

Abstract
Bruton’s tyrosine kinase (Btk) is essential for normal B lymphocyte development and function. The activity of Btk is partially regulated by transphosphorylation within its kinase domain by Src family kinases at residue Tyr-551 and subsequent autophosphorylation at Tyr-223. Activation correlates with Btk association with cellular membranes. Based on specific loss of function mutations, the Btk pleckstrin homology (PH) domain plays an essential role in this activation process. The Btk PH domain can bind in vitro to several lipid end products of the phosphatidylinositol 3-kinase (PI 3-kinase) family including phosphatidylinositol 3,4,5-trisphosphate. Activation of Btk as monitored by elevation of phosphotyrosine content and a cellular transformation response was dramatically enhanced by coexpressing a weakly activated allele of Src (E378G) and the two subunits of PI 3-kinase-γ. This activation correlates with new sites of phosphorylation on Btk identified by two-dimensional phosphopeptide mapping. Activation of Btk was dependent on the catalytic activity of all three enzymes and an intact Btk PH domain and Src transphosphorylation site. These combined data define Btk as a downstream target of PI 3-kinase-γ and Src family kinases.
Bruton’s tyrosine kinase (Btk) is a nonreceptor tyrosine kinase that contains a pleckstrin homology (PH) domain but no apparent lipid modification motif (1). Btk is critical for development and signaling. Btk mutations are associated with the genetic diseases human X-linked agammaglobulinemia (XLA) and murine X-linked immunodeficiency (Xid; refs. 2–5). XLA patients have a dramatic decrease in the number of mature B cells and circulating Ig levels (6). Xid mice or mice with a targeted disruption of Btk have diminished B cell numbers and levels of certain Ig classes (7–9).
PH domains are primarily involved in protein–protein or protein–lipid interactions and regulate enzyme function by controlling interacting partners or cellular localization (10, 11). The N-terminal PH domain of Btk is essential for its activation and biological activity. A mutation in the Btk PH domain causes Xid (R28C; refs. 4 and 5), and other mutations within the PH domain also result in XLA (12, 13). In contrast, a Glu-to-Lys mutation (E41K, BTK*) in the PH domain activates Btk and increases membrane association (14). These gain or loss of function mutations suggest that the PH domain is a critical regulatory domain for Btk activation but give little information regarding specific signaling mechanisms.
The PH domain of Btk was recently shown to bind the phosphatidylinositol 3-kinase (PI 3-kinase) lipid product phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3] (15, 16) and inositol 3-phosphates in vitro (17). Computer modeling identified several residues within the Btk PH domain including Lys-12, Phe-25, and Arg-28, which are thought to be essential for binding these lipid molecules (15, 16, 18, 19). Interestingly, mutation of these residues results in human XLA (e.g., F25S and R28H; ref. 12) or murine Xid (R28C; ref. 4). These data strongly suggest binding of PI (3,4,5)P3 to the Btk PH domain is critical for Btk activation.
PI 3-kinase isoforms are regulated by either receptor tyrosine kinases or G protein-coupled receptors (20, 21). The type IA subfamily signals downstream of receptor tyrosine kinases and is ubiquitously expressed. This subfamily is composed of heterodimers containing one regulatory and one catalytic subunit. The best studied type IA member is p85/PI 3-kinase, which consists of a p110 catalytic subunit and a p85 regulatory subunit (22). The p85 SH2 domain binds phosphotyrosyl residues on activated tyrosine kinases leading to increased PI 3-kinase activity. A G protein-regulated type IB PI 3-kinase, PI 3-kinase-γ, was recently cloned from human bone marrow (23) and pig neutrophils (24). The catalytic subunit of PI 3-kinase-γ, p110γ, can dimerize with a p101 regulatory subunit (24). Complex formation between p110γ and p101 makes p110γ more sensitive to activation by the G protein β-γ heterodimer (24). The specific binding of the Btk PH domain with PI (3,4,5)P3 prompted us to investigate whether there is a functional interaction between PI 3-kinase and Btk.
The biological function of Btk is influenced by Src family kinases, which directly activate Btk (25–28). Src family kinases transphosphorylate Btk on tyrosine 551 (Tyr-551), which is homologous to the conserved activation loop tyrosine of the human insulin receptor tyrosine kinase (27, 29). Phosphorylation of Btk Tyr-551 subsequently activates the kinase activity of Btk. Btk then autophosphorylates tyrosine 223 (Tyr-223) within the SH3 domain (30).
To analyze the interaction of PI 3-kinase and Btk in cellular signaling, we expressed both enzymes in rodent fibroblasts. Previous studies showed that this system is a useful surrogate to analyze Btk activation (14, 27, 28). Btk activation in fibroblasts by Src family kinases is similar to activation by Src family kinases in B cells stimulated through B cell receptors (27). This report provides evidence for Btk serving as a downstream target for the joint action of PI 3-kinase and Src family kinases, which are both essential for full activation.
MATERIALS AND METHODS
Plasmids and Virus Stocks.
