that PglB, a Stt3p homolog in Campylobacter jejuni bacteria, catalyzes N-linked glycosylation activity by itself. Moreover, the expression of PglB in E. coli could enable N-linked glycosylation activity in the E. coli host unless point mutations were introduced into the WWDYG motif in PglB (15). Similarly, expression of the Leishmania major Stt3p homolog in yeast not only complements the yeast STT3 deletion, but also is able to replace the whole OT complex of yeast (16, 17).
We report here the cloning, overexpression, purification and biophysical characterization of the C-terminal domain of Stt3p. To our knowledge, this is the first report of recombinant overexpression of a eukaryotic OT subunit in E. coli. Pure and homogeneous recombinant C-terminal domain of Stt3p was produced at a level of 60∼70 mg/L of bacterial culture. Particularly, we introduced a novel and simple, yet robust purification protocol, involving His-Tag Nickel Affinity chromatography without the use of any imidazole. This novel protocol has the potential to become a versatile method for the purification of other IMPs. We would also like to emphasize here that the C-terminal domain of Stt3p (466-718) is only soluble in detergent micelles and behaves like a membrane protein. It was previously reported that this domain is a hydrophilic lumenal domain (10, 18) based on the results of topology reporter studies. However, several other TM predication programs such as DAS (19), TopPred (20), TMpred (21) and SPLIT 4.0 (22) predict residues 564-584 to be a TM domain both for full length and the C-terminal domain (466-718) of Stt3p. The Kyte-Doolittle algorithm predicts the presence of at least one transmembrane region (564-580) located within the C-terminal domain of Stt3p. It is also very clear from our protein purification work that this domain (466-718) is unusually hydrophobic in character. Whether this particular domain (466-718) contains any transmembrane helices or only a few membrane embedded residues will be clear when we determine the topology and 3D solution structure.
Biophysical characterization of the C-terminal domain of Stt3p indicates that sodium dodecyl sulfate (SDS) is the most suitable detergent for the structure/function studies of this protein. The C-terminal domain of Stt3p in SDS micelles is highly helical with a stable tertiary structure. It is clear from the 2D {1H, 15N}- heteronuclear single quantum coherence (HSQC) spectrum that structural characterization on the C-terminal domain of Stt3p is amenable by solution NMR.
The highly conserved WWDYG motif (516-520), in the C-terminal domain of Stt3p has been reported to play a central role in the glycosylation process. Moreover, point mutations in this motif either eliminate or sharply reduce OT activity (11). Of particular interest is the conserved D518E mutation that results in a lethal phenotype with complete loss of N-linked glycosylation function. The extra methylene group in the glutamate side chain renders the enzyme complex completely nonfunctional (11, 14). It is clear from our biophysical data that Asp518 plays a structural role in maintaining the conformational geometry of the active site of the Stt3p for the catalysis to occur. Results of circular dichroism (CD), fluorescence and NMR indicate that although the D518E mutant has an overall similar secondary structure to that of the wild-type protein, its tertiary structure is remarkably different. These observations unequivocally prove that the D518E mutation does result in a dramatic conformational change in the protein, thus interfering with its function.
Furthermore, the interaction study of the C-terminal domain of Stt3p with the acceptor substrate peptide containing the N-X-T/S consensus motif was also investigated by Saturation Transfer Difference (STD) and NMR titration by HSQC experiments to confirm the biological activity of the protein.
Materials and Methods
The detergents used in this study were sodium dodecyl sulfate (SDS) (Sigma), dodecylphosphocholine (DPC) (Anatrace), lauryl dimethylamine oxide (LDAO) (Anatrace), octyl-β-glucoside (OG) (Sigma), n-dodecyl-β-D-maltoside (DDM) (Anatrace) and digitonin (Calbiochem). For NMR studies, predeuterated detergents were used when commercially available. The perduterated detergents used were SDS (Sigma, 98% atom D) and DPC (Cambridge Isotope Laboratories, D38, >98%).
Subcloning
To clone only the C-terminal Stt3p domain protein (residues 466-718), the open reading frame (ORF) sequence was amplified using the following primers: sense, 5′-GGAATTCCATATGTCTACTTGGGTAACAAGAACTGC-3′ and antisense, 5′-CCGCTCGAGTTAGACTCTCAAGCCTAATTCAGG-3′. The yeast plasmid pRS316 containing the STT3 gene was used as the template. The fragments obtained by PCR amplification were purified, digested by Nde1 and Xho1, and cloned into the Nde1 and Xho1 restriction sites of the pET28c vector with a His tag at the N-terminus (Nde1 site). The orientation and sequence of the C-terminal Stt3p cDNA in the expression vector was confirmed by restriction mapping analysis and sequencing.
Mutagenesis
Oligonucleotides were specifically designed to introduce the D518E mutation. QuickChange site-directed mutagenesis kit (Stratagene, USA) was used to incorporate the desired mutation. The following sense and antisense primers were used wherein the sites of the mutation are italicized and underlined: 5′-GTTGCAGCGTGGTGGGAATACGGTTACCAAATGG-3′(sense), 5′-CCAATTTGGTAACCGTATTCCCACCACGCTGCAAC -3′ (antisense). Incorporation of the mutation was verified by DNA sequencing.
Overexpression of Wild-type and Mutant Proteins
The C-terminal domain (residues 466-718) of the yeast Stt3p was produced in Escherichia coli BL21(DE3)-CodonPlus cells (Strategene) using pET28c vector (Invitrogen). Expression of the N-terminal His6-tagged Stt3p in the pET28c vector was under the control of an IPTG (isopropyl-β-D-thiogalactopyranoside) -inducible promoter. Briefly, the overnight starter culture was diluted to an OD600 of 0.1 in fresh LB medium containing 50 μg/L kanamycin and were grown at 37 °C to an OD600 of 0.4-0.6. At that point, the temperature was reduced to 30 °C and protein production was induced by the addition of IPTG to a final concentration of 0.5 mM. After 4 hours, the cells were harvested by centrifugation (10,000×g) for 20 min at 4 °C, and frozen at -80°C until needed. The same protocol was followed to overexpress the D518E mutant and the overexpression level was nearly identical to that of wild-type C-terminal domain of Stt3p.
For the production of 15N-labeled C-terminal domain of Stt3p, cells were grown in M9 minimal media culture containing 0.12% 15NH4Cl (Cambridge Isotope Laboratories). All the rest of the procedures were same except that cells were grown for 8 hours after induction with IPTG before harvesting.
Preparation of Inclusion Bodies
The E. coli cells containing C-terminal domain of Stt3p wild-type (or D518E mutant) were passed through 4 cycles of freeze-thaw using liquid nitrogen and ice respectively before resuspended in B-PER solution (Pierce). The cells were then subjected to sonication (10×15s); the supernatant was removed after centrifugation at 10,000 × g for 30min. The pellet was resuspended once with 10% B-PER solution, sonicated and centrifuged again as above. The inclusion bodies were stored at -20 °C until needed.
Purification and Simultaneous Refolding of Wild-type and Mutant Proteins
The C-terminal domain of Stt3p (wild-type or D518E mutant) inclusion bodies were dissolved in denaturing buffer containing 6 M guanidine hydrochloride (Gnd-HCl), 500 mM NaCl, 25 mM imidazole in 20 mM phosphate buffer at pH 7.4. The insoluble materials were removed by centrifugation. The supernatant containing solubilized C-terminal domain of Stt3p was loaded onto the Ni-NTA column (GE Healthcare) which was pre-equilibrated with binding buffer (500 mM NaCl, 25 mM imidazole, 20 mM phosphate buffer, pH 7.4). Impurities were removed using a washing buffer (20 mM phosphate buffer, pH 7.4, 500 mM NaCl, 200 mM imidazole, and 1% triton X-100 (v/v)) several times with shaking. In order to remove imidazole and NaCl from the protein sample before elution, a final wash was followed with 20 mM phosphate buffer, pH 6.5. The absorbance of the washing was monitored by measuring OD280 until there was no apparent reading. The elution and simultaneous refolding were carried out by loading elution buffer (50 mM SDS, 1% glycerol, 20 mM phosphate buffer, pH 6.5) to the column followed by shaking for at least 2 hours. To keep the protein concentration high, the volume of elution buffer added was kept to a minimum (< 1 ml). The elution was continued until there was no more absorbance as monitored by OD280 readings. Protein concentration was calculated from the A280 using an extinction coefficient of 63083 M-1 cm-1 (23). The purity of the protein in each elution was assessed by SDS-PAGE analysis. The pure protein samples were kept at room temperature away from light.
Matrix-Assisted Laser Desorption Ionization (MALDI)-Time of Flight Mass Spectrometry
The protein sample for MALDI-TOF measurement was in 10mM SDS, 20mM ammonium acetate. The target was spotted as follows: protein sample was mixed with the matrix (10 mg/ml sinapinic acid (SA) in 4:6 methanol:water, 0.1% trifluoroacetic acid) in a 1:4 ratio for a total of 1μL and the entire solution applied to target and allowed to air dry. MALDI mass spectra were acquired on an Autoflex II TOF mass spectrometer from Bruker. The spectra were acquired in linear mode using the following settings: laser power 55%, ion source 1: 20 kV, ion source 2: 18 kV, lens: 6.50 kV, number of shots: 50, detection: 10,000 to 100000.
NMR Sample Preparation
Each sample for NMR measurement was concentrated to 0.2 mM using an Amicon Ultra-15 (MWCO = 5 kDa) centrifugal ultrafiltration cartridge. The final NMR sample was in 20 mM phosphate buffer, pH 6.5, 1 mM EDTA, 100 mM SDS and 10 % D2O (v/v). In this study, besides SDS, five other detergents were screened to find out the most suitable membrane mimetic for C-terminal Stt3p domain. These detergents are: DPC, LDAO, OG, DDM, and Digitonin. The protein samples in the above detergents were prepared by buffer exchange of the protein in SDS detergent to the desired detergent by using Amicon ultrafiltration device with a MWCO of 5 kDa. Typically, 500 μl desired detergent solution was added to 500 μl SDS-containing protein sample in Amicon Ultra-15 tube and centrifuged until there was approximately 500 μl solution left. This process was repeated 10 times for complete detergent exchange.
NMR Measurement
{1H, 15N}-HSQC spectra were acquired for both wild-type and D518E mutant of the C-terminal domain of Stt3p. NMR measurements were conducted at 308 K. Data were collected on a Bruker Avance600 MHz spectrometer fitted with a cryogenic triple-resonance probe equipped with z-axis pulsed field gradients. The data were acquired with 256 and 2048 complex points in the t1 time domain (15N dimension) and t2 time domain (1H dimension) respectively. The data were zero-filled to 512 × 4096 and apodized using a Gaussian window function prior to Fourier transformation using NMRPipe (24).
