

Abstract
The PKC1–MPK1 pathway in yeast functions in the maintenance of cell wall integrity and in the stress response. We have identified a family of genes that are putative regulators of this pathway. WSC1, WSC2, and WSC3 encode predicted integral membrane proteins with a conserved cysteine motif and a WSC1–green fluorescence protein fusion protein localizes to the plasma membrane. Deletion of WSC results in phenotypes similar to mutants in the PKC1–MPK1 pathway and an increase in the activity of MPK1 upon a mild heat treatment is impaired in a wscΔ mutant. Genetic analysis places the function of WSC upstream of PKC1, suggesting that they play a role in its activation. We also find a genetic interaction between WSC and the RAS–cAMP pathway. The RAS–cAMP pathway is required for cell cycle progression and for the heat shock response. Overexpression of WSC suppresses the heat shock sensitivity of a strain in which RAS is hyperactivated and the heat shock sensitivity of a wscΔ strain is rescued by deletion of RAS2. The functional characteristics and cellular localization of WSC suggest that they may mediate intracellular responses to environmental stress in yeast.
Stress challenges all organisms and a response has evolved to protect cellular components and to repair damage (1, 2). In the yeast Saccharomyces cerevisiae, the PKC1–MPK1 pathway regulates cell wall biosynthesis during periods of polarized growth such as budding and mating projection formation and is necessary for a normal response to stress (3–8). PKC1 controls the activity of a mitogen-activated protein kinase cascade that is composed of BCK1/SLK1 (6, 9), MKK1 and MKK2 (10), and MPK1/SLT2 (7, 11). PKC1 is a target of the GTPase RHO1 (12, 13), which is regulated by the phosphatidylinositol 3-kinase homolog TOR2 (14). Null mutations in the PKC1–MPK1 pathway result in a lysis defect that can be rescued by osmotic stabilizers, such as 1M sorbitol. This phenotype is thought to be a consequence of a defect in polarization of the actin cytoskeleton that may result in the inability to recruit vesicles necessary for biosynthesis of the cell wall (15). RHO1 functions in the organization of the actin cytoskeleton (16, 17) possibly by regulation of the PKC1–MPK1 cascade (12, 13) and directly regulates cell wall biosynthesis by activation of β-glucan synthase, an enzyme that synthesizes one of its major components (18, 19). Mutants in the PKC1–MPK1 pathway have defects in their response to stress as measured by their inability to acquire thermotolerance and by lack of activation of MPK1 upon treatment with mild heat (20). Acquisition of thermotolerance is a response in which cells are able to withstand an otherwise lethal heat shock (50–55°C), if they are pretreated with mild heat (37°C) or if they are starved (21, 22).
Defects in acquisition of thermotolerance are also associated with mutations that constitutively activate the RAS–cAMP pathway (21–23). RAS regulates the activity of adenylate cyclase to produce cAMP, which activates cAMP-dependent protein kinase (PKA). Deletion of IRA1 or IRA2, which encode GTPase-activating proteins, result in a heat-shock-sensitive phenotype because activated GTP-bound RAS accumulates in the cell (23). We reasoned that we could identify inhibitors of the activity of RAS or its targets, by screening for genes that suppress the heat shock sensitivity of an ira1Δ strain. By this approach, we isolated two genes with sequence homology, WSC1 and WSC2 (for cell wall integrity and stress response component). We identified another homolog, WSC3, by searching the GenBank database. Interestingly, the characterization of the function of these genes indicate that they may be components of the PKC1–MPK1 signaling pathway and that they function in the heat shock response and in the maintenance of cell wall integrity in yeast.
MATERIALS AND METHODS
Media and Strains.
The composition of the media and manipulation of yeast for transformation was as described (24). Strains used in this study are as follows: SP1 (25), IR-1 (26), IR2.53 (27), TF1.5prC (28), JF36A (29), DJ13 (30), DL251 (9), and KT626, MATa leu2 ura3 his4 (Kelly Tatchell, North Carolina State University). ALHWT MATa leu2 his3 ura3 trp1 ade8; ALH7 MATa leu2 his3 ura3 trp1 ade8 wsc1::ADE8; ALH18 MATa leu2 his3 ura3 trp1 ade8 wsc2::URA3; ALH15 MATa leu2 his3 ura3 trp1 ade8 wsc3::TRP1; ALH715 MATa leu2 his3 ura3 trp1 ade8 wsc1::ADE8 wsc3::TRP1; ALH718 MATa leu2 his3 ura3 trp1 ade8 wsc1::ADE8 wsc2::URA3; ALH758 MATa leu2 his3 ura3 trp1 ade8 wsc1::ADE8 wsc2::URA3 wsc3::TRP1; HRB718, MATa leu2 his3 ura3 trp1 ade8 wsc1::ADE8 wsc2::HIS3; JV758Δras, MATa leu2 his3 ura3 trp1 ade8 wsc1::ADE8 wsc2::URA3 wsc3::TRP1 ras2::LEU2 (this study).
Genetic Screen.
The ira1Δ strain (IR-1) was transformed with a yeast genomic library cloned into a high-copy-number plasmid (30). Transformants were replica-plated and heat-shocked at 55°C for 15 min. Surviving colonies were subjected to segregation analysis (31). Clones were mapped for regions of complementation and sequenced. pIRIS7 contains the gene that we named WSC1. pIRIS18 and pIRIS22 contain WSC2. To study the function of WSC3, we amplified the gene from a yeast genomic library. The sequence of these genes are accessible as a result of the S. cerevisiae genome project. The locus for WSC1 is YOR008C, for WSC2 is YNL283C, for WSC3 is YOL105C, and for WSC4 is YHL028W.