Wild type Btk cDNA and Btk mutants (R28C, ΔSH3, K430R, and Y551F) were subcloned into the retroviral expression vector pSRαMSVtkneo (2, 14). Myc-epitope-tagged PI 3-kinase-γ catalytic subunit p110γ was subcloned into the retroviral expression vector pSRαMSVtkneo vector by using a unique NotI site 3′ of the long terminal repeat promoter. Kinase-inactive p110γ mutant R947P was generated by in vitro mutagenesis and subcloned into the NotI site of the pSRαMSVtkneo vector. Myc-epitope-tagged PI 3-kinase-γ regulatory subunit p101 was subcloned into the XbaI site of the pSRαMSV/IRES-XbaI vector. The pSRαMSV/IRES-XbaI vector was derived from pSRαMSVtkneo by replacement of the tkneo fragment with an internal ribosome entry site (IRES) sequence excised from the pLNEPN vector (a pyrine nucleotide phosphorylase retroviral expression vector using encephalomyocarditis virus translation, a gift from Dusty Miller’s laboratory; ref. 31), using EcoRI/XbaI double digestion. A double-header retroviral construct (pSRαBtkIRESmycp101) encoding both Btk and p101 was made by cloning the Btk cDNA into the unique NotI site 3′ of the long terminal repeat promoter and the myc-tagged p101 cDNA into the unique XbaI site downstream of the IRES sequence in the pSRαMSV/IRES-XbaI vector.
High titer helper-free retroviruses were generated by transient cotransfection of 293T cells (32) with retroviral constructs and psi minus ectopic packaging plasmid (33). The 293T cell line was kindly provided by David Baltimore (MIT). Supernatants from transfected 293T cells were collected 24–60 h posttransfection. For retroviruses encoding wild type Btk or Btk mutants, viral stocks were normalized for equivalent levels of Btk protein expression by Western blot analysis.
Antibodies, Western Blotting, and Immunoprecipitation.
Polyclonal antibody against Btk was used for Btk immunoprecipitation and Western blotting as described (2). Supernatant containing monoclonal antibody against myc epitope (9E10) was collected from hybridoma 9E10 cell culture (from ATCC) and used in Western blotting. Monoclonal antibody specific for phosphotyrosine (4G10) was purchased from Upstate Biotechnology (Lake Placid, NY).
Phosphopeptide Mapping.
Btk was coexpressed in the absence or presence of PI 3-kinase-γ as described above. Culture media were replaced with 2 ml of phosphate-free DMEM plus 0.1% dialyzed FCS and 3 mCi/ml 32PO4. After 3 h, cells were washed with PBS, lysed, and then Btk was immunoprecipitated. Immunoprecipitated Btk proteins were analyzed by tryptic phosphopeptide mapping (27, 30).
Soft Agar Fibroblast Transformation Assays.
Soft agar assays were performed as described (14, 28, 33). In all superinfections, virus titer was normalized by using the same amount of pre-titered retroviruses in 3 ml for a 10-cm tissue culture dish. Forty-eight hours postinfection, cells were plated in soft agar at a density of 1 × 104 cells per 6-cm tissue culture dish. Samples were plated in duplicate in media containing 20% fetal calf serum. Colonies equal to and larger than 0.5 mm in diameter were scored 10–12 days after plating.
RESULTS
Ectopic Expression of PI 3-Kinase-γ Promotes Tyrosine Phosphorylation of Btk.
The predominant hematopoietic tissue expression of the PI 3-kinase-γ isoform led us to evaluate its role in Btk activation. Our strategy involved simultaneous introduction of up to four separate genes into fibroblasts, which required high titer retroviruses. Generation of retroviral titers on the order of 106 infectious units per ml are readily achievable with a 293T cell transfection system (32). Btk is tyrosine phosphorylated in Rat-2 fibroblasts expressing a weakly activated allele of Src (SrcE378G; ref. 34). SrcE378G transforms avian but not rodent fibroblasts. SrcE378G strongly synergizes with activated but not wild type Btk in transformation of rodent cells (28). Tyrosine phosphorylation of immunoprecipitated Btk in the presence of SrcE378G was not enhanced by expression of either PI 3-kinase-γ subunit alone (Fig. 1). However, when both PI 3-kinase-γ subunits were coexpressed with Btk, the tyrosine phosphorylation on Btk was increased at least 10-fold (Fig. 1).
Figure 1.