Cicular Dichrosim (CD) Spectropolarimetry
All CD experiments were performed on a JASCO J-810 automatic recording spectropolarimeter using a 0.05 cm path length quartz cell at room temperature. Wild-type C-terminal domain of Stt3p was recorded in both SDS and DPC micelles, while D518E was only recorded in SDS micelles. The buffer used was 20 mM phosphate buffer (pH 6.5). The protein concentration was 10 μM for far-UV CD measurement and 89 μM for near-UV CD measurement. Data were averaged over 100 scans with a response time of 1s, and scan speed of 100 nm min-1. CD data were converted to mean residual ellipticity (θ) by standard procedures.
Fluorescence
All fluorescence spectra were recorded on a Perkin Elmer Precisely LS 55 Luminescence spectrofluorometer. All experiments were carried out in 10 mM phosphate buffer, pH 6.5 containing 1 μM protein at 25 °C. The data were recorded by monitoring intrinsic tryptophan fluorescence (excitation at 280 nm and emission 300–500 nm).
Ligand Binding Studies by Saturation Transfer Difference (STD) NMR Spectroscopy
For STD studies, the methyl-protonated {Ile(δ1 only), Leu(13CH3, 12CD3), Val(13CH3, 12CD3)} U-{15N, 13C, 2H} labeled C-terminal domain of Stt3p was overexpressed by using the same cell lines and vectors as described earlier. Briefly, the transformed cells picked from LB agar plate were grown in 3 mL of LB medium at 37 °C for 3 h, then transferred to 25 mL of unlabeled minimal M9/H2O medium, and grown until an OD600 of ∼ 0.5. The cells were then separated from the medium by centrifugation at 3,000 rpm for 15 minutes and transferred to 100 mL of M9/D2O culture containing 0.12% (m/v) of 15NH4Cl as the sole nitrogen source and 0.4% (m/v) of 13C, 2H –glucose (Cambridge Isotope Laboratory, Andover, MA) as the sole carbon source. At OD600 ∼ 0.5, the culture was diluted to 500 mL with M9/D2O. One hour prior to induction, 35 mg of 2-keto-3,3-d2-1,2,3,4-13C-butyrate (Sigma Aldrich) and 60 mg of 2-keto-3-methyl-d3-3-d1-1,2,3,4-13C-butyrate (Sigma Aldrich) were added to medium. The expression of the protein was induced at OD600 ∼ 0.4 with 0.5 mM IPTG, and the culture was allowed to grow for an additional 11-12 h at 30 °C (final OD600 ∼ 2.0), at which point the cells were harvested by centrifugation. The protocols for cell lysis and purification were the same as described previously.
The six-residue peptide (Tyr-Asn-Ser-Thr-Ser-Cys-Am, purity >99%) was custom synthesized by Biomatik USA, LLC. Protein NMR sample for STD experiment was prepared in 20 mM phosphate buffer (pH 6.5), containing 100 mM perdeuterated SDS and 10% D2O. Protein and substrate peptide concentrations in the NMR sample were 30 μM and 300 μM respectively.
The STD measurements were performed on a Bruker Avance 600 MHz spectrometer fitted with a cryogenic triple-resonance probe equipped with z-axis pulsed field gradients at 308 K. The irradiation power was set to (γ/2π)B1 = 20 Hz, which was applied on-resonance at 0.738 ppm where no peptide signals were present, or off-resonance at 100 ppm, where no protein signals were present. In order to efficiently saturate the entire protein by spin diffusion, the saturation time was set to 10 s. A 50-ms spin-lock pulse (T1ρ filter) was used to eliminate the background protein resonances to facilitate analysis. The spectra were collected in an interleaved pseudo-2D fashion to reduce temporal fluctuations. AU program “stdsplit” from TOPSPIN 2.1 (Bruker) was used to subtract the unprocessed on- and off-resonance spectra.
Ligand Binding Studies by NMR HSQC Titrations
In order to measure dissociation constants (KD), a series of 2D {1H, 15N} HSQC spectra were collected with progressive additions of substrate peptide (Asn-Asp-Thr-NH2) to 15N-labled C-terminal Stt3p to attain molar ratios of protein to peptide of 1:0, 1:0.5, 1:1, 1:5, 1:10, 1:20, 1:35, 1:50, 1:75 and 1:100. The starting sample contained 170 μM protein in 20 mM phosphate buffer, pH 6.5, 100 mM SDS, 5% D2O, 1% glycerol and 5 mM Mg2+. The peptide with > 95 % purity was custom synthesized by Genemed Synthesis, Inc. (South San Francisco, CA, USA). NMR data collection and processing were the same as previously described. The chemical shift changes of the affected residues of the protein were plotted against the peptide concentration and fitted by Hill model in Origin 7.0 (Microcal). Chemical shift perturbations were calculated as [(δ1H)2 + (δ15N/5)2]1/2, in which δ1H and δ15N are changes in chemical shift for 1H and 15N, respectively.
Results
Overexpression and Purification of Wild-type and Mutant Proteins
It is imperative to produce milligram quantities of pure protein for structural characterization of any protein. This requirement along with the necessity of a suitable membrane mimetic, are formidable obstacles to structural characterization of IMPs. To date, there are no reports of recombinant expression of any eukaryotic OT subunit in E. coli or in any other heterologous system. This deficiency seriously impedes any biophysical and/or biochemical research on N-linked glycosylation. After many trials, we were able to overexpress C-terminal domain of Stt3p with an N-terminal His6-tag in pET-28c vector using BL21(DE3) CodonPlus E. coli strain (Figure 1). Protein expression was optimized using different E. coli strains, temperature, and IPTG concentration (see methods and materials for details). The target protein was expressed as inclusion bodies, which is quite common for eukaryotic membrane proteins since these proteins are often not incorporated well into the plasma membrane of E. coli.
Figure 1.
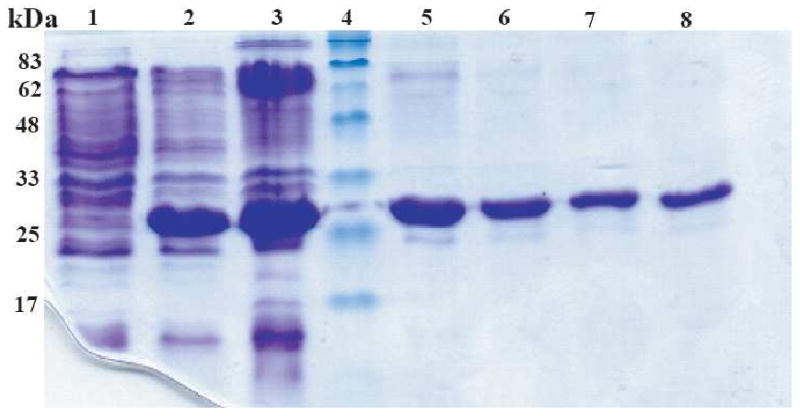
Coomassie-stained SDS-PAGE of samples from a typical C-terminal Stt3p expression and purification run. The mobility of the His-tagged C-terminal Stt3p in the SDS-PAGE gel is compatible with its molecular mass (31.4 kDa). Lane 1, before induction; Lane 2, 4 hours after induction with 0.5mM IPTG; Lane 3, inclusion body; Lane 4, protein molecular weight markers; Lane 5-8, protein purified by “SDS Elution” where elution fractions: 1, 2, 3 and 4 are shown.
Inclusion bodies were solubilized by using 6 M Gdn-HCl, followed by binding to Ni (II) metal ion affinity resin and washing off all of the impurities. Detergent was used during purification processes since the C-terminal Stt3p domain was found to be a water insoluble protein. The standard protocol is the use of imidazole to compete off the His-tagged protein from the Ni-NTA resin. However, this simple procedure did not work well for the C-terminal Stt3p domain. Most of the protein remained bound to the Ni-NTA resin even when the imidazole concentration was increased to ∼ 2 M in the elution buffer containing digitonin (as analyzed by SDS PAGE). It was clear that there are other interactions most likely between the hydrophobic regions of the protein and the resin that are playing a major role in the binding. This observation was proved to be correct when the His-tagged C-terminal domain was able to efficiently bind to even EDTA treated Ni2+ depleted resin. Thus, developing a method for the elution of the protein off the resin was unavoidable in our case.
Here, we report a novel, simple, yet robust purification protocol to purify C-terminal Stt3p domain without using imidazole. The protein bound to the Ni-NTA resin was efficiently eluted off of the column with buffer containing 50 mM SDS in 20 mM phosphate buffer at pH 6.5 after 2 hours of shaking at room temperature. Indeed, the first several elutions (500 μL of each eluted fraction) contained ∼200 μM of pure protein (Figure 1). However, C-terminal Stt3p domain could also be eluted off the Ni-NTA column with SDS concentration as low as 10 mM. In fact, this method also worked for the elution of the protein from Ni2+ depleted resin. SDS was exchanged freely to any other detergent by following the protocol described under experimental procedures.
The D518E mutant was expressed and purified following the same protocol as that of the wild-type protein. The level of overexpression and yield of pure protein obtained for the mutant were comparable to that of the wild-type protein.
Mass Determination by MALDI-TOF Mass Spectroscopy
Relative mobilities of polypeptide chains in SDS PAGE can be used to assign tentative molecular weights, however this is purely an empirical procedure. Thus, MALDI-TOF mass spectroscopy was utilized to confirm the mass of the purified protein. However, this simple approach was complicated by the presence of SDS. Several reports have demonstrated that SDS is detrimental to MALDI-MS (25-27). After numerous optimizations, eventually, the appropriate conditions, including solvent system, matrix type and concentration of SDS, were determined to obtain reliable MALDI signals. Ammonium acetate buffer was found to be essential and SDS concentration was optimized to 10 mM. The mass spectrum of the purified protein showed a molecular ion at m/z 31493.1 (Figure 2). This is in accordance with the calculated mass of 31502.3 Da for His-tagged C-terminal domain of Stt3p as calculated by the ProtParam tool of expert protein analysis system (ExPASy) (28). With MALDI-TOF data, an error margin of 0.15% is acceptable (29), however for our data, the error margin was only 0.03%.
Figure 2.