Gene Disruptions.
The details of the constructs used for deletion of the WSC genes are available upon request from the authors. The plasmids were used to conduct sequential gene replacement experiments (32) in the diploid strain DJ13. Southern blot hybridization was used to verify the deletions, generating the strain DJ13–7/18/15. Tetrad analysis was performed to isolate the individual mutant strains that we named ALH (see Table 1). The HRB718 strain was obtained by deletion of WSC2 in a wsc1Δ strain. The JV758Δras strain was generated by deletion of RAS2 (33) in the strain ALH758.
Table 1.
Phenotypes caused by deletion of WSC
| Genotype | Growth on YPD 28°C | Growth on YPD 37°C | Growth on SC 28°C | Growth on SC 37°C | Caffeine sensitivity SC 28°C | Glycogen accumulation | Staurosporine sensitivity SC 28°C | Nitrogen starvation sensitivity |
|---|---|---|---|---|---|---|---|---|
| Wild type | + | + | + | + | − | + | − | − |
| wsc1Δ | + | − | + | + | − | + | − | − |
| wsc1wsc3Δ | + | − | + | − | + | − | ± | − |
| wsc1wsc2Δ | − | − | + | − | + | − | ± | − |
| wsc1wsc2wsc3Δ | − | − | + | − | + | − | + | ± |
YPD, 1% yeast extract, 2% peptone, 2% dextrose; SC, synthetic complete medium containing yeast nitrogen base at 0.67 g/liter, 2% dextrose, and amino acid supplements. Cells were streaked on plates to test for growth at the designated temperatures, sensitivity to caffeine (3 mM), and staurosporine (1 μg/ml). Plates were scored after 2 days. Glycogen accumulation was determined by iodine staining of patches on SC plates. Nitrogen starvation was tested after 2 weeks of incubation on plates lacking a nitrogen source.
Treatment of Cells and Preparation of Cell Extracts.
To measure activation of MPK1 after a mild heat treatment, we followed published procedures (20) except that 40 μg of protein was loaded onto an 8% gel and immunoblotted with phospho-specific p44/42 mitogen-activated protein kinase antibody (New England Biolabs, product 9101).
To prepare extracts from cells expressing the WSC1–green fluorescence protein (GFP) fusion protein, we grew cultures in glucose or galactose containing medium. Cells were washed and lysed by vortexing with glass beads in buffer A (50 mM Tris⋅HCl and protease inhibitors; ref. 27). Extracts were centrifuged at 1,000 rpm in an Eppendorf centrifuge for 10 sec, and the supernatant was removed and centrifuged at 14,000 rpm in an Eppendorf centrifuge for 30 min. The supernatant was removed and used as the cytosolic fraction. The remaining pellet was washed, centrifuged, and resuspended in buffer A containing 1% Nonidet P-40 followed by incubation on ice for 10 min and centrifugation at 14,000 rpm for 30 min. The supernatant from this centrifugation was used as the membrane fraction.
RESULTS AND DISCUSSION
Sequence Comparison of the WSC Proteins.
We have recently identified other proteins related to WSC. One is from S. cerevisiae and we named the gene WSC4. A second gene, Hp-WSC, encodes a partial hypothetical protein in the 3′ region of LEU2 (34) of the related yeast Hansenula polymorpha. The sequence of all WSC proteins and their comparison is shown in Fig. 1A. The WSC proteins are 50% similar and 35% identical. WSC2 and WSC3 show the highest degree of conservation, 61% similar and 50% identical. The sequence does not suggest a known enzymatic activity but predicts that they are transmembranous. The WSC proteins have similar characteristics (Fig. 1B). A hydrophobic domain at the N terminus, which may be a signal peptide, is followed by a cysteine motif. A serine/threonine-rich domain that is variable in length may be sites for glycosylation and is located between the cysteine motif and the predicted transmembrane domain. The C terminus is also variable in length and is highly charged. It is predicted to be intracellular and has the highest degree of divergence among the WSC family, with the exception of WSC2 and WSC3. There are, however, two conserved sequences in the S. cerevisiae WSC proteins, a KXYQ after the transmembrane domain and a DXXD at the end of the C terminus (Fig. 1A). These amino acids are not present in Hp-WSC, but the sequence of the gene is incomplete. These blocks may be important for the function of the proteins, with the tyrosine residue being a putative site for phosphorylation.
Figure 1.
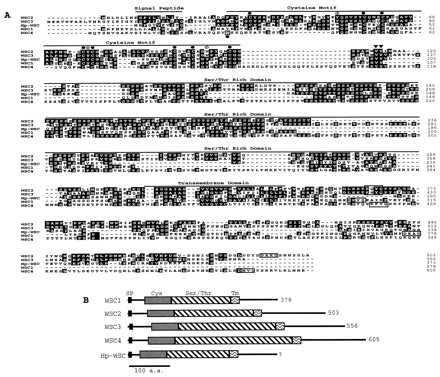
Sequence comparison of the WSC protein family. (A) Identical amino acids in at least two of the proteins are highlighted in white with a dark background. Gaps were used to maximize the alignment generated with the GCG software package. Different domains in the proteins are labeled. The conserved cysteine residues in the cysteine motif are designated by a solid circle, and the aromatic amino acids are designated by the symbol Φ. The last amino acids of this motif VY are designated by inverted solid triangles. Conserved sequences in the C terminus, KXYQ and DXXD, are boxed. (B) Schematic diagrams of the WSC family. SP, signal peptide; Cys, cysteine motif; Ser/Thr, serine and threonine-rich domain; Tm, transmembrane domain.