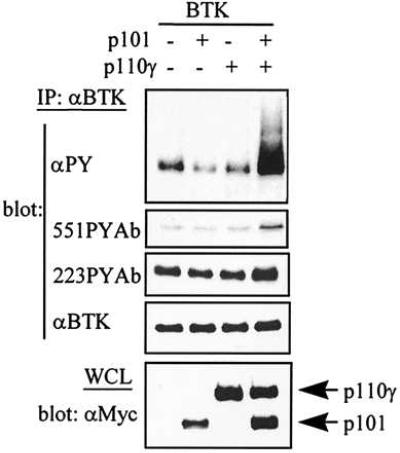
Increased PI 3-kinase-γ dosage elevates Btk tyrosine phosphorylation in fibroblast cells. Rat-2 cells harboring the SrcE378G mutants infected with retroviruses encoding Btk, Btk, and p101, Btk and p110γ, or Btk with p101 and p110γ were lysed, and Btk protein was immunoprecipitated with anti-Btk antibodies. The immunoprecipitates were subjected to 8% SDS/PAGE and analyzed by immunoblotting. The figure shows the immunoblots incubated with anti-phosphotyrosine antibodies (4G10). The blot was subsequently stripped using Western stripping buffer and probed with polyclonal anti-phosphorylated Tyr-551 (551PYAb), anti-phosphorylated Tyr-223 (223PYAb), or anti-Btk antibody followed by horseradish peroxidase-conjugated goat anti-rabbit secondary antibodies and detected by ECL. A small aliquot (2 × 105 cells) of the whole cell lysate (WCL) was loaded on SDS/PAGE, analyzed for p101 or p120 expression using an anti-myc epitope tag antibody (9E10), followed by horseradish peroxidase-conjugated goat-anti-mouse antibody, and visualized by ECL.
PI 3-Kinase-γ Induces New Phosphorylation Sites on Btk.
Btk activation results in the sequential increase of tyrosine phosphorylation at two known sites, Tyr-551 and Tyr-223. Increase of phosphorylation on these two residues correlates with activation of Btk catalytic activity (27). The phosphorylation of certain Btk tyrosine residues can be detected using newly developed antibodies that bind specifically to either phosphorylated Tyr-551 (551PYAb) or phosphorylated Tyr-223 (223PYAb) (36). Btk from control and PI 3-kinase-γ expressing fibroblasts was immunoblotted with 551PYAb or 223PYAb. PI 3-kinase-γ coexpression induced increases in phosphorylation of both regulatory tyrosines (Fig. 1). Comparison of Tyr-223 and Tyr-551 phosphorylation increases with the total level of phosphotyrosine on Btk suggested that increased phosphorylation of Tyr-551 and Tyr-223 in the presence of PI 3-kinase-γ does not totally account for the marked enhancement of total phosphorylation.
Cells expressing Btk in the absence or presence of PI 3-kinase-γ were metabolically labeled with 32PO4. Btk was immunoprecipitated, isolated by gel electrophoresis, and digested with trypsin, and the resulting phosphopeptides were visualized after two-dimensional electrophoresis and chromatography. The total 32P incorporated into Btk was slightly enhanced (∼50%) by PI 3-kinase-γ (data not shown). Comparison of the Btk tryptic phosphopeptide maps (Fig. 2) revealed the presence of two novel phosphopeptides, I and II, in the presence of PI 3-kinase-γ.
Figure 2.
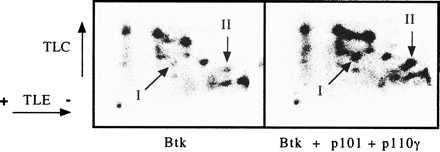
Influence of PI 3-kinase-γ on the phosphopeptide map of Btk. Cells were radiolabeled with 32PO4, and Btk was immunoprecipitated and digested with trypsin as described in Materials and Methods. The phosphopeptides were separated by thin-layer electrophoresis and thin-layer chromatography and then visualized by autoradiography. Phosphopeptides I and II are indicated with arrows.
The tryptic digests were passed over immobilized anti-phosphotyrosine antibody. The non-bound phosphopeptides were recovered, and the phosphotyrosine-containing peptides were eluted from the antibody matrix. Comparison of the two-dimensional phosphopeptide maps of non-bound and eluted samples revealed that phosphopeptide II contains phosphotyrosine (data not shown). PI 3-kinase-γ induces phosphorylation of at least two new distinct sites on Btk, one of which is a tyrosine residue.
Activation of Btk Requires the Catalytic Activity of PI 3-Kinase-γ.
Activation of Btk by coexpression of PI 3-kinase-γ could be due to protein–protein interaction of PI 3-kinase-γ with Btk or the lipid kinase activity of PI 3-kinase-γ. Mutation of Arg-947 located within the conserved catalytic domain blocks the ability of p110γ to phosphorylate lipid substrates in vitro (A.E., L.S., P.H., unpublished data). High titer retroviruses encoding either p110γ or p110γ-R947P were generated with similar titers. Rat-2 cells expressing SrcE378G were coinfected with retroviruses doubly encoding both p101 and Btk and with retroviruses encoding either p110γ or p110γ-R947P. Coexpression of p101 and p110γ-R947P in these cells did not elevate Btk tyrosine phosphorylation (Fig. 3A), indicating that the catalytic activity of PI 3-kinase-γ is required for activation of Btk.
Figure 3.