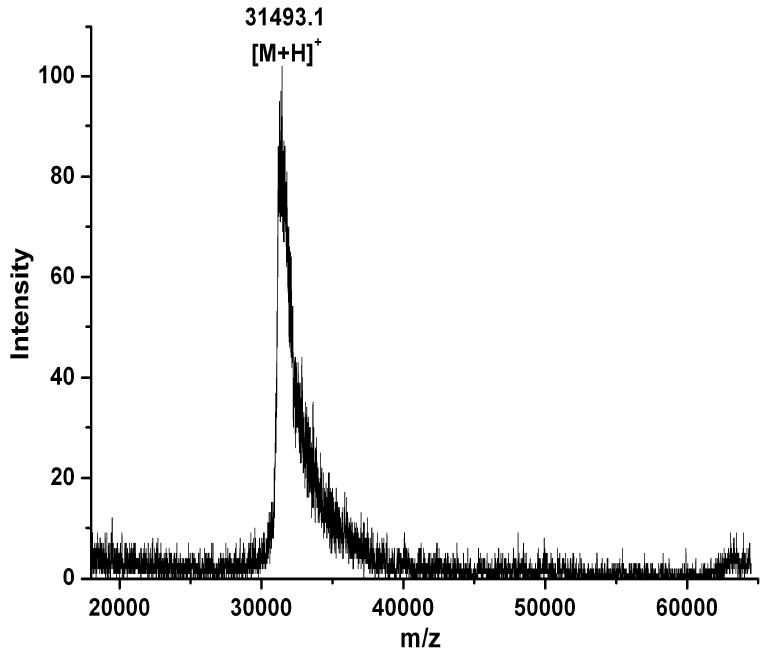
MALDI-TOF analysis of molecular mass of the purified His-tagged C-terminal domain of Stt3p. The mass spectrum of the purified protein showed a molecular ion at m/z 31413.1, which is in accordance with the calculated value (31422.3 Da) for His-tagged C-terminal domain of Stt3p.
Detergent Screening by NMR Spectroscopy
Since it is not currently possible to determine the best detergent a priori, in this study, a number of different detergents including SDS, DPC, LDAO, OG, Digitonin, and DDM were screened. During the detergent exchange process to LDAO, the protein precipitated indicating that LDAO is not a proper detergent to solubilize the C-terminal domain of Stt3p. The five detergents were screened by NMR spectroscopy to determine their suitability for reconstitution of the C-terminal domain of Stt3p. The 2D HSQC spectrum provides both qualitative and quantitative information for the evaluation of whether a protein is well folded and exists in a single conformation. The quality and the number of peaks present in 2D HSQC NMR spectrum reveals whether a protein is monomeric or exists in oligomeric forms. This information is vital to assess the feasibility of further solution NMR based structural characterizations. As shown in Figure 3 and Figure 4, the quality of HSQC spectra varies markedly as a function of detergent. The HSQC spectrum of the C-terminal domain of Stt3p in DPC micelles, a detergent often found to provide high quality NMR spectra for membrane proteins (30), showed very broad line widths and a number of missing resonances. Digitonin and DDM micelles produced poorly resolved spectra (Figure 3). These observations clearly demonstrate that the C-terminal domain of Stt3p is oligomerized under the above micellar environments. Oligomerization leads to slower tumbling and rapid transverse relaxation rates, which substantially broaden and weaken the resonances, thus dramatically reducing spectral resolution. In the case of the detergent OG, more peaks were observed than expected in the HSQC spectrum, indicating the presence of multiple conformations or oligomeric equilibria. Among all the five detergents tested, SDS was determined to be the best for further NMR based structural characterization. It produced a far superior spectrum (Figure 4) with favorable dispersion and narrow line widths, which indicated that the C-terminal domain of Stt3p was folded into a single stable conformation under the experimental condition. Furthermore, out of 263 non-proline residues, 245 resolved peaks with relatively uniform intensity were counted.
Figure 3.
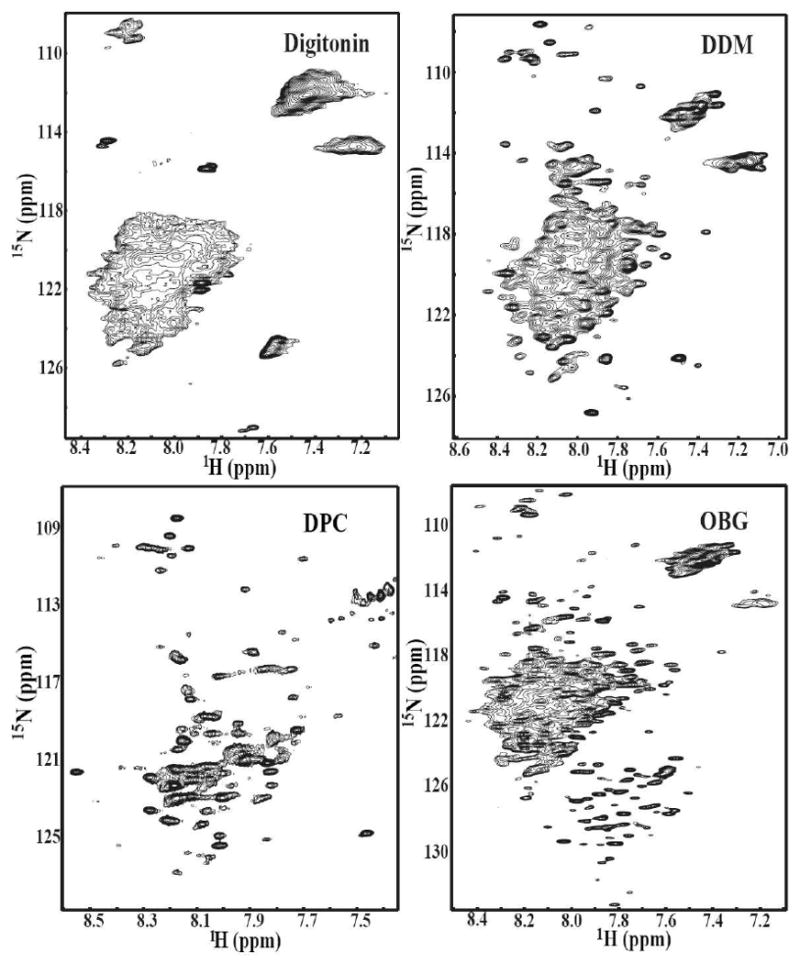
2D NMR [1H, 15N] HSQC spectra of the purified [U-15N] His-tagged C-terminal domain of Stt3p at concentration of ∼0.2 mM in 20 mM phosphate buffer containing 5% D2O, pH 6.5 in different detergent micelles. The concentrations of detergents were as follows: (A) 1.5% Digitonin, (B) 1% DDM, (C) 300 mM DPC, and (D) 150 mM OG.
Figure 4.
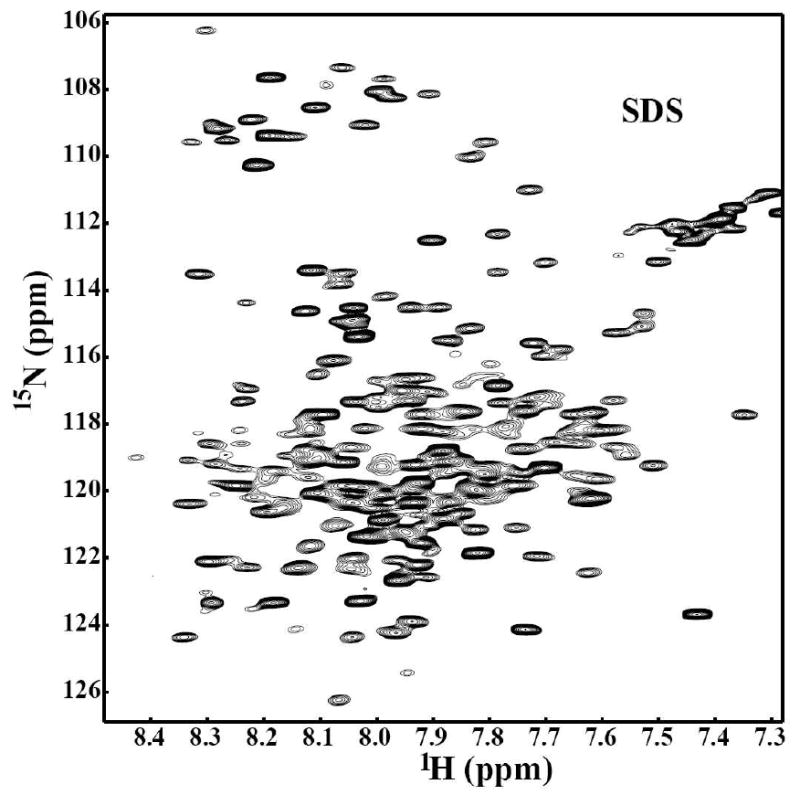
2D NMR [1H, 15N] HSQC spectrum of the purified [U-15N] His-tagged C-terminal domain of Stt3p at concentration of ∼0.2 mM in 20 mM phosphate buffer containing 5% D2O, pH 6.5 containing 100 mM SDS.
The optimum concentration of SDS was determined by thoroughly investigating the effect of SDS on protein conformation by NMR. Our data indicated that the HSQC spectra were well resolved and closely resembled one another when SDS concentration is in the range of 50-200 mM (Figure 5 A-C). However, the spectra started to lose its resolution at a concentration above 250mM. When SDS concentration was increased to 400 mM or above the resonance dispersion became very narrow with many missing peaks indicating that the protein had partially denatured (Figure 5D). Taken together, 100 mM SDS was chosen as the working condition for further characterization.
Figure 5.
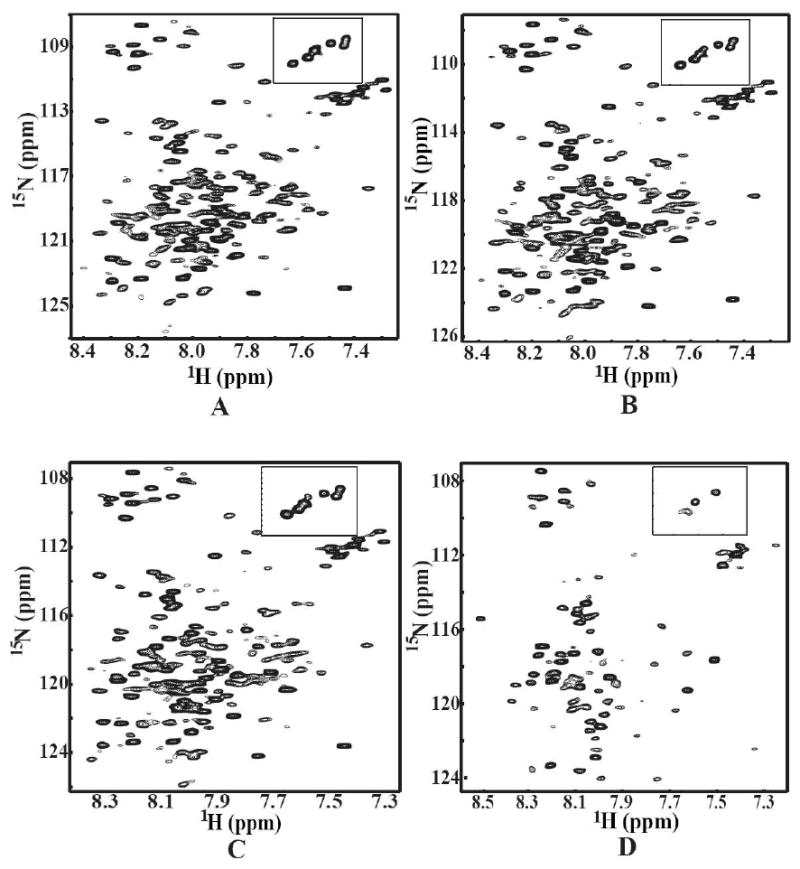
2D NMR [1H, 15N] HSQC spectra of the purified [U-15N] His-tagged C-terminal domain of Stt3p (in 20 mM phosphate buffer, 5% D2O, pH 6.5) as a function of SDS concentration. The inner figure is close-up view of the tryptophan indole amide proton region from the same spectrum. The concentrations of SDS were as follows: (A) 50 mM SDS; (B) 100 mM SDS; (C) 200 mM SDS and (D) 400 mM SDS.