Significantly, the most highly conserved domain among the WSC proteins is the cysteine motif which is predicted to be extracellular. The motif is as follows: C1-X-S-X12–16-Φ-Q-S-X3-C2-X3-C3-X5–8-A-L(I)-X5–6-C4-Φ-C5-X12–17-C6-X3-C7-X-G-Φ-X4-C8-G-X6(30)-VY, where the cysteine (C) residues are numbered 1 to 8. Cysteine motifs occur in receptors, transcription factors, and many proteins with diverse functions. They are involved in ligand binding, dimerization, and coordination with zinc ions (35–37). The cysteine residues stabilize a protein fold that is necessary for protein–protein or for protein–DNA interactions. The presence of the cysteine motif in the WSC proteins suggests that it plays an important role in their function. Searches of this motif in the GenBank database result in no significant matches.
WSC4, which may be functionally related to the other WSC genes, was identified as a suppressor (YFW1) of the sensitivity to alkylating agents of an Escherichia coli alkB mutant (38). alkB is one of a group of genes induced by alkylating agents. The significance of this finding awaits further characterization of the function of alkB in E. coli and the function of WSC4 in yeast.
Cellular Localization of WSC1.
To establish cellular localization, we expressed a fusion of WSC1 and GFP (Fig. 2A). GFP expressed alone is seen as a diffuse signal throughout the cell, whereas the fusion protein (WSC–GFP) is located in the periphery of the cell. The same distribution is seen in the absence of the cell wall. By using antibodies to GFP, we established that GFP partitions equally between a crude membrane fraction and the soluble fraction, whereas the WSC–GFP protein fractionates mostly with the crude membrane fraction (Fig. 2B). This indicates that WSC1 is localized to the plasma membrane.
Figure 2.
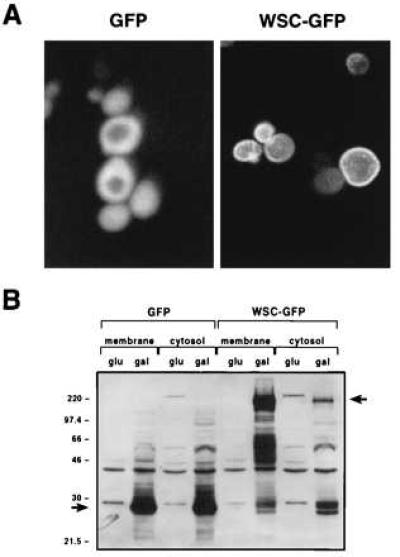
Cellular localization of WSC1. WSC1 was amplified by PCR and cloned into pMTS395 (39) to generate pMTS395-WSC1. This is a single-copy plasmid that contains a GFP (S65T) mutant gene, expressed under the control of a galactose-inducible promoter. The WSC1 gene was cloned 5′ to GFP. A functional test of this construct is shown in Fig. 6. (A) The strain KT626 was transformed with the GFP or the WSC1–GFP fusion construct. Transformants were selected on medium containing glucose and then grown for 12 h on galactose-containing medium. Cells were fixed in 4% paraformaldehyde followed by analysis using fluorescence confocal microscopy. (B) Cultures from A were diluted in glucose (glu)- or galactose (gal)-containing medium and grown for 12 h. Cells extracts were prepared and 20 μg of protein from membrane or cytosolic fractions was loaded unto a 10% gel. Immunoblot analysis was performed with a GFP monoclonal antibody (CLONTECH, product 8362–1).
Phenotypes Caused by the Deletion of the WSC Genes.
Deletion of WSC2 and WSC3, individually or in combination, do not cause phenotypes that are different from the wild type. Deletion of WSC1 results in a cell lysis defect that is also observed in mutants of the PKC1–MPK1 pathway (3–12). A wsc1Δ mutant has a thermosensitive growth defect at 37°C on YPD (Table 1). It grows at all temperatures in SC medium. Deletion of WSC2 and/or WSC3 exacerbates the phenotype of the wsc1Δ strain, suggesting that the WSC genes may be partially redundant. Double deletion mutants become temperature sensitive in SC medium at 37°C and deletion of the three genes results in inability to grow on YPD at any temperature. The growth defect is due to cell lysis, determined by its suppression by an osmotic stabilizer (Fig. 3) or by assaying alkaline phosphatase activity upon incubation at 37°C on SC medium.
Figure 3.
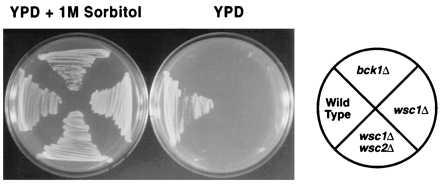
Suppression of the lysis defect of the wscΔ strains by sorbitol. Yeast strains containing deletions of WSC1 (wsc1Δ) or WSC1 and WSC2 (wsc1Δwsc2Δ) were streaked and scored for growth after 2 days on YPD or YPD supplemented with 1 M Sorbitol at 37°C. A bck1Δ strain was used for comparison.