Kinase-inactive p110γ (R947P) and wortmannin treatment block increased Btk tyrosine phosphorylation induced by PI 3-kinase-γ. (a) Rat-2 cells harboring SrcE378G mutant infected with Btk retroviruses, double header retroviruses encoding Btk and p101 in addition to p110γ retroviruses or retroviruses encoding p110γ-R947P mutant were lysed, and Btk was immunoprecipitated with anti-Btk antibody. The immunoprecipitated Btk was run on 8% PAGE. Btk was immunoblotted with monoclonal anti-phosphotyrosine antibody (4G10) or polyclonal anti-Btk antibody, followed by horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit antibodies. Whole cell extracts were immunoblotted using an anti-Myc monoclonal antibody (9E10) and visualized with ECL (Amersham). (b) Rat-2 cells harboring the SrcE378G mutant infected with double header viruses encoding Btk and p101 plus retroviruses encoding p110γ were treated with 10 nM, 100 nM, 1 μM, or 10 μM wortmannin 40 h postinfection for 1.5 h before lysis. Tyrosine phosphorylation of immunoprecipitated Btk from wortmannin-treated cells was compared with Btk from untreated Rat-2 cells harboring the SrcE378G mutant and expressing either Btk alone or Btk, p101, and p110γ. The anti-phosphotyrosine blots or anti-Btk blots were produced and detected as described in a.
Previous work has shown that endogenous or ectopic PI 3-kinase-γ activity can be blocked by wortmannin (24, 37). Rat-2 cells simultaneously coexpressing SrcE378G, BTK, and PI 3-kinase-γ were treated with varying concentrations of wortmannin. Levels over 100 nM efficiently blocked increased BTK tyrosine phosphorylation induced by PI 3-kinase-γ (Fig. 3B), suggesting that the lipid product of PI 3-kinase is essential for Btk activation.
PI 3-Kinase-γ and Src Synergize with Wild Type Btk to Transform Fibroblasts.
Tyrosine phosphorylation of Btk correlates with functional activation. Synergy in fibroblast transformation by Btk* (E41K) and the mildly activated c-Src mutant, E378G, has been used to quantitate the in vivo interaction of Btk and Src in cellular signaling (28). A 10-fold increase in colony number is observed when wild type Btk is coexpressed with both subunits of PI 3-kinase-γ in Rat-2 cells expressing SrcE378G (Fig. 4).
Figure 4.
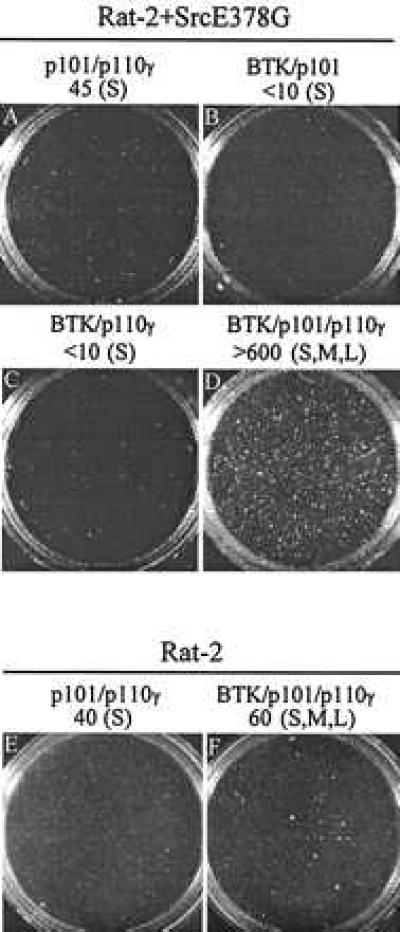
Btk and PI 3-kinase-γ synergize in fibroblast transformation. Rat-2 cells expressing SrcE378G were coinfected with the following combination of retroviruses: p101 and p110γ (A), Btk and p101 (B), Btk and p110γ (C), and Btk with p101 and p110γ (D); Rat-2 cells without SrcE378G were coinfected with p101 and p110γ (E) or Btk with p101 and p110γ (F) as controls. Forty-eight hours postinfection, 10,000 cells from each infection were plated in duplicate soft agar dishes (6 cm diameter). Colonies equal to or larger than 0.5 mm were counted and photographed after 12 days of incubation, and numbers are reflective of at least two different experiments.. Colonies are ∼0.5 mm, small (S); 0.5 to 1.0 mm, medium (M); or larger than 1.0 mm, large (L).
Wild type Btk does not strongly synergize with either SrcE378G (ref. 28 and data not shown) or PI 3-kinase-γ in Rat-2 cells alone (Fig. 4). Coexpression of Btk and PI 3-kinase-γ does result in approximately 60 colonies, but added Src is required for efficient transformation synergy. Btk does not synergize with kinase-inactive PI 3-kinase-γ (p101 and p110γ-R947P) in Rat-2 cells coexpressing SrcE378G (data not shown). This is consistent with earlier results demonstrating that the catalytic activity of PI 3-kinase-γ is required for increased tyrosine phosphorylation on Btk (Fig. 3A). These combined results strongly suggest that both Src and PI 3-kinase-γ activities are necessary but not sufficient for full activation of Btk in fibroblasts.