Characterization of the C-terminal Domain of Stt3p by far-UV and near-UV CD Spectroscopy
Far-UV and near-UV CD spectroscopy were employed to probe the secondary and tertiary structure of the C-terminal domain of Stt3p in 100 mM SDS micelles and 400 mM DPC micelles for comparison. The far-UV CD spectra (Figure 6A) both in SDS and DPC micelles had the characteristics of a typical α-helical protein with CD minima at 208 and 222 nm. This observation is consistent with what was seen in the 2D HSQC spectrum i.e. relatively narrow proton dispersion, which is another indication of a helical protein. These results are also consistent with the crystal structure of its archaea homolog reported recently (11), even though there is only limited sequence similarities between these two proteins.
Figure 6.
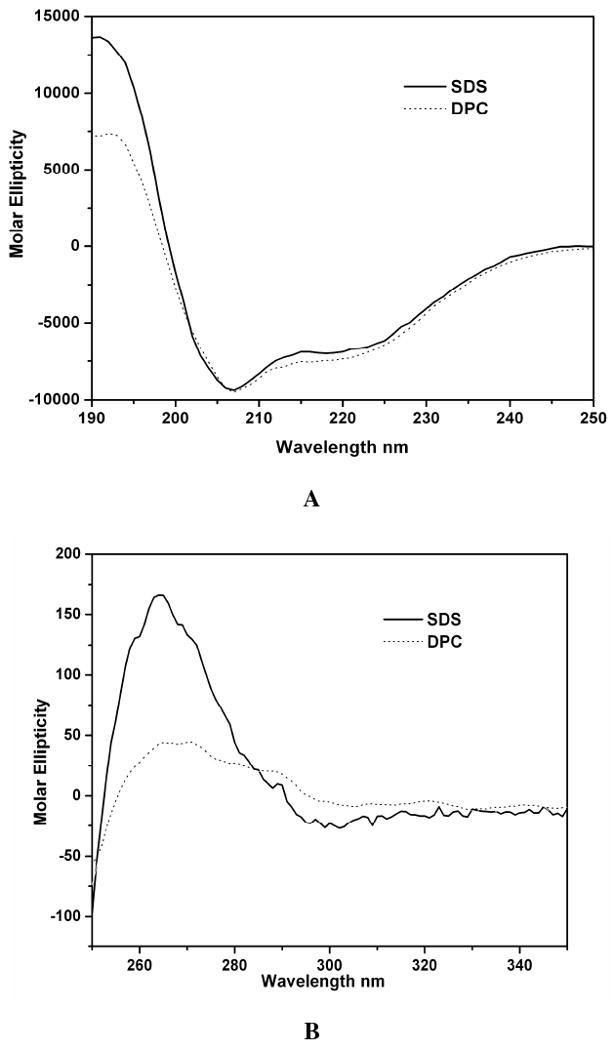
Circular dichroism (CD) spectroscopic analysis of the C-terminal domain of Stt3p at room temperature. (A) far-UV CD spectra of the C-terminal domain of Stt3p in 300 mM DPC and 100 mM SDS detergent micelles. The protein concentration was 10 μM in 20 mM phosphate buffer, pH 6.5. The characteristic double minima at 208 and 222 nm are indicative of significant α-helical content. (B) near-UV CD spectra of the C-terminal domain of Stt3p in 300 mM DPC and 100 mM SDS detergent micelles. The protein concentration was 89 μM, and the buffer conditions were same as for A.
Near-UV (250-350 nm) CD spectrum are due to the dipole absorption of the aromatic residues and disulfide bonds (if present), which depends upon the orientation and nature of the surrounding environment of these chromophores, and is therefore sensitive to the overall tertiary structure of a protein. For a protein in an unfolded or molten globule state, one of the classical spectroscopic signatures is the absence of a near-UV signal (31). In other words, the presence of significant near-UV signals is a strong indication that the protein is folded into a well-defined structure (32-34). The presence of near-UV CD signal for the C-terminal domain of Stt3p in 100 mM SDS (Figure 6B) indicates that the protein has a well defined tertiary structure. Interestingly, the tertiary structure appears to be disrupted in DPC micelles (Figure 6B), which is consistent with the NMR data (Figure 3C) despite the widely accepted notion that DPC is usually a “milder” detergent that generally doesn't denature proteins. Close inspection of the near-UV CD spectrum in SDS micelles reveals that there are three humps, from left to right, which can be attributed to the absorption of phenylalanine, tyrosine and tryptophan respectively.
Intrinsic Tryptophan Fluorescence
Intrinsic fluorescence, especially with tryptophan as a probe, provides a powerful analytical tool for membrane protein studies due to its sensitivity and simplicity (35). The fluorescence emission spectrum of the C-terminal domain of Stt3p (containing 8 tryptophan residues) upon excitation at 280 nm showed a broad emission spectrum with λmax ranging from 330 nm to 350 nm (Figure 7C). This result indicates that of the 8 tryptophan residues, some are totally buried into the hydrophobic core; some are partially exposed to water, while the rest are completely exposed to water. This result is not surprising at all since in integral membrane proteins, tryptophan residues have been found to show preferential clustering at the membrane interface (36-41). The exact location and orientation of each tryptophan residue can only be clear once the high resolution 3D structure of the C-terminal domain of Stt3p is solved.
Figure 7.
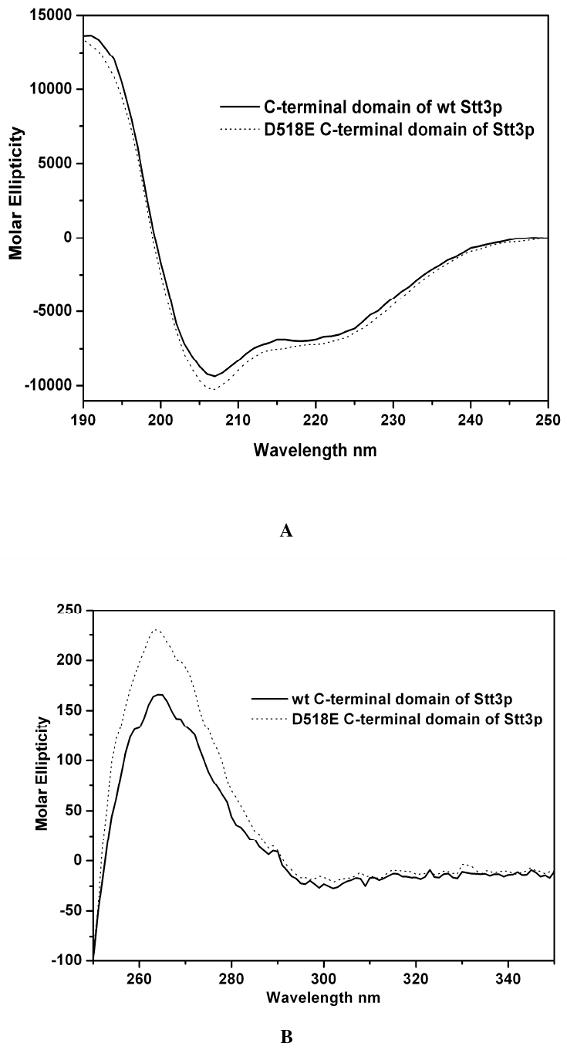
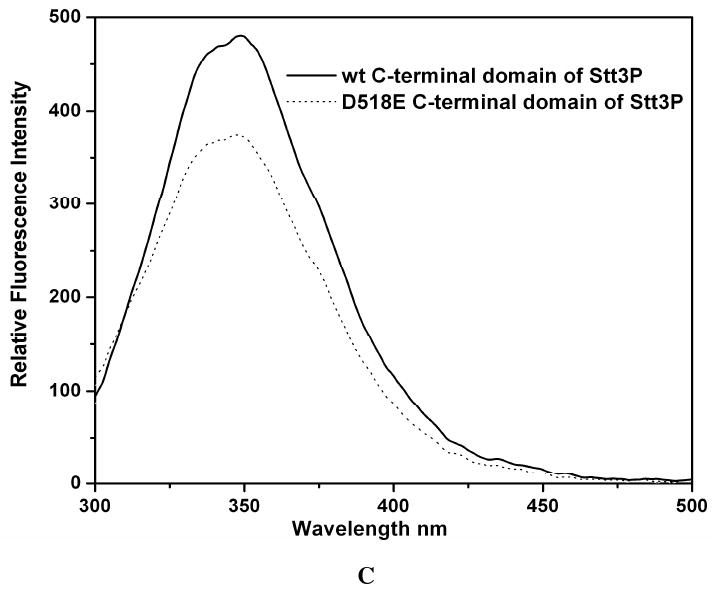
CD spectra of the wild-type and D518E mutant of the C-terminal domain of Stt3p. The data were collected under the same conditions. (A) far-UV CD spectra. The protein concentrations were 10 μM in 20 mM phosphate buffer, pH 6.5, 100 mM SDS. (B) near-UV CD spectra. The protein concentrations were 89 μM in 20 mM phosphate buffer, pH 6.5, 100 mM SDS. (C) intrinsic tryptophan fluorescence spectra. The protein concentrations were 1 μM in 10 mM phosphate buffer, pH 6.5, 100 mM SDS. The introduction of the mutation leads to an intensity quench and blue shift of the spectrum.
Comparison of Wild-type and Mutant Protein
In the C-terminal domain of Stt3p, residues 516-520, which make up the WWDYG motif, are highly conserved through several branches on the evolutionary tree. This motif is thought to be directly involved in the glycosylation site recognition and / or in the catalytic glycosylation process based on co-immunoprecipitation, photoaffinity labeling, and both block and single mutational analysis (11). Furthermore, subtle substitution of a single residue such as Asp518 to Glu renders the enzyme completely inactive causing cell death in yeast, Saccharomyces cerevisiae (11, 14). This observation demonstrates that there may be a strict geometric or conformational requirement for the enzyme to catalyze the N-linked glycosylation reaction. To investigate whether Asp518 acts only as a catalytic base as proposed previously (11), or has any other role in the conformational geometry required for catalysis, we carried out biophysical characterization of both the wild-type and the D518E mutant under identical conditions. The far-UV CD spectra (Figure 7A) of the wild-type Stt3p C-terminal domain and that of D518E mutant are very similar, suggesting that there is no significant change in the secondary structure upon point mutation. In contrast, there are significant differences in the near-UV CD spectra, which reveal that both of the proteins have distinct tertiary structures (Figure 7B). This evidence is further supported by measurements of their tryptophan fluorescence spectra. The D518E mutation led to an apparent blue-shift as well as quenching of the actual intensity of the fluorescence emission of the wild-type protein (Figure 7C), indicating the change in the microenvironments of the tryptophan residues. This observation demonstrates that the D518E mutation did change the structure of the C-terminal domain of Stt3p affecting the microenvironment and solvent exposure of some tryptophan residues, most likely neighboring W516 and W517 (36, 42).