Because mutants in the PKC1–MPK1 pathway in yeast have other phenotypes (6, 40), we tested whether deleting the WSC genes has the same effects (Table 1). Strains with deletion of WSC1, WSC2, or WSC3 are wild type in these responses. The wsc1Δ strain, but not the wsc2Δ or the wsc3Δ, shows sensitivity to caffeine (a phosphodiesterase inhibitor) when the concentration is increased from 3 mM to 10 mM. Deletion of WSC2 or WSC3 in a wsc1Δ background results in sensitivity to 3 mM caffeine, in a weak sensitivity to staurosporine (a kinase inhibitor), and in a defect in glycogen accumulation. Deletion of the three WSC genes increases the sensitivity to staurosporine and results in sensitivity to nitrogen starvation. These results suggest that the WSC genes are required for maintenance of cell wall integrity and that their function is similar to the PKC1–MPK1 pathway in yeast.
Heat Shock Sensitivity of the wsc1wsc2wsc3Δ Mutant.
The wsc1wsc2wsc3Δ mutant is more sensitive than a wild type to a direct exposure to a strong heat shock (Fig. 4). Preexposure of the wild-type strain to a mild heat treatment results in a high survival rate after the strong heat shock (72% at 20 min). In contrast, in the wscΔ mutant, the survival rate is very low (3% at 20 min) and similar to a wild-type strain that has not been preexposed to the mild heat treatment. Although the survival rate of the wscΔ mutant is extremely low, they acquire some thermotolerance. This can be seen by comparing the survival rate of the wscΔ culture that has not been preexposed to mild heat to the wscΔ culture that has (Fig. 4). These results indicate that the WSC genes are required for a normal response to heat shock.
Figure 4.
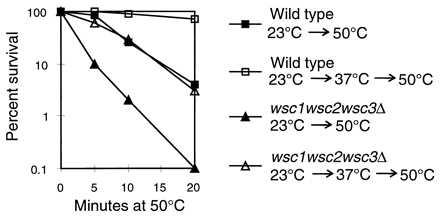
Heat shock sensitivity of a wsc1wsc2wsc3Δ mutant. Cells were grown on YPD containing 1 M sorbitol at 30°C until they reached an OD600 of 1. Cultures were washed and resuspended in SC medium. They were shifted to 50°C for the indicated periods of time or first shifted to 37°C for 30 min followed by the shift to 50°C. After heat shock, the cells were plated on YPD containing 1 M sorbitol and scored for growth after 2–3 days at 30°C. The results are shown as percent survival relative to the cultures before treatment at 50°C. The strains are SP1, wild type, or the ALH758 wscΔ strain. Results are from three experiments.
Genetic Interaction of WSC and the RAS–cAMP Pathway.
We determined the effect of inhibiting the activity of RAS in the heat shock sensitivity of a wscΔ mutant. Fig. 5A shows that overexpression of IRA2, which encodes a GTPase-activating protein that inhibits RAS (23), can suppress the heat shock sensitivity of the wscΔ mutant. Significantly, deletion of RAS2 rescues the heat shock sensitive phenotype of the wscΔ strain (Fig. 5B). Deletion of RAS2 alone does not make the cells more resistant to a direct exposure to the strong heat shock. These results indicate that there is a functional relation between WSC and RAS.
Figure 5.
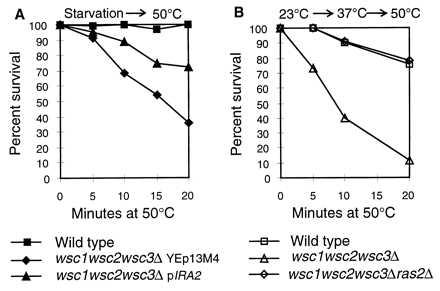
Suppression of the heat shock sensitivity of the wsc1wsc2wsc3Δ strain by overexpression of IRA2 or by deletion of RAS2. (A) The wsc1wsc2wsc3Δ (ALH758) strain was transformed with IRA2 or a control plasmid. Transformants were grown for 2 days and were then shifted to 50°C for the indicated times. Cells were diluted, plated, and grown as in Fig. 4. The wild-type strain (SP1) was used for comparison. (B) The wild type (SP1), wsc1wsc2wsc3Δ (ALH758), and wsc1wsc2wsc3ras2Δ (JV758Δras) strains were grown and treated as in Fig. 4.
Two models can be proposed to explain this genetic interaction. In one, WSC and the RAS–cAMP pathway may be acting on the same downstream target with opposing results. In the second, WSC could be releasing the negative effect that RAS imposes on the target by inactivating the RAS–cAMP pathway. To distinguish between these models, we determined at what point in the RAS–cAMP pathway the WSC genes are exerting their effects by testing for their ability to suppress the heat shock sensitivity of strains with different mutations in the RAS–cAMP pathway (Table 2).
Table 2.
Suppression of the heat shock sensitivity of mutants in the RAS–cAMP pathway
| CDC25 | |||
|---|---|---|---|
| GDP-RAS ⇄ GTP-RAS → CYR1/CAP → cAMP → BCY1/ | |||
| IRA1/IRA2TPK1, TPK2, TPK3 | |||
|
| |||
| Mutant strains | Genotype | WSC1 | WSC2 |
| IR2.53 | ira1Δira2ΔR2Y64 | + | + |
| JF36A | ras1Δras2ΔpCYR1 | + | − |
| TF1.5prC | bcy1Δtpk2Δtpk3Δtpk1w | − | − |
Cells were transformed with high-copy-number/plasmids expressing WSC1 or WSC2 and tested for heat shock sensitivity at 55°C. +, Growth; −, no growth after heat shock treatment.