Btk Domain Requirements for Activation by PI 3-Kinase-γ.
Mutations in Btk were evaluated for their effect on activation of Btk by PI 3-kinase-γ in fibroblasts. The Xid mutation (R28C) of the Btk PH domain was reported to block the binding of PI (3,4,5)P3 with the Btk PH domain in vitro (15, 16). Cells were superinfected with retroviruses encoding either Btk or Btk R28C and PI 3-kinase-γ. Tyrosine phosphorylation of Btk R28C was lower than wild type Btk in Rat-2 cells coexpressing SrcE378G. When both subunits of PI 3-kinase-γ were coexpressed with the Xid mutant, PI 3-kinase-γ failed to elevate Btk tyrosine phosphorylation (Fig. 5B).
Figure 5.
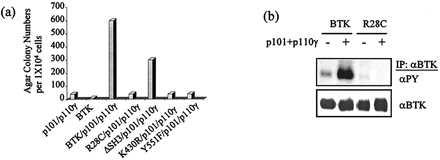
Fibroblast transformation by PI 3-kinase-γ and Btk or Btk mutants. (a) Ten-thousand Rat-2 cells harboring SrcE378G and expressing PI 3-kinase-γ (p101 plus p110γ) and either wild type Btk or one of the Btk mutants (R28C, ΔSH3, K430R, or Tyr-551) were plated in duplicate into agar (6 cm). This figure does not show the colony number formed by each Btk mutant, which is the same as that formed by wild type Btk alone (less than 10 colonies). Colonies equal to or larger than 0.5 mm were counted and photographed 12 days postplating. Expression of p101, p110γ, and Btk was analyzed by immunoblots using anti-Btk or anti-myc epitope antibodies (data not shown). (b) Approximately 4 × 106 Rat-2 cells expressing Btk, Btk/p101/p110γ, BtkR28C mutant, or BtkR28C/p101/p110γ were lysed, and Btk was immunoprecipitated using anti-Btk antibody. Immunoprecipitates were run on 8% SDS-PAGE gels and analyzed by immunoblotting with anti-phosphotyrosine (4G10) or anti-Btk antibodies, followed by either horseradish peroxidase-conjugated goat-anti-mouse or horseradish peroxidase-conjugated goat-anti-rabbit antibodies, respectively. Proteins were visualized by ECL.
The relative decrease of tyrosine phosphorylation on Btk R28C suggested that the Xid mutation would block cellular transformation. Rat-2 cells expressing SrcE378G and coexpressing Btk R28C with PI 3-kinase-γ gave low numbers of small colonies in agar (Fig. 5A). These data combined with the data showing a requirement for the PI 3-kinase enzymatic activity (Fig. 3a) suggests that the lipid product of PI 3-kinase is interacting with the Btk PH domain. This interaction could then facilitate the transphosphorylation on Btk by Src family kinases and lead to an activated Btk kinase.
Previous data showed that the Btk kinase activity but not its SH3 domain is critical for Btk activation by SrcE378G (30). Rat-2 cells expressing SrcE378G with either PI 3-kinase-γ alone or PI 3-kinase-γ with kinase-inactive Btk K430R formed low numbers of small agar colonies (Fig. 5A). In contrast, coexpression of the BtkΔSH3 mutant with PI 3-kinase-γ resulted in numerous and large colonies (Fig. 5A). The BtkΔSH3 virus titer was lower than wild type Btk, perhaps explaining the 2-fold decrease in the number of agar colonies. These data show that the SH3 domain of Btk is not essential for activation by PI 3-kinase-γ.
The major transphosphorylation site of Btk, Tyr-551, was evaluated to provide a genetic test as to whether PI 3-kinase-γ requires Src family kinases to efficiently activate Btk. Expression of either PI 3-kinase-γ alone or with the Btk Y551F mutant formed few, small agar colonies (Fig. 5A). These results correlate with the observation that the Y551F mutant blocks Btk hyperphosphorylation induced by PI 3-kinase-γ in Rat-2 cells coexpressing SrcE378G (data not shown). These data are also consistent with the previous finding that the Tyr-551 site is required for activation of Btk by Src family kinases (27, 28, 38).
DISCUSSION
Model for Btk Activation by PI 3-Kinase-γ and Src Family Kinases.
Our data provide evidence that PI 3-kinase-γ coactivates Btk in vivo through its PH domain in concert with Src family kinases (Fig. 6). The primary product of PI 3-kinase, PI (3,4,5)P3, likely binds to the Btk PH domain in vitro, supporting a role for PI 3-kinase as a proximal activator of Btk (15). The lipid interaction with the PH domain may activate Btk by recruiting it from the cytoplasm to the membrane, where it is subsequently activated by Src family kinases. We were unable to demonstrate an increased membrane association of Btk in Rat-2 cells overexpressing PI 3-kinase-γ (data not shown). However, we cannot exclude the membrane targeting model to explain the activation of Btk by PI 3-kinase-γ because the fraction of Btk that is recruited by PI (3,4,5) P3 onto the membrane may be relatively small or the association of Btk with the membrane may be transient. In both cases, it would be difficult to detect the change of Btk subcellular localization affected by the dosage increase of PI 3-kinase-γ activity.