NMR is an extremely robust technique to monitor the changes in the conformation of a protein sample due to change in pH, temperature, salt or addition of a ligand. The 2D {1H, 15N} HSQC spectrum represents the fingerprint region of a protein. This region is extremely sensitive and any perturbation in the chemical shifts or resonances from the original positions is an indication of the change in the conformation of the protein. This change can be local, involving few residues or a global conformational change involving most of the residues in the protein. In the present study, HSQC spectra were collected to compare the fingerprint region of the wild-type and the D518E mutant in SDS micelles under identical conditions. It is clear from Figure 8 that the D518E point mutation induced drastic changes in the chemical shift positions of a number of peaks indicating that wild-type and the D518E mutant have distinctly different conformations. This conformational change can not be attributed to the change of the local environment around Asp518 since chemical shift perturbation is dramatic for most of the peaks in the HSQC spectrum. In fact, some of the resonances observed in the wild-type HSQC spectrum did disappear in the spectrum of the mutated protein. This observation demonstrates that the D518E mutation, indeed affects both conformation and dynamics of the wild-type protein, which may have bearing on OT function.
Figure 8.
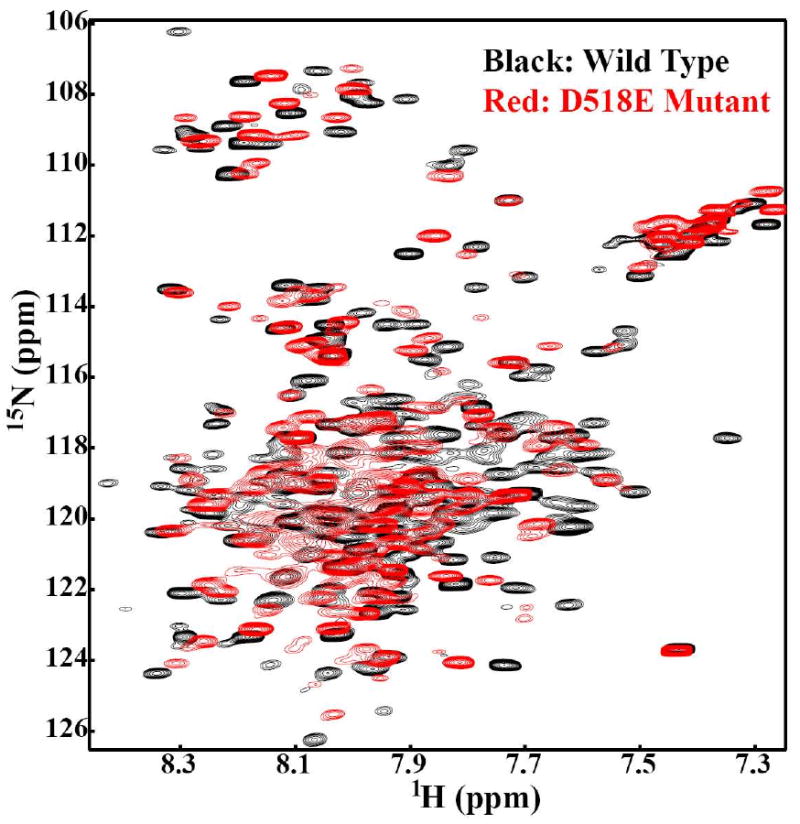
The impact of the D518E mutation on 2D [1H, 15N] -HSQC spectrum. The black spectrum represents the wild-type, while the superimposed red spectrum is of the D518E mutant of the C-terminal domain of Stt3p.
Acceptor Substrate Binding Studies by STD NMR Spectroscopy
To investigate the interactions of acceptor substrate of OT with the C-terminal domain of Stt3p, binding studies were carried out with a six-residue peptide containing the consensus N-linked glycosylation sequon by saturation transfer difference (STD) NMR spectroscopy. STD has been proven to be a powerful method to probe low affinity interactions (KD ≈ 10-8 to 10-3 M) of small molecules with proteins (43-48). In the STD technique, selective saturation of a protein resonance leads to a rapid spread of the magnetization over the entire protein via spin diffusion, and intermolecular transfer of magnetization from protein to ligand leads to changes in NMR signal intensity of the ligand. However, for interaction studies involving proteins and peptides, attention should be paid to make sure a well separated peak in the protein is picked for STD experiment. Thus the saturation resonance must exclusively belong to protein. Moreover, the resulting signals in STD spectra must exclusively belong to peptide ligand. The latter is especially true if incomplete protein signal suppression occurs.
To overcome these, here, a methyl-protonated {Ile(δ1 only), Leu(13CH3, 12CD3), Val(13CH3, 12CD3)} U-{15N, 13C, 2H} labeled sample of the C-terminal domain of Stt3p was prepared by using biosynthetic precursors (49). This labeling pattern is extremely desirable for STD studies since in these labeled proteins, except for water-exchangeable protons, only the methyl groups of the Ile (δ1 only), Leu and Val residues are protonated. On one hand, the commonly used regions for irradiation of protein remain, such as the upfield region (about 0 ppm) or downfield region (about 10 ppm). On the other hand, the simplified protein spectrum facilitates the data analysis process significantly and reduces the risk of having the pseudo-positive effect resulted from incomplete elimination of background protein signals. As shown in Figure 9, the C-terminal domain of Stt3p and peptide ligand complex was irradiated at 0.738 ppm, where no peptide NMR signal was present. The peaks a, b, c, d, e, and f in STD spectrum exclusively correspond to the peaks 1, 2, 3, 4, 5 and 6 respectively in the NMR spectrum of acceptor peptide (Figure 9B and 9C). The appearance of the NMR peaks of the peptide ligand in the difference spectrum unequivocally indicates that the acceptor peptide ligand is bound to the C-terminal domain of Stt3p. More importantly, close inspection of the difference spectrum reveals that the amide protons (peak a, which has a chemical shift of 7.13 ppm) on the side chain of Asn residue, the N-glycosylaiton site, are significantly affected by the saturation pulse (Figure 9C), which strongly suggest that the side-chain of Asn residue is directly involved in the protein-substrate recognition process.
Figure 9.
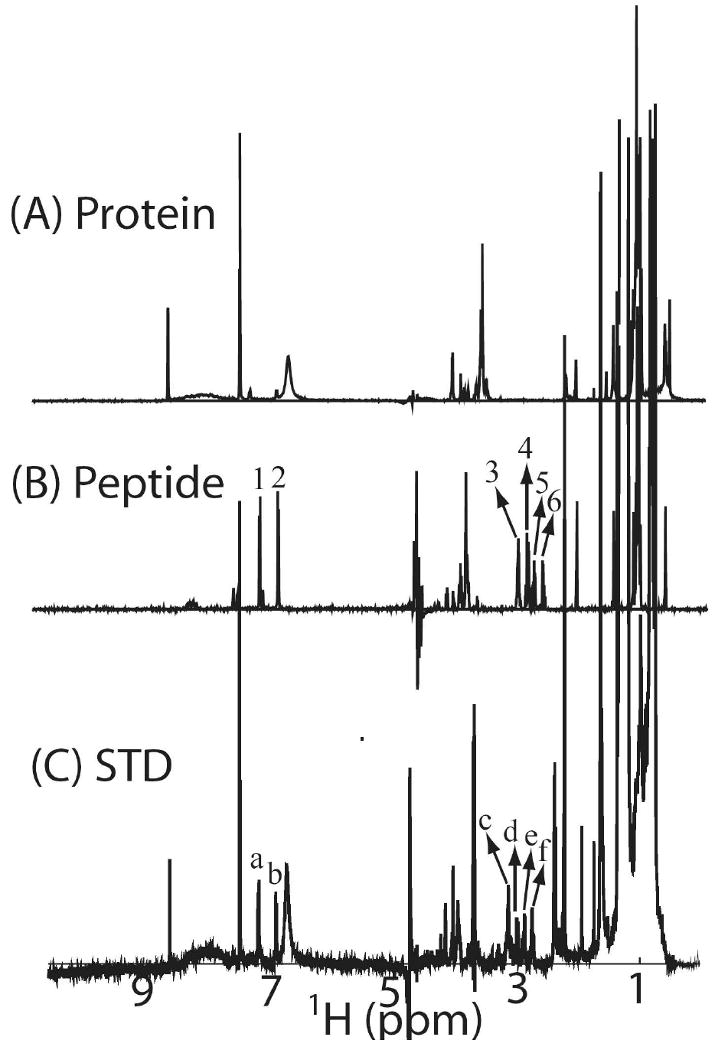
(A) 1D NMR spectrum of methyl-protonated {Ile(δ1 only), Leu(13CH3, 12CD3), Val(13CH3, 12CD3)} U-{15N, 13C, 2H} labeled sample of the C-terminal domain of Stt3p (30 μM) in phosphate buffer (20 mM, pH 6.5) and 100 mM deuterated SDS. (B) 1D NMR spectrum of 300 μM peptide ligand, Tyr-Asn-Ser-Thr-Ser-Cys-Am, in phosphate buffer (20 mM, pH 6.5) and 100 mM deuterated SDS. (C) STD NMR spectrum of the complex of methyl-protonated {Ile (δ1 only), Leu(13CH3, 12CD3), Val(13CH3, 12CD3)} U-{15N, 13C, 2H} labeled sample of the C-terminal domain of Stt3p and acceptor peptide substrate (Tyr-Asn-Ser-Thr-Ser-Cys-Am) in phosphate buffer (20 mM, pH 6.5) and 100 mM deuterated SDS. The protein and peptide concentrations are 30 μM and 300 μM, respectively. The spectrum was recorded with a T1ρ filter, consisting of a 50-ms spin-lock pulse, to eliminate the resonances of the protein. The appearance of peaks a, b, c, d, e and f in STD spectrum, which correspond to the peaks 1, 2, 3, 4, 5 and 6 in the NMR spectrum of acceptor peptide, reveals the C-terminal domain of Stt3p binds to the acceptor substrate of OT.
Affinity Studies by NMR Titrations
To further determine the affinity of acceptor substrate (of OT) with the recombinant C-terminal domain of Stt3p, titration studies were carried out with Asn-Asp-Thr-NH2 acceptor peptide containing the consensus N-linked glycosylation sequon. Substrate binding was followed by monitoring the changes in chemical shift positions in the fingerprint region of the protein in 2D HSQC spectra as shown in Figure 10 (A). The chemical shift perturbation of four representative peaks were fitted to Hill model by using Origin® 7.0 software and the plot is shown in Figure 10 (B). It is clear from the fit that the C-terminal domain of Stt3p exhibits a sigmoidal saturation curve, and binds to the acceptor peptide with an apparent KD of 9.97 ± 0.44 mM. The Hill coefficient “n” is 1.70 suggesting positive cooperativity (as n > 1) in binding with a relatively low affinity.