The effects of WSC are not mediated by IRA1 or IRA2 because they both suppress the heat shock sensitivity of an ira1ira2Δ strain expressing a dominant interfering form of RAS2, which attenuates its strong heat shock sensitivity (41).
The effect of WSC1 and WSC2 seem to be mediated by PKA because they do not suppress the heat shock sensitivity of a strain in which the activity of PKA cannot be regulated. In this strain (Table 2) the regulatory subunit of PKA, BCY1, and two catalytic subunits, TPK2 and TPK3, have been deleted. It also contains a mutation, tpk1w, that attenuates the activity of the kinase but leaves it strong enough to make the cells heat shock sensitive (28).
To determine whether the WSC genes require RAS to mediate their effects, we used a strain with a deletion of both RAS genes (Table 2). This strain is heat-shock-sensitive because it is overexpressing adenylate cyclase (CYR1) in a high-copy-number vector (29). Overexpression of WSC1 but not WSC2 suppresses the heat shock sensitivity of this strain. This suggests that WSC1 acts downstream of RAS perhaps modulating the activity of adenylate cyclase or the activity of PKA. The inability of WSC2 to suppress the heat shock sensitivity of the ras1ras2 pCYR1 strain indicates that WSC2 may function to regulate the activity of RAS. This discrepancy suggests that WSC1 and WSC2 may have multiple targets, consistent with the variability of their C termini and their partial redundancy. Also consistent is the alternative model that the WSC genes act parallel to the RAS–cAMP pathway but their input is weak, explaining their inability to suppress the heat shock sensitivity of the bcy1Δtpk2Δtpk3Δtpk1w mutant or, in the case of WSC2, the heat shock sensitivity of the ras1Δras2ΔpCYR1 strain. Further studies will be necessary to establish the molecular basis of the interaction between WSC and the RAS–cAMP pathway but this interaction seems to be restricted to the heat shock response. Overexpression or deletion of WSC1 does not rescue the lethal phenotype of a ras1Δras2ts strain, which suggests that WSC does not mediate the effects of RAS in cell cycle progression. Conversely, the role of the WSC genes in the maintenance of cell wall integrity seems to be independent of the RAS–cAMP pathway because deletion or overexpression of RAS2 does not rescue the cell lysis defect of the wsc1wsc2wsc3Δ strain. There is evidence that RAS in yeast has other functions. One such function is linked to the regulation of the actin cytoskeleton because the cyclase-associated protein (CAP) that is required for RAS activation of adenylate cyclase is also required for the normal distribution of actin (42–47). The lysis defect of the wscΔ mutants may be in part a result of a defect in the function of CAP. We have not found evidence of a genetic interaction between WSC and CAP, but because the role of CAP in the function of the actin cytoskeleton and the mechanism by which CAP is regulated are not known, a possibility for a functional relation between WSC and CAP cannot be discarded.
Genetic Interaction of WSC, PKC1, RHO1, and RHO3.
PKC1 expressed in a multicopy vector suppresses the lysis defect of a wsc1wsc3Δ mutant (Fig. 6A) and a wsc1wsc2wsc3Δ strain. Overexpression of BCK1 or MPK1 does not rescue the lysis defect of the wscΔ strains. A single copy of an activated mutant allele, BCK1–20, suppresses the defect very weakly and overexpression of BCK1 potentiates the suppression by PKC1. These results are consistent with the observation that BCK1 does not suppress the lysis defect of a pkc1Δ strain, but the activated mutant allele BCK1–20 does (9).
Figure 6.
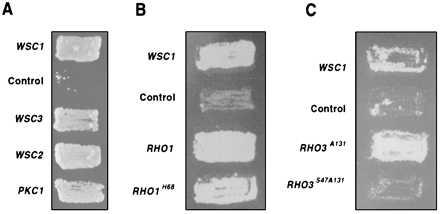
Suppression of the lysis defect of the wscΔ strains by overexpression of PKC1, RHO1, RHO3, and WSC. (A) The strain ALH715 (wsc1wsc3Δ) was transformed with various multicopy plasmids: pAD4Δ (control) (26), pIRIS7 (WSC1), pIRIS18(WSC2), pAD4Δ-WSC3, and PKC1 (3). Patches were made on selective medium with 1 M sorbitol, grown for 2 days, replica-plated onto YPD plates, and then incubated at 37°C for 3 days. The WSC3 gene (1.5 kb) was amplified by PCR and is expressed under the control of the alcohol dehydrogenase promoter (ADH). (B) The HRB718 (wsc1wsc2Δ) was transformed with plasmids expressing RHO1 or the RHO1H68 mutant (19) under the control of a galactose-inducible promoter and the plasmids pMTS395 and pMTS395-WSC1 described in Fig. 2. Patches were made on medium containing glucose and 1 M sorbitol and after 2 days, the plates were replica-plated onto YPgal plates (yeast extract 1%/peptone 2%/galactose 2%). They were then incubated at 30°C for 3 days and scored for growth. (C) Same as in B, but the cells were transformed with plasmids expressing the mutant RHO3A131, or RHO3S47A131 (48).
Overexpression of the WSC genes suppress the lysis defect of the wsc1wsc3Δ mutant (Fig. 6A) or the wsc1wsc2wsc3Δ mutant strain, further demonstrating that they have overlapping functions. Overexpression of WSC1 or WSC2 in a pkc1Δ or in a bck1Δ background does not suppress the lysis defect. This epistasis analysis places the function of WSC upstream of PKC1.