Figure 6.
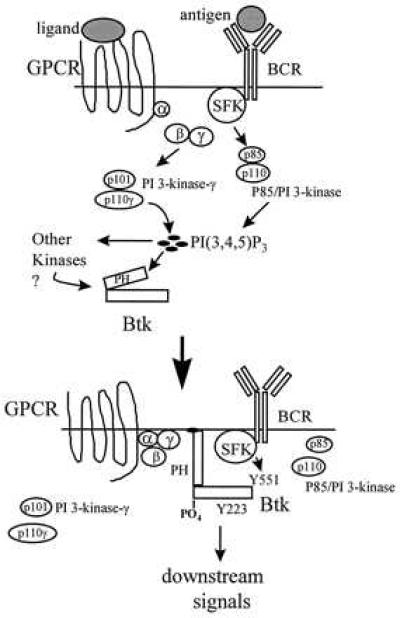
A proposed model for Btk activation by PI 3-kinase-γ and Src family kinases. PI 3-kinase-γ (p101 and p110γ) is activated by dissociated G protein β, γ subunits as a result of ligand binding to G protein-coupled receptors (GPCR). P85/PI 3-kinase is activated by BCR. The product of p85/PI 3-kinase and PI 3-kinase-γ, PI(3,4,5)P3, then binds the Btk PH domain directing BTK localization to the membrane. Alternatively, one or several unknown kinases dependent on PI(3,4,5)P3 may activate Btk by transphosphorylation. In both cases, Btk becomes available for the activation by Src family kinases (SFK) associated with the BCR. Src family kinases then transphosphorylate Btk on Tyr-551 to induce Btk activation. Btk subsequently autophosphorylates Tyr-223 within its SH3 domain and effects a downstream signal. Both PI 3-kinase and Src family kinases are required for the efficient activation of Btk and induction of Btk downstream signaling.
Another mechanism of activation is transphosphorylation by a secondary serine/threonine or tyrosine kinases. An example of this is PI (3,4,5)P3 binding to the PH domain of Akt, increasing its membrane association (39). PI (3,4,5)P3 also activates downstream serine/threonine kinases to transphosphorylate Akt (39). A PI (3,4,5)P3-dependent kinase, PDK1, binds, phosphorylates, and activates Akt in vitro (40). It is conceivable that a PDK1-related enzyme could control Btk activity in an analogous manner. PI 3-kinase also activates a number of PKC isoforms in vitro (41–44). Isoforms of PKC bind the Btk PH domain with high affinity (45, 46). One or several of the PI 3-kinase-dependent PKC family members may modulate Btk activity via the Btk PH domain.
We have demonstrated an increase in phosphorylation of two novel sites on Btk in addition to the increased phosphorylation of Tyr-551 and Tyr-223. One phosphopeptide was identified to contain phosphotyrosine, and the other is likely phosphoserine (data not shown) and may be a target of PKC or PDK1 regulation. Identification of the specific phosphorylated residues will be critical for characterizing the mechanism of Btk activation by PI 3-kinase-γ.
Interaction of Btk and Isoforms of PI 3-Kinase in B Lymphocytes.
There are two known PI 3-kinases in B cells, P85/PI 3-kinase and PI 3-kinase-γ. Both isoforms can activate Btk in fibroblasts albeit to differing degrees. Tyrosine phosphorylation of ectopically expressed Btk was blocked by 50 nM wortmannin, a specific inhibitor of PI 3-kinases (ref. 47 and data not shown), indicating that endogenous PI 3-kinase is involved in Btk activation by Src family kinases. Vaccinia viral overexpression of p110*, an activated subunit of p85/PI 3-kinase (48), increased the phosphotyrosine content on Btk (Z.L., D. J. Rawlings., and O.N.W., unpublished data). Btk and p110* also synergize to transform fibroblasts (Z.L. and O.N.W., unpublished data) and flux calcium in B cells and increase cellular PI(3,4,5)P3 (A. Scharenberg et al., unpublished data). These data suggest that all PI 3-kinase isoforms can contribute to Btk activation.
P85/PI 3-kinase is stimulated upon B cell antigen receptor (BCR), CD38, and CD40 ligation (49–53). PI 3-kinase-γ associates with and is activated by heterotrimeric G proteins, but the identity of the upstream G protein-coupled receptor in B lymphocytes is unknown. Our results and prior data suggest that Btk may be activated by different isoforms of PI 3-kinase, each associated with distinct signaling pathways. A recent report has demonstrated that G protein-coupled receptors can activate Btk in a certain avian B cell line (54).