Figure 10.
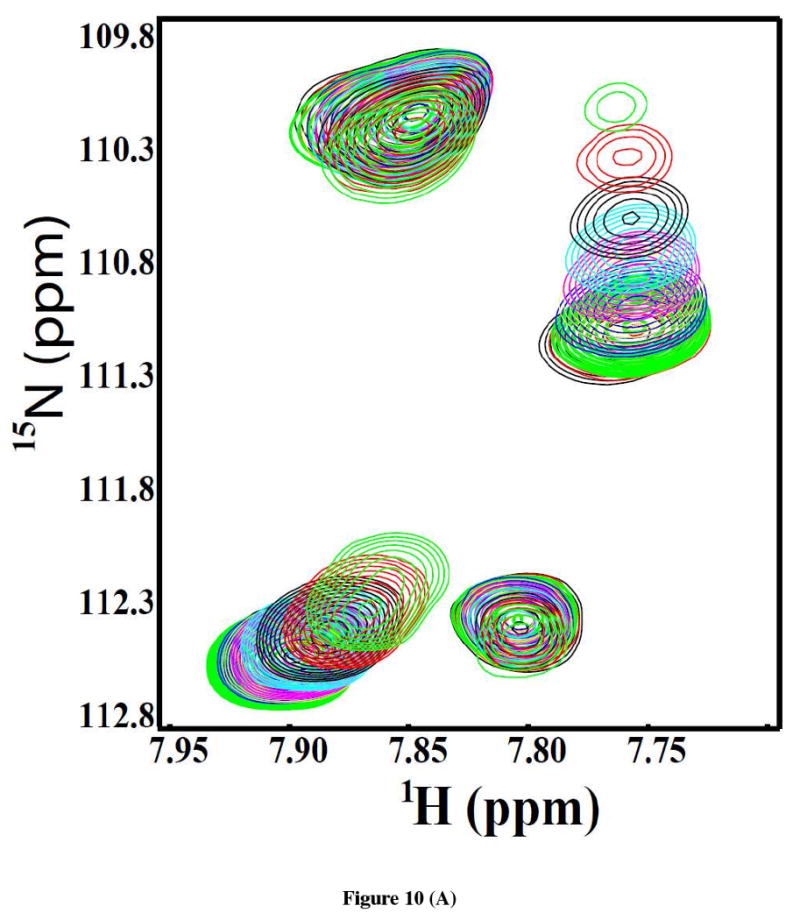
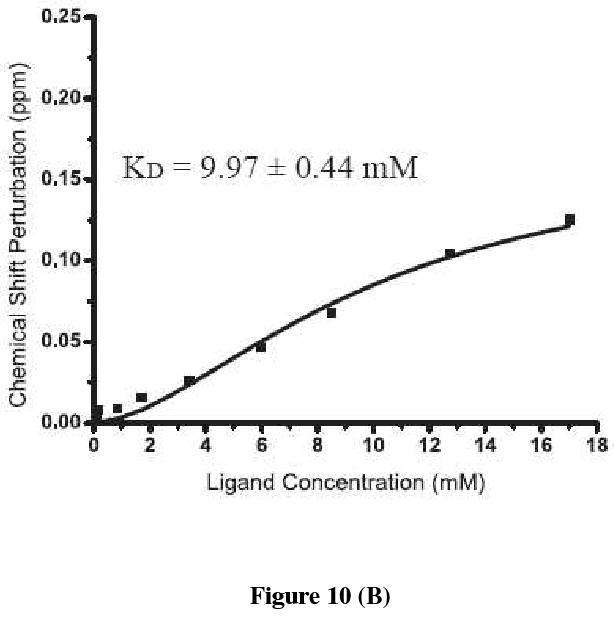
(A) An expanded region of the overlay of 2D [1H, 15N]-HSQC spectra of the [U-15N]-labeled C-terminal domain of Stt3p (170 μM) showing changes in the chemical shift positions upon addition of increasing concentration of the substrate peptide. Ratios of protein to peptide are: 1:0 (black), 1:0.5 (red), 1:1 (green), 1:5 (blue), 1:10 (yellow), 1:20 (purple), 1:35 (cyan), 1:50 (black), 1:75 (red) and 1:100 (green). (B) The chemical shift perturbation average of four representative resonances are plotted as a function of the concentration of the substrate peptide and fitted using Hill model of Origin® 7.0 software.
Discussion
High-level Protein Expression and Purification
Although N-linked glycosylaiton is an essential, critical and highly conserved process in all eukaryotes, very little structural and functional information on the OT enzyme complex is known. The primary reason being the difficulties in the production of milligram quantities of integral membrane proteins (IMPs) for structural or functional characterization. Recombinant expression of IMPs in E. coli, the primary machine for large-scale protein production for structural studies, has had very limited success (50). As a result, there is only one example of recombinant expression of an OT subunit from prokaryotic sources as of now, which is the water-soluble C-terminal domain of Stt3p homolog from archaea P. furiosus (51).
We report here a high-level recombinant expression in E. coli and purification of the C-terminal domain of Stt3p from the yeast, Saccharomyces cerevisiae. This is the first report of heterologous expression of a eukaryotic OT subunit. This high level production of pure C-terminal domain of Stt3p makes isotopic labeling for structural characterization either by solution NMR or by X-ray crystallography straightforward, affordable and most importantly, possible.
After many unsuccessful attempts to refold denatured C-terminal domain of Stt3p in aqueous solution without the use of any detergents, we were convinced that a membrane mimetic environment is necessary for its purification and reconstitution. This evidence suggests that the C-terminal domain of Stt3p may contain at least one TM helix or several membrane embedded residues. Purification and reconstitution of membrane proteins are notoriously difficult tasks. Indeed, reports of successful isolation and refolding of IMPs from inclusion bodies have thus far been limited to a small number of proteins (52-54). In an elegant work, Page and co-workers have reported two methods for isolation and purification of helical integral membrane proteins: ‘Detergent Exchange’ and ‘Reconstitution’. Both of these methods use standard protocols for detergent mediated purification via Ni2+ affinity chromatography (30). Here, we report a novel method for the one-step purification and reconstitution of the C-terminal domain of Stt3p that we have named ‘SDS Elution’. Using our novel method, we were able to obtain very high yield of purified protein (60-70 mg of protein per liter of bacterial culture) in a single step. To evaluate the efficiency of our method, we also produced 15N-labeled C-terminal Stt3p following the “reconstitution” method of Page et al. (30). After the precipitated proteins were reconstituted in 100 mM SDS and 300mM DPC micelles as per the “reconstitution” method (30), the HSQC spectra were collected and compared with the spectra of the samples prepared by our “SDS Elution” method. Although the HSQC spectra were overall similar (data not shown), the quality of the spectra for the protein obtained by “reconstitution” method appeared to be deteriorated possibly due to aggregation. Moreover, the protein yield obtained by the “SDS elution” method was much higher. Indeed, in principle, “SDS Elution” combines both “Detergent Exchange” and “Reconstitution” together, but greatly simplifies the protocols.
This novel methodology has several advantages over the conventional methods. First and foremost, with our method, purification and reconstitution are achieved simultaneously, which dramatically shortens the sample preparation process. Secondly, since there is no imidazole in elution buffer, the conventional method of removal of imidazole through buffer exchange is not required, which prevent the otherwise unavoidable loss and dilution of the protein samples. Moreover, since the detergent is not introduced until the last step, it saves the amount of detergents used. This is especially true for NMR sample preparations where the use of deuterated detergents is necessary and deuterated detergents are generally very expensive. We are currently investigating the versatility of this method using other integral membrane proteins.
Feasibility of Structural Determination by Solution NMR
To carry out structural determination of any membrane protein by solution NMR, detergent screening to find the suitable membrane mimetic is an essential prerequisite. The suitability of a detergent micelle is determined by taking into account the protein solubility and stability along with the quality of the 2D HSQC NMR spectrum. The 2D HSQC spectrum correlates the amide proton and the corresponding nitrogen pair of each residue within a protein and provides a map of the finger print region. It also serves as a building block for a multitude of multidimensional NMR experiments upon which the resonance assignments and the determination of the 3D structure of a protein rest. Thus, obtaining high quality, i.e. sufficiently resolved HSQC spectra is imperative for structural characterization by solution NMR.
To find a suitable detergent for obtaining a homogeneous sample of the C-terminal domain of Stt3p, six detergents were screened. These include digitonin, which has been successfully used to extract and reconstitute the OT complex in microsomes (55); SDS and DPC, which are commonly used for solution NMR; LDAO, DDM and OG, the common detergents for membrane protein crystallization. As expected, digitonin gave unresolvable HSQC spectra due to its large micellar sizes (70 kDa). For all the rest of the detergents except OG and SDS, the protein appeared to be oligomerized leading to poorly resolved spectra with broad linewidths and missing resonances. For OG, while it has a small micellar size (25 kDa), it seems that its short alkyl chain (C8) jeopardizes the protein conformational stability since the number of HSQC peaks is much higher than expected. In contrast, SDS micelles yielded an HSQC spectrum that was far superior in quality in comparison to all the rest of the detergents that were screened in all of the aspects: number of resonances, signal to noise ratio, dispersion, linewidths and uniformity of signal intensities. In fact, SDS has served as one of the most popular detergents for IMPs studies (56), and has been widely used as a membrane mimetic for membrane protein structural and functional studies (9, 57-61). The high quality HSQC spectra along with the CD, and fluorescence data suggest that the C-terminal domain of Stt3p in SDS micelles, is well-folded producing a homogeneous sample. These support the feasibility of conducting solution NMR-based structural studies.
In fact, the quality of the 2D HSQC spectrum is much better than what would be expected for such a large protein–detergent complex, implying relatively small relaxation rate. The 15N T1, T2 relaxation measurements show that the rotational correlation time for the C-terminal domain of Stt3p in SDS micelles is surprisingly short- ∼10 ns, a value expected for a 20 kDa protein tumbling isotropically in solution (data not shown). This, however, is consistent with the results reported by Krueger-Koplin, et al (9), where a survey of seven membrane proteins in different detergent micelles showed a rather short rotational correlation time ranging from 8 to 12 ns. According to these authors, this phenomenon can be attributed to the fluid property of detergents which allows rotation of the proteins within the confines of the micelle. In the case of the C-terminal domain of Stt3p, an alternative explanation is its flexible dynamic property. The high flexibility of the C-terminal domain of Stt3p is reasonable since N-linked glycosylaiton is co-translational. Therefore, only a flexible active site can recognize glycosylatable sequons rapidly and efficiently in all different types of growing polypeptide chains. This ensures the rapid product discharge from the active site. Furthermore, the flexibility of this domain is supported by the cryo-electron microscopy structure of the yeast OT, which shows a flexible groove formed between the lumenal domains of Ost1p, Wbp1p, and Stt3p (62). This groove is proposed to thread and scan the unfolded nascent polypeptide chain (62).