To further characterize the genetic interaction between WSC and the PKC1–MPK1 pathway, we tested the phenotypes of strains with deletion of the WSC genes in combination with a deletion of PKC1 or BCK1. We deleted the PKC1 gene in a diploid strain with deletions in WSC1, WSC2, and WSC3 and performed tetrad analysis. Deletion of the WSC genes individually or in combination are not synthetic lethal with pkc1Δ. We tested the effect of deletion of the WSC genes (all combinations) on the growth of a pkc1Δ strain on YPD medium containing 0.5 M sorbitol at room temperature and at 30°C and on YPD medium containing 1 M sorbitol at 33°C. Under these conditions a pkc1Δ mutant grows poorly (ref. 48 and this study), whereas the wscΔ strains grow normally. Deletion of the WSC genes does not exacerbate the growth defect of the pkc1Δ strain. In addition, the lysis defect of a wsc1wsc2Δ strain is not exacerbated by deletion of bck1Δ. These results strongly suggest that the WSC genes function in the PKC1–MPK1 pathway in yeast.
Because RHO1 acts upstream of PKC1 (12, 13), we tested whether its overexpression can rescue the lysis defect of a wscΔ mutant strain. Expression of RHO1 or the constitutively active mutant RHO1H68 (19) rescues the lysis defect (Fig. 6B), further supporting that the WSC genes function upstream of the PKC1–MPK1 pathway.
RHO3 and RHO4 encode GTPases required for bud formation and organization of the actin cytoskeleton and their deletion results in a lysis defect. They do not complement defects of a rho1Δ strain and RHO1 does not suppress the lysis defect of a rho3rho4Δ strain (49, 50). RHO3 does not regulate β-glucan synthase activity (18). An activated allele, RHO3A131, is able to rescue the defect of the wscΔ mutant (Fig. 6C). Expression of this allele with a second mutation (RHO3S47–131) is unable to suppress the lysis defect of the wscΔ strain. This mutation is analogous to mutations in the effector domain of mammalian Ha-ras and has been shown to abolish the activity of RHO3A131 (49). These results suggest that the wscΔ mutants have defects that affect other pathways in addition to the RHO1-regulated PKC1–MPK1 pathway.
Requirement of WSC for the Activation of the PKC1–MPK1 Cascade.
Activation of the PKC1–MPK1 kinase cascade can be assessed by measuring the activity of MPK1 in response to a mild heat shock (20). The catalytic activity of MPK1 increases upon phosphorylation by the dual specificity kinases MKK1/MKK2. We measured activation of MPK1 in a wscΔ strain by using a specific antibody that recognizes only the phosphorylated active form of MPK1. A mild heat shock treatment of the wild-type strain results in a strong signal in both endogenous and epitope-tagged MPK1–hemagglutinin (HA; Fig. 7A, lanes 1 and 2). Previous analysis of the phosphorylation state of MPK1–HA in response to changes in osmolarity (51) showed that the lower molecular weight band is not present in mpk1Δ strains. We confirmed that the upper band is the HA-epitope-tagged protein by immunoprecipitation. In contrast to the wild type, the wscΔ shows only a small increase in phosphorylation of MPK1–HA (Fig. 7A, lanes 3 and 4) in cells that are all expressing equivalent amounts of the MPK1–HA (Fig. 7B). This indicates that the WSC genes are required for signaling in the PKC1–MPK1 pathway, further supporting results from the epistasis analyses. Similar results have been obtained in a wsc1Δ (called HCS77) strain (52). The residual MPK1 phosphorylation that is seen in Fig. 7B suggest that these cells have other mechanisms to activate the PKC1–MPK1 pathway upon exposure to a mild heat treatment and is consistent with the residual ability of these strains to acquire thermotolerance.
Figure 7.
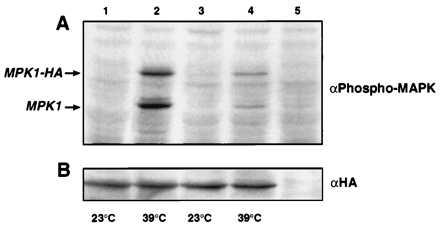
Phosphorylation of MPK1 in response to a mild heat treatment in the wscΔ mutant. (A) A wild-type (ALHWT; lanes 1 and 2) or a mutant wsc1wsc2wsc3Δ (ALH758; lane 3 and 4) strain overexpressing MPK1–HA were incubated at 23°C (lanes 1 and 3) or 39°C (lanes 2 and 4) for 30 min. Phosphorylated MPK1–HA or endogenous MPK1 are marked with an arrow and were detected by immunoblotting with a phospho-specific p44/42 mitogen-activated protein kinase antibody. Lane 5 contains an untreated cell extract that is not expressing MPK1–HA. (B) Cell extracts identical to those in A were immunoblotted with the anti-HA antibody.
In summary, the WSC family of genes are components of the stress response in yeast. Environmental stress affects all organisms and there are multiple elements of this response that have been conserved in evolution, including the involvement of small GTPases and mitogen-activated protein kinase cascades (53, 54). Perhaps, this conservation also extends to the presence of proteins that may function like WSC in animal cells.