One of the functions of Btk is to induce calcium flux in B cells upon BCR ligation (Z.L., unpublished results, and ref. 55). A wortmannin-sensitive PI 3-kinase activity is also required for calcium entry into B cells from the extracellular space upon BCR stimulation (56). Our data showing a functional connection between Btk and PI 3-kinase signaling suggest that Btk and PI 3-kinase may interact to regulate the critical process of calcium flux in B cells upon BCR stimulation. It will be important to investigate whether Btk and PI 3-kinase functionally interact in this process and are regulated by G protein-coupled receptors.
Acknowledgments
We gratefully acknowledge Dr. Randolph Mohr for manuscript editing and Julia Shimaoka for manuscript preparation; we also acknowledge Drs. Larry Zipursky, Steve Smale, David Rawlings, Anne Satterthwaite, and James Li for critical reading of the manuscript. We thank Drs. Simon Cooke and Sylvia Braselman of Onyx Pharmaceuticals for helpful discussions and sharing information on their unpublished data; we also thank Drs. Anke Klippel and Lewis Williams of Chiron for kindly providing monoclonal antibody to p110α. Z.L. is a Research fellow of the National Cancer Institute of Canada supported by the Terry Fox Run. M.I.W. is supported by the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation Fellowship (DRG-086). O.N.W. is an Investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS
- PH
pleckstrin homology
- Btk
Bruton’s tyrosine kinase
- PI 3-kinase
phosphatidylinositol 3-kinase
- XLA
X-linked agammaglobulinemia
- Xid
X-linked immunodeficiency
- PI(3
4,5)P3, phosphatidylinositol 3,4,5-trisphosphate
- IRES
internal ribosome entry site
- PKC
protein kinase C
- BCR
B cell antigen receptor
Note Added in Proof
After this manuscript was submitted, August et al. (57) published their results that down-regulation of p85/PI 3-kinase activity blocks the activation of ITK, a Btk homologue present in T lymphocytes.
References
- 1.Rawlings D J, Witte O N. In: Immunological Reviews. Moller G, editor. Vol. 138. Copenhagen: Munksgaard; 1994. pp. 105–119. [DOI] [PubMed] [Google Scholar]
- 2.Tsukada S, Saffran D C, Rawlings D J, Parolini O, Allen R C, Klisak I, Sparkes R S, Kubagawa H, Mohandas T, Quan S, Belmont J W, Cooper M D, Conley M E, Witte O N. Cell. 1993;72:279–290. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 3.Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, Hammarström L, Kinnon C, Levinsky R, Bobrow M, Smith C I E, Bentley D R. Nature. 1993;361:226–233. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 4.Rawlings D J, Saffran D C, Tsukada S, Largaespada D A, Grimaldi J C, Cohen L, Mohr R N, Bazan J F, Howard M, Copeland N G, Jenkins N A, Witte O N. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 5.Thomas J D, Sideras P, Smith C I E, Vorechovsky I, Chapman V, Paul W E. Science. 1993;261:355–358. doi: 10.1126/science.8332900. [DOI] [PubMed] [Google Scholar]
- 6.Bruton O C. Pediatrics. 1952;9:722–727. [PubMed] [Google Scholar]
- 7.Wicker L S, Scher I. Curr Top Microbiol Immunol. 1986;124:87–101. doi: 10.1007/978-3-642-70986-9_6. [DOI] [PubMed] [Google Scholar]
- 8.Khan W N, Alt F W, Gerstein R M, Malynn B A, Larsson I, Rathbun G, Davidson L, Müller S, Kantor A B, Herzenberg L A, Rosen F S, Sideras P. Immunity. 1995;3:283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- 9.Kerner J D, Appleby M W, Mohr R N, Chien S, Rawlings D J, Maliszewski C R, Witte O N, Perlmutter R M. Immunity. 1995;3:301–312. doi: 10.1016/1074-7613(95)90115-9. [DOI] [PubMed] [Google Scholar]
- 10.Musacchio A, Gibson T, Rice P, Thompson J, Saraste M. Trends Biochem Sci. 1993;18:343–348. doi: 10.1016/0968-0004(93)90071-t. [DOI] [PubMed] [Google Scholar]
- 11.Lemmon M A, Ferguson K M, Schlessinger J. Cell. 1996;85:621–624. doi: 10.1016/s0092-8674(00)81022-3. [DOI] [PubMed] [Google Scholar]
- 12.Vihinen M, Zvelebil M J, Zhu Q, Brooimans R A, Ochs H D, Zegers B J, Nilsson L, Waterfield M D, Smith C I. Biochemistry. 1995;34:1475–1481. doi: 10.1021/bi00005a002. [DOI] [PubMed] [Google Scholar]
- 13.Vihinen M, Brooimans R A, Kwan S-P, Lehvaslaiho H, Litman G W, Ochs H D, Resnick I, Schwaber J H, Vorechovsky I, Smith C I E. Immunol Today. 1996;17:502–506. doi: 10.1016/0167-5699(96)30058-3. [DOI] [PubMed] [Google Scholar]