One striking feature of the C-terminal domain of Stt3p is that it is highly conserved in eukaryotes. Actually, the sequence alignment shows that, from yeast to humans, the sequence identity is over 50% (10). The strictly conserved “WWDYG” motif is believed to be the catalytic and/or acceptor protein recognition site. The aspartate residue (Asp518) of this conserved motif was thought to function as a catalytic base (11). However, it appears that the role of Asp518 is more than simply to act as a base in the catalysis since the D518E mutation results in a complete loss of enzyme activity, even though both Asp and Glu residues have similarly charged side chains. If the role of Asp is just to act as a base, then how can the loss of activity for D518E be explained?
To address the above question, comprehensive biophysical characterizations of the D518E mutant and wild-type C-terminal domain of Stt3p were carried out. Interestingly, while both the wild-type and D518E mutant share nearly identical secondary structural contents, they have distinctly different tertiary structures as revealed by near-UV CD, fluorescence and NMR spectroscopies. The most direct evidence for this conclusion has come from the comparison of their 2D HSQC spectra. The replacement of Asp518 with the longer Glu side chain leads to large global changes in the structure involving nearly all of the amino acid residues (Figure 8). This observation led to the conclusion that the residue Asp518 is critical to maintain the catalytically active conformational geometry of the C-terminal domain of Stt3p.
Additionally, the apparent disruption of the active conformation after a point mutation strongly suggests that the C-terminal domain of Stt3p has folded into its native conformation in SDS micelles. This is based on the fact that it is very unlikely to change the ‘structure’ of a protein that is denatured or in a molten globule state by the replacement of one residue with a structurally similar residue.
The loss of enzyme activity by mutation of Asp→Glu is not that common, but OT is not unique in this regard (10). In fact, for the enzyme Ca2+-ATPase, mutation of D601E and D707E result in an inactive enzyme (63). More importantly, the residues Asp601and Asp707 have been proposed to play structural but not catalytic or substrate recognition roles. It is therefore likely that OT and ATPase may have similar mechanisms of function.
In vitro Functional probing of the C-terminal domain of Stt3p
In the present study, an elaborate STD NMR experiment and HSQC titrations were carried out to probe the in vitro protein-substrate interaction studies. Our results demonstrate that the C-terminal domain of Stt3p interacts with the acceptor peptide substrate containing the N-linked glycosylation recognition motif. The strong signals exclusively belonging to the acceptor peptide were observed in the STD spectrum while chemical shift perturbations were observed in the HSQC experiments upon addition of the substrate peptide. These observations provide direct experimental proof that the C-terminal domain of Stt3p contains the recognition site for the N-glycosylation acceptor substrate even though the affinity is relatively low (KD = ∼ 10 mM). One explanation could be that SDS micelles may not mimic the native lipid bilayer, which may impair the activity of the protein to some extent. Additionally, since the functional OT complex is composed of eight different subunits, it is more likely that while the C-terminal domain of Stt3p possesses the substrate recognition site, the other subunit(s) may facilitate the binding process (62).
The C-terminal domain of Stt3p in SDS micelles has a short rotational correlation time of ∼ 10 ns suggesting that it is a monomer under the experimental conditions. However, the sigmoidal saturation curve observed upon acceptor substrate binding indicates that this monomeric protein is allosterically activated, suggesting that it may contain more than one binding site. The binding of a peptide substrate (allosteric activator) to the activator site results in increased affinity in the second site (active site). The detailed regulatory mechanism can only be addressed by further structure–function studies.
Conclusion
We have reported the over-expression of the C-terminal domain of Stt3p subunit from the yeast Saccharomyces cerevisiae. A robust, novel yet simple purification protocol called ‘SDS Elution” has been developed. It is likely that this novel purification method may work for the purification and reconstitution of other recombinant integral membrane proteins. Far-UV CD data show C-terminal Stt3p is a helical protein while near-UV CD, fluorescence and NMR experiments demonstrate the presence of a stable tertiary structure. In addition, our biophysical data suggest that the critical residue Asp518 plays a structural role in maintaining the strict conformational geometry necessary for the catalytic activity of the enzyme. Our acceptor substrate binding studies indicate that this C-terminal domain Stt3p contains the acceptor peptide recognition site. The high resolution 3D structure determinations of C-terminal Stt3p both by solution NMR and X-ray crystallography methods are well underway in our laboratory. The high resolution structure will be instrumental in our understanding of the structure/function and mechanisms of catalysis for this enzyme complex.
Acknowledgments
The authors are grateful to Prof. William J. Lennarz, distinguished professor at Stony Brook University, NY for the original Stt3p clone in yeast. The authors acknowledge Dr. David Zoetewey for critical reading of the manuscript.
This research was financially supported by USDA PECASE (Presidential Early Career Award for Scientists and Engineers) award 2003-35302-12930, NSF grant IBN-0628064, and NIH grant DK082397 (to S. M). C. H. was supported by the ACHE-GRSP (Alabama Commission on Higher Education Graduate Research Scholars Program) Scholarship.
Abbreviations
- OT
oligosaccharide transferase
- IMP
integral membrane protein
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single quantum coherence
- CD
circular dichroism
- SDS
sodium dodecyl sulfate
- DPC
dodecylphosphocholine
- LDAO
lauryl dimethylamine oxide
- OG
octyl-β-glucoside
- DDM
n-dodecyl-β-D-maltoside
- am
amidated
- STD
saturation transfer difference
Footnotes
Author contributions: S. M. designed research, cloning, expression and method development; C. H. protein production, and collected all biophysical data; S. M. and C. H. analyzed data; S. M. and C. H. prepared the manuscript.
References
- 1.Knauer R, Lehle L. The Oligosaccharyltransferase Complex from Saccharomyces cerevisiae. Biochim Biophys Acta. 1999;1426:259–273. doi: 10.1016/s0304-4165(98)00128-7. [DOI] [PubMed] [Google Scholar]
- 2.Silberstein S, Gilmore R. Biochemistry, molecular biology, and genetics of the oligosaccharyltransferase. FASEB J. 1996;10:849–858. [PubMed] [Google Scholar]
- 3.Welply JK, Shenbagamurthi P, Lennarz WJ, Naider F. Substrate recognition by oligosaccharyltransferase. Studies on glycosylation of modified Asn-X-Thr/Ser tripeptides. J Biol Chem. 1983;258:11856–11863. [PubMed] [Google Scholar]
- 4.Dempski RE, Jr, Imperiali B. Oligosaccharyl transferase: gatekeeper to the secretory pathway. Curr Opin Chem Biol. 2002;6:844–850. doi: 10.1016/s1367-5931(02)00390-3. [DOI] [PubMed] [Google Scholar]
- 5.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 6.Marquardt T, Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr. 2003;162:359–379. doi: 10.1007/s00431-002-1136-0. [DOI] [PubMed] [Google Scholar]
- 7.Zubkov S, Lennarz WJ, Mohanty S. Structural basis for the function of a minimembrane protein subunit of yeast oligosaccharyltransferase. Proc Natl Acad Sci. 2004;10:3821–3826. doi: 10.1073/pnas.0400512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Igura M, Maita N, Kamishikiryo J, Yamada M, Obita T, Maenaka K, Kohda D. Structure-guided identification of a new catalytic motif of oligosaccharyltransferase. EMBO J. 2008;27:234–243. doi: 10.1038/sj.emboj.7601940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krueger-Koplin R, Sorgen P, Krueger-Koplin S, Rivera-Torres I, Cahill S, Hicks D, Grinius L, Krulwich T, Girvin M. An evaluation of detergents for NMR structural studies of membrane proteins. J Biomol NMR. 2004;28:43–57. doi: 10.1023/B:JNMR.0000012875.80898.8f. [DOI] [PubMed] [Google Scholar]
- 10.Zufferey R, Knauer R, Burda P, Stagljar I, te Heesen S, Lehle L, Aebi M. STT3, a highly conserved protein required for yeast oligosaccharyl transferase activity in vivo. EMBO J. 1995;14:4949–4960. doi: 10.1002/j.1460-2075.1995.tb00178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan Q, Lennarz WJ. Studies on the Function of Oligosaccharyl Transferase Subunits. J Biol Chem. 2002;277:47692–47700. doi: 10.1074/jbc.M208136200. [DOI] [PubMed] [Google Scholar]
- 12.Kelleher DJ, Karaoglu D, Mandon EC, Gilmore R. Oligosaccharyltransferase isoforms that contain different catalytic STT3 subunits have distinct enzymatic properties. Mol Cell. 2003;12:101–111. doi: 10.1016/s1097-2765(03)00243-0. [DOI] [PubMed] [Google Scholar]
- 13.Nilsson I, Kelleher DJ, Miao Y, Shao Y, Kreibich G, Gilmore R, von Heijne G, Johnson AE. Photocross-linking of nascent chains to the STT3 subunit of the oligosaccharyltransferase complex. J Cell Biol. 2003;161:715–725. doi: 10.1083/jcb.200301043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glover KJ, Weerapana E, Numao S, Imperiali B. Chemoenzymatic Synthesis of Glycopeptides with PglB, a Bacterial Oligosaccharyl Transferase from Campylobacter jejuni. Chem Biol. 2005;12:1311–1315. doi: 10.1016/j.chembiol.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feldman MF, Wacker M, Hernandez M, Hitchen PG, Marolda CL, Kowarik M, Morris HR, Dell A, Valvano MA, Aebi M. Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc Natl Acad Sci. 2005;102:3016–3021. doi: 10.1073/pnas.0500044102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nasab FP, Schulz BL, Gamarro F, Parodi AJ, Aebi M. All in One: Leishmania major STT3 Proteins Substitute for the Whole Oligosaccharyltransferase Complex in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19:3758–3768. doi: 10.1091/mbc.E08-05-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hese K, Otto C, Routier FH, Lehle L. The yeast oligosaccharyltransferase complex can be replaced by STT3 from Leishmania major. Glycobiololy. 2009;19:160–171. doi: 10.1093/glycob/cwn118. [DOI] [PubMed] [Google Scholar]