Acknowledgments
We thank Joe Gray and Ira Herskowitz for communicating results before publication; L. Rodgers and M. Riggs for sequencing; G. Paravicini, D. Levin, A. Thierry, B. Dujon, J. Imai, Y. Matsui, and J. Field for plasmids, cosmids, and yeast strains; and B. Matsumoto for help with the microscope. This work was supported by funds from the National Science Foundation and an American Cancer Society Junior Faculty Award to R.B. This work was initiated in the laboratory of M. Wigler, who is supported by funds from the National Institutes of Health. R.B. thanks him for his inspiration and support.
ABBREVIATIONS
- PKA
cyclic AMP-dependent protein kinase
- HA
hemagglutinin
- GFP
green fluorescence protein
- CAP
cyclase-associated protein
References
- 1.Minowada G, Welch W J. J Clin Invest. 1995;95:3–12. doi: 10.1172/JCI117655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruis H, Schüller C. Bioessays. 1995;17:959–965. doi: 10.1002/bies.950171109. [DOI] [PubMed] [Google Scholar]
- 3.Paravicini J, Cooper M, Friedli L, Smith D J, Carpentier J-L, Klig L S, Payton M A. Mol Cell Biol. 1992;12:4896–4905. doi: 10.1128/mcb.12.11.4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levin D E, Bartlett-Heubusch E. J Cell Biol. 1992;116:1221–1229. doi: 10.1083/jcb.116.5.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levin D E, Bowers B, Chen C-Y, Kamada Y, Watanabe M. Cell Mol Biol Res. 1994;40:229–239. [PubMed] [Google Scholar]
- 6.Costigan C, Gehrung S, Snyder M. Mol Cell Biol. 1992;12:1162–1178. doi: 10.1128/mcb.12.3.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazzoni C, Zarzov P, Rambourg A, Mann C. J Cell Biol. 1993;123:1821–1833. doi: 10.1083/jcb.123.6.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zarzov P, Mazzoni C, Mann C. EMBO J. 1996;15:83–91. [PMC free article] [PubMed] [Google Scholar]
- 9.Lee K S, Levin D E. Mol Cell Biol. 1992;12:172–182. doi: 10.1128/mcb.12.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irie K, Takase M, Lee K S, Levin D E, Araki H, Matsumoto K, Oshima J. Mol Cell Biol. 1993;13:3076–3083. doi: 10.1128/mcb.13.5.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee K S, Irie K, Gotoh Y, Watanabe Y, Araki H, Nishida E, Matsumoto K, Levin D E. Mol Cell Biol. 1993;13:3067–3075. doi: 10.1128/mcb.13.5.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nonaka H, Tanaka K, Hirano H, Fujiwara T, Kohno H, Umikawa M, Mino A, Takai Y. EMBO J. 1995;14:5931–5938. doi: 10.1002/j.1460-2075.1995.tb00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamada Y, Qadota H, Python C P, Anraku Y, Ohya Y, Levin D E. J Biol Chem. 1996;271:9193–9196. doi: 10.1074/jbc.271.16.9193. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt A, Bickle M, Beck T, Hall M N. Cell. 1997;88:531–542. doi: 10.1016/s0092-8674(00)81893-0. [DOI] [PubMed] [Google Scholar]
- 15.Cid V J, Durán A, del Rey F, Snyder M P, Nombela C, Sánchez M. Microbiol Rev. 1995;59:345–386. doi: 10.1128/mr.59.3.345-386.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamochi W, Tanaka K, Nonaka H, Maeda A, Musha T, Takai T. J Cell Biol. 1994;125:1077–1093. doi: 10.1083/jcb.125.5.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang T, Bretscher A. Mol Biol Cell. 1995;6:1011–1024. doi: 10.1091/mbc.6.8.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drgonová J, Drgon T, Tanaka K, Kollár R, Chen G-C, Ford R A, Chan C S M, Takai Y, Cabib E. Science. 1996;272:277–279. doi: 10.1126/science.272.5259.277. [DOI] [PubMed] [Google Scholar]
- 19.Qadota H, Python C P, Inoue S B, Arisawa M, Anraku Y, Zheng Y, Watanabe T, Levin D E, Ohya Y. Science. 1996;272:279–281. doi: 10.1126/science.272.5259.279. [DOI] [PubMed] [Google Scholar]
- 20.Kamada Y, Jung U S, Piotrowski J, Levin D E. Genes Dev. 1995;9:1559–1571. doi: 10.1101/gad.9.13.1559. [DOI] [PubMed] [Google Scholar]
- 21.Piper P W. FEMS Microbiol Rev. 1993;11:339–356. doi: 10.1111/j.1574-6976.1993.tb00005.x. [DOI] [PubMed] [Google Scholar]
- 22.Werner-Washburne M, Braun E, Johnston G C, Singer R. Microbiol Rev. 1993;57:383–401. doi: 10.1128/mr.57.2.383-401.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broach J R, Deschenes R J. Adv Cancer Res. 1990;54:79–139. doi: 10.1016/s0065-230x(08)60809-x. [DOI] [PubMed] [Google Scholar]
- 24.Rose M D, Winston F, Hieter P, editors. Methods in Yeast Genetics: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1990. [Google Scholar]