- 18.Kim H, von Heijne G, Nilsson I. Membrane Topology of the STT3 Subunit of the Oligosaccharyl Transferase Complex. J Biol Chem. 2005;280:20261–20267. doi: 10.1074/jbc.M412213200. [DOI] [PubMed] [Google Scholar]
- 19.Cserzo M, Wallin E, Simon I, von Heijne G, Elofsson A. Prediction of transmembrane alpha-helices in procariotic membrane proteins: the Dense Alignment Surface method. Prot Eng. 1997:673–676. doi: 10.1093/protein/10.6.673. [DOI] [PubMed] [Google Scholar]
- 20.von Heijne G. Membrane protein structure prediction. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 21.Hofmann K, Stoffel W. TMBASE - A database of membrane spanning protein segments. Biol Chem Hoppe-Seyler. 1993;374:166. [Google Scholar]
- 22.Juretic D, Zoranic L, Zucic D. Basic charge clusters and predictions of membrane protein topology. J Chem Inf Comput Sci. 2002;42:620–632. doi: 10.1021/ci010263s. [DOI] [PubMed] [Google Scholar]
- 23.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 25.Rosinke B, Strupat K, Hillenkamp F, Rosenbusch J, Dencher N, Kruger U, Galla H. Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) of membrane proteins and non-covalent complexes. J Mass Spectrom. 1995;30:1462–1468. [Google Scholar]
- 26.Galvani M, Hamdan M. Electroelution and passive elution of γ-globulins from sodium dodecyl sulphate polyacrylamide gel electrophoresis gels for matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2000;14:721–723. doi: 10.1002/(SICI)1097-0231(20000430)14:8<721::AID-RCM927>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 27.Jeannot MA, Jing Z, Li L. Observation of sodium gel-induced protein modifications in dodecylsulfate polyacrylamide gel electrophoresis and its implications for accurate molecular weight determination of gel-separated proteins by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. J Am Soc Mass Spectrom. 1999;10:512–520. doi: 10.1016/s1044-0305(99)00022-7. 1999. [DOI] [PubMed] [Google Scholar]
- 28.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chatterjee S, Schoepe J, Lohmer S, Schomburg D. High level expression and single-step purification of hexahistidine-tagged L-2-hydroxyisocaproate dehydrogenase making use of a versatile expression vector set. Protein Expr Purif. 2005;39:137–143. doi: 10.1016/j.pep.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Page RC, Moore JD, Nguyen HB, Sharma M, Chase R, Gao FP, Mobley CK, Sanders CR, Ma L, Sönnichsen FD, Lee S, Howell SC, Opella SJ, Cross TA. Comprehensive evaluation of solution nuclear magnetic resonance spectroscopy sample preparation for helical integral membrane proteins. J Struct Func Genom. 2006;7:51–64. doi: 10.1007/s10969-006-9009-9. [DOI] [PubMed] [Google Scholar]
- 31.Demarest SJ, Boice JA, Fairman R, Raleigh DP. Defining the core structure of the α-lactalbumin molten globule state. J Mol Biol. 1999;294:213–221. doi: 10.1006/jmbi.1999.3228. [DOI] [PubMed] [Google Scholar]
- 32.Batenjany MM, Mizukami H, Salhany JM. Near-UV circular dichroism of band 3. Evidence for intradomain conformational changes and interdomain interactions. Biochemistry. 1993;32:663–668. doi: 10.1021/bi00053a035. [DOI] [PubMed] [Google Scholar]
- 33.Taylor RM, Zakharov SD, Bernard HJ, Girvin ME, Cramer WA. Folded state of the integral membrane colicin E1 immunity protein in solvents of mixed polarity. Biochemistry. 2000;39:12131–12139. doi: 10.1021/bi000206c. [DOI] [PubMed] [Google Scholar]
- 34.Turk E, Gasymov OK, Lanza S, Horwitz J, Wright EM. A reinvestigation of the secondary structure of functionally active vSGLT, the vibrio sodium/galactose cotransporter. Biochemistry. 2006;45:1470–1479. doi: 10.1021/bi052160z. [DOI] [PubMed] [Google Scholar]
- 35.Ladokhin AS, Jaysainghe S, White SH. How to Measure and Analyze Tryptophan Fluorescence in Membranes Properly, and Why Bother? Anal Biochem. 2000;285:235–245. doi: 10.1006/abio.2000.4773. [DOI] [PubMed] [Google Scholar]
- 36.Reshetnyak YK, Koshevnik Y, Burstein EA. Decomposition of Protein Tryptophan Fluorescence Spectra into Log-Normal Components. III. Correlation between Fluorescence and Microenvironment Parameters of Individual Tryptophan Residues. Biophys J. 2001;81:1735–1758. doi: 10.1016/S0006-3495(01)75825-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reithmeier RA. Characterization and modeling of membrane sequence analysis. Curr Opin Struct Biol. 1995;5:491–500. doi: 10.1016/0959-440x(95)80034-4. [DOI] [PubMed] [Google Scholar]
- 38.Deber CM, Goto NK. Folding proteins into membranes. Nat Struc Biol. 1996;3:815–818. doi: 10.1038/nsb1096-815. [DOI] [PubMed] [Google Scholar]
- 39.Landolt-Marticorena C, Williams KA, Deber CM, Reithmeier RA. Non-random distribution of amino acids in the transmemrbane segments of human type I single span membrane proteins. J Mol Biol. 1993;229:602–608. doi: 10.1006/jmbi.1993.1066. [DOI] [PubMed] [Google Scholar]
- 40.Eftink MR. Methods of Biochemical Analysis. John Wiley; New York: 1991. pp. 127–205. [DOI] [PubMed] [Google Scholar]
- 41.Lakowicz JR. Principles of Fluorescence Spectroscopy. Kluwer-Plenum; New York: 1999. [Google Scholar]
- 42.Vivian JT, Callis PR. Mechanisms of Tryptophan Fluorescence Shifts in Protein. Biophys J. 2001;80:2093–2109. doi: 10.1016/S0006-3495(01)76183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mayer M, Meyer B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem Int Ed Engl. 1999;38:1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 44.Mayer M, Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 45.Peng JW, Lepre CA, Fejzo J, Abdul-Manan N, Moore JM. Nuclear magnetic resonance-based approaches for lead generation in drug discovery. Methods Enzymol. 2001;338:202–230. doi: 10.1016/s0076-6879(02)38221-1. [DOI] [PubMed] [Google Scholar]
- 46.Stockman BJ, Dalvit C. NMR screening techniques in drug discovery and drug design. Prog Nucl Magn Reson Spectrosc. 2002;41:187–231. [Google Scholar]
- 47.Meinecke R, Meyer B. Determination of the binding specificity of an integral membrane protein by saturation transfer difference NMR: RGD peptide ligands binding to integrin αIIbβ3. J Med Chem. 2001;44:3059–3065. doi: 10.1021/jm0109154. [DOI] [PubMed] [Google Scholar]
- 48.Streiff JH, Juranic NO, Macura SI, Warner DO, Jones KA, Perkins WJ. Saturation Transfer Difference Nuclear Magnetic Resonance Spectroscopy as a Method for Screening Proteins for Anesthetic Binding. Mol Pharmacol. 2004;66:929–935. doi: 10.1124/mol.66.4.. [DOI] [PubMed] [Google Scholar]
- 49.Tugarinov V, Kay LE. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J Am Chem Soc. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- 50.Tate CG. Overexpression of mammalian integral membrane proteins for structural studies. FEBS Lett. 2001;504:94–98. doi: 10.1016/s0014-5793(01)02711-9. [DOI] [PubMed] [Google Scholar]
- 51.Mayumi I, Nobuo M, Obita T, Kamishikiryo J, Maenaka K, Kohda D. Purification, crystallization and the preliminary X-ray diffraction studies of the soluble domain of the oligosaccharyltransferase STT3 subunit from the thermophilic archaeon Pyrococcus furiosus. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:798–801. doi: 10.1107/S1744309107040134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rogl H, Kosemund K, Kuhlbrandt W, Collinson I. Refolding of Escherichia coli produced membrane protein inclusion bodies immobilised by nickel chelating chromatography. FEBS Lett. 1998;432:21–26. doi: 10.1016/s0014-5793(98)00825-4. [DOI] [PubMed] [Google Scholar]
- 53.Gorzelle BM, Nagy JK, Oxenoid K, Lonzer WL, Cafiso DS, Sanders CR. Reconstitutive refolding of diacylglycerol kinase, an integral membrane protein. Biochemistry. 1999;38:16373–16382. doi: 10.1021/bi991292n. [DOI] [PubMed] [Google Scholar]
- 54.Baneres JL, Martin A, Hullot P, Girard JP, Rossi JC, Parello J. Structure-based Analysis of GPCR Function: Conformational Adaptation of both Agonist and Receptor upon Leukotriene B4 Binding to Recombinant BLT1. J Mol Biol. 2003;329:801–814. doi: 10.1016/s0022-2836(03)00438-8. [DOI] [PubMed] [Google Scholar]
- 55.Kelleher DJ, Kreibich G, Gilmore R. Oligosaccharyltransferase activity is associated with a protein complex composed of ribophorins I and II and a 48 kD protein. Cell. 1992;69:55–65. doi: 10.1016/0092-8674(92)90118-v. [DOI] [PubMed] [Google Scholar]
- 56.Baleja JD. Structure determination of membrane-associated proteins from NMR data. Anal Biochem. 2001;288:1–15. doi: 10.1006/abio.2000.4815. [DOI] [PubMed] [Google Scholar]
- 57.Chill JH, Louis JM, Miller C, Bax A. NMR study of the tetrameric KcsA potassium channel in detergent micelles. Protein Sci. 2006;15:684–698. doi: 10.1110/ps.051954706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jaroniec CP, Kaufman JD, Stahl SJ, Viard M, Blumenthal R, Wingfield PT, Bax A. Structure and Dynamics of Micelle-Associated Human Immunodeficiency Virus gp41 Fusion Domain. Biochemistry. 2005;44:16167–16180. doi: 10.1021/bi051672a. [DOI] [PubMed] [Google Scholar]
- 59.Howell SC, Mesleh MF, Opella SJ. NMR Structure Determination of a Membrane Protein with Two Transmembrane Helices in Micelles: MerF of the Bacterial Mercury Detoxification System. Biochemistry. 2005;44:5196–5206. doi: 10.1021/bi048095v. [DOI] [PubMed] [Google Scholar]
- 60.Lee S, Mesleh MF, Opella SJ. Structure and dynamics of a membrane protein in micelles from three solution NMR experiments. J Biomol NMR. 2003;26:327–334. doi: 10.1023/a:1024047805043. [DOI] [PubMed] [Google Scholar]
- 61.Mascioni A, Porcelli F, Ilangovan U, Ramamoorthy A, Veglia G. Conformational preferences of the amylin nucleation site in SDS micelles: an NMR study. Biopolymers. 2003;69:29–41. doi: 10.1002/bip.10305. [DOI] [PubMed] [Google Scholar]
- 62.Li H, Chavan M, Schindelin H, Lennarz WJ, Li H. Structure of the Oligosaccharyl Transferase Complex at 12 Å Resolution. Structure. 2008;16:432–440. doi: 10.1016/j.str.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 63.Clarke DM, Loo TW, MacLennan DH. Functional consequences of alterations to amino acids located in the nucleotide binding domain of the Ca2+-ATPase of Sarcoplasmic Reticulum. J Biol Chem. 1990;265:22223–22227. [PubMed] [Google Scholar]