- 25.Toda T, Uno I, Ishikawa T, Powers S, Kataoka T, Broek D, Cameron S, Broach J, Matsumoto K, Wigler M. Cell. 1985;40:27–36. doi: 10.1016/0092-8674(85)90305-8. [DOI] [PubMed] [Google Scholar]
- 26.Ballester R, Michaeli T, Ferguson K, Xu H-P, McCormick F, Wigler M. Cell. 1989;59:681–68. doi: 10.1016/0092-8674(89)90014-7. [DOI] [PubMed] [Google Scholar]
- 27.Gutmann D H, Boguski M, Marchuk D, Wigler F, Collins, Ballester R. Oncogene. 1993;8:761–769. [PubMed] [Google Scholar]
- 28.Cameron S, Levin L, Zoller M, Wigler M. Cell. 1988;53:555–566. doi: 10.1016/0092-8674(88)90572-7. [DOI] [PubMed] [Google Scholar]
- 29.Field J, Nikawa J-I, Broek D, MacDonald B, Rodgers L, Wilson I A, Lerner R A, Wigler M. Mol Cell Biol. 1988;8:2159–2165. doi: 10.1128/mcb.8.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nikawa J-I, Cameron S, Toda T, Ferguson K M, Wigler M. Genes Dev. 1987;1:931–937. doi: 10.1101/gad.1.9.931. [DOI] [PubMed] [Google Scholar]
- 31.Rose M D, Broach J R. Methods Enzymol. 1991;194:195–230. doi: 10.1016/0076-6879(91)94017-7. [DOI] [PubMed] [Google Scholar]
- 32.Rothstein R. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- 33.Kataoka T, Powers S, McGill C, Fasano O, Strathern J, Broach J, Wigler M. Cell. 1984;37:437–445. doi: 10.1016/0092-8674(84)90374-x. [DOI] [PubMed] [Google Scholar]
- 34.Agaphonov M O, Poznyakovski A I, Bogdanova A I, Ter-avanesyan M D. Yeast. 1994;10:509–513. doi: 10.1002/yea.320100410. [DOI] [PubMed] [Google Scholar]
- 35.Klug A, Schwabe J W R. FASEB J. 1995;9:597–604. [PubMed] [Google Scholar]
- 36.Saurin A J, Borden K L B, Boddy M N, Freemont P S. Trends Biochem Sci. 1996;21:208–214. [PubMed] [Google Scholar]
- 37.Wells J A. Curr Biol. 1994;6:163–173. doi: 10.1016/0955-0674(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 38.Wei Y-F, Chen B J, Samson L. J Bacteriol. 1995;177:5009–5015. doi: 10.1128/jb.177.17.5009-5015.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marschall L G, Jeng R L, Mulholland J, Stearns T. J Cell Biol. 1996;134:443–454. doi: 10.1083/jcb.134.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshida S, Ikeda E, Uno I, Mitsuzawa H. Mol Gen Genet. 1992;101:337–344. doi: 10.1007/BF00292700. [DOI] [PubMed] [Google Scholar]
- 41.Jung V, Wei W, Ballester R, Camonis J, Mi S, van Aelst L, Wigler M, Broek D. Mol Cell Biol. 1994;14:3707–3718. doi: 10.1128/mcb.14.6.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Field J, Vojtek A, Ballester R, Bolger G, Colicelli J, Ferguson K, Gerst J, Kataoka T, Michaeli T, Powers S, Riggs M, Rodgers L, Wieland I, Wheland B, Wigler M. Cell. 1990;61:319–327. doi: 10.1016/0092-8674(90)90812-s. [DOI] [PubMed] [Google Scholar]
- 43.Fedor-Chaiken M, Deschenes R J, Broach J R. Cell. 1990;61:329–340. doi: 10.1016/0092-8674(90)90813-t. [DOI] [PubMed] [Google Scholar]
- 44.Vojtek A, Haarer B, Field J, Gerst J, Pollard T D, Brown S, Wigler M. Cell. 1991;66:497–505. doi: 10.1016/0092-8674(81)90013-1. [DOI] [PubMed] [Google Scholar]
- 45.Gerst J E, Ferguson K, Vojtek A, Wigler M, Field J. Mol Cell Biol. 1991;11:1248–1257. doi: 10.1128/mcb.11.3.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freeman N L, Chen Z, Horenstein J, Weber A, Field F. J Biol Chem. 1995;270:5680–5685. doi: 10.1074/jbc.270.10.5680. [DOI] [PubMed] [Google Scholar]
- 47.Freeman N L, Lila T, Mintzer K A, Chen Z, Pahk A J, Ren R, Drubin D G, Field J. Mol Cell Biol. 1996;16:548–556. doi: 10.1128/mcb.16.2.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee K S, Hines L K, Levin D E. Mol Cell Biol. 1993;13:5843–5853. doi: 10.1128/mcb.13.9.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Imai J, Toh-e A, Matsui Y. Genetics. 1996;142:359–369. doi: 10.1093/genetics/142.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsui Y, Toh-e A. Mol Cell Biol. 1992;12:5690–5699. doi: 10.1128/mcb.12.12.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davenport K R, Sohaskey M, Kamada Y, Levin D E, Gustin M C. J Biol Chem. 1995;270:30157–30161. doi: 10.1074/jbc.270.50.30157. [DOI] [PubMed] [Google Scholar]
- 52.Gray J, Ogas J, Kamada Y, Stone M, Levin D, Herskowitz I. EMBO J. 1997;16:4924–4937. doi: 10.1093/emboj/16.16.4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Denhardt D T. Biochem J. 1996;318:729–747. doi: 10.1042/bj3180729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woodgett J R, Kyriakis J M, Avruch J, Zon L I, Zanke B, Templeton D J. Philos Trans R Soc Lond-Biol Sci. 1996;351:135–142. doi: 10.1098/rstb.1996.0009. [DOI] [PubMed] [Google Scholar]