- 14.Li T, Tsukada S, Satterthwaite A, Havlik M H, Park H, Takatsu K, Witte O N. Immunity. 1995;2:451–460. doi: 10.1016/1074-7613(95)90026-8. [DOI] [PubMed] [Google Scholar]
- 15.Salim K, Bottomley M J, Querfurth E, Zvelebil M J, Gout I, Scaife R, Margolis R L, Gigg R, Smith E I E, Driscoo P C, Waterfield M D, Panayotou G. EMBO J. 1996;15:6241–6250. [PMC free article] [PubMed] [Google Scholar]
- 16.Rameh L E, Arvidsson A-K, Carraway K L, Couvillon A D, III, Rathbun G, Crompton A, VanRenterghem B, Czech M P, Ravichandran K S, Burakoff S J, Wang D-S, Chen C-S, Cantley L C. J Biol Chem. 1997;272:22059–22066. doi: 10.1074/jbc.272.35.22059. [DOI] [PubMed] [Google Scholar]
- 17.Fukuda M, Kojima T, Kabayama H, Mikoshiba K. J Biol Chem. 1996;271:30303–30306. doi: 10.1074/jbc.271.48.30303. [DOI] [PubMed] [Google Scholar]
- 18.Ferguson K M, Lemmon M A, Schlessinger J, Sigler P B. Cell. 1995;83:1037–1046. doi: 10.1016/0092-8674(95)90219-8. [DOI] [PubMed] [Google Scholar]
- 19.Hyvonen M, Saraste M. EMBO J. 1997;16:3396–3404. doi: 10.1093/emboj/16.12.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toker A, Cantley L C. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 21.Vanhaesebroeck B, Leevers S J, Panayotou G, Waterfield M D. Trends Biochem Sci. 1997;22:267–272. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- 22.Carpenter C L, Duckworth B C, Auger K R, Cohen B, Schaffhausen B S, Cantley L C. J Biol Chem. 1990;265:19704–19711. [PubMed] [Google Scholar]
- 23.Stoyanov B, Volinia S, Hanck T, Rubio I, Loubtchenkov M, Malek D, Stoyanova S, Vanhaesebroeck B, Dhand R, Nurnberg B, Giershik P, Seedorf K, Hsuan J J, Waterfield M D, Wetzker R. Science. 1995;269:690–693. doi: 10.1126/science.7624799. [DOI] [PubMed] [Google Scholar]
- 24.Stephens L R, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka A S, Thelen M, Cadwallader K, Tempst P, Hawkins P T. Cell. 1997;89:105–114. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 25.Cheng G, Ye Z-S, Baltimore D. Proc Natl Acad Sci USA. 1994;91:8152–8155. doi: 10.1073/pnas.91.17.8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahajan S, Fargnoli J, Burkhardt A L, Kut S A, Saouaf S J, Bolen J B. Mol Cell Biol. 1995;15:5304–5311. doi: 10.1128/mcb.15.10.5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rawlings D J, Scharenberg A M, Park H, Wahl M I, Lin S, Kato R M, Fluckiger A C, Witte O N, Kinet J P. Science. 1996;271:822–825. doi: 10.1126/science.271.5250.822. [DOI] [PubMed] [Google Scholar]
- 28.Afar D E H, Park H, Howell B W, Rawlings D J, Cooper J, Witte O N. Mol Cell Biol. 1996;16:3465–3471. doi: 10.1128/mcb.16.7.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hubbard S R, Wei L, Ellis L, Hendrickson W A. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 30.Park H, Wahl M I, Afar D E, Turck C W, Rawlings D J, Tam C, Scharenberg A M, Kinet J-P, Witte O N. Immunity. 1996;4:515–525. doi: 10.1016/s1074-7613(00)80417-3. [DOI] [PubMed] [Google Scholar]
- 31.Miller A D, Rosman G J. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- 32.Pear W S, Nolan G P, Scott M L, Baltimore D. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muller A J, Young J C, Pendergast A-M, Pondel M, Landau N R, Littman D R, Witte O N. Mol Cell Biol. 1991;11:1785–1792. doi: 10.1128/mcb.11.4.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levy J B, Hanafusa H, Iba H. Proc Natl Acad Sci USA. 1986;83:4228–4232. doi: 10.1073/pnas.83.12.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seidel-Dugan C, Meyer B E, Thomas S M, Brugge J S. Mol Cell Biol. 1992;12:1835–1845. doi: 10.1128/mcb.12.4.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wahl M I, Fluckiger A-C, Kato R M, Park H, Witte O N, Rawlings D J. Proc Natl Acad Sci USA. 1997;94:11526–11533. doi: 10.1073/pnas.94.21.11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stephens L, Smrcka A, Cooke F T, Jackson T R, Sternweis P C, Hawkins P T. Cell. 1994;77:83–93. doi: 10.1016/0092-8674(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 38.Kurosaki T, Kurosaki M. J Biol Chem. 1997;272:15595–15598. doi: 10.1074/jbc.272.25.15595. [DOI] [PubMed] [Google Scholar]
- 39.Stokoe D, Stephens L R, Copeland T, Gaffney P R J, Reese C B, Painter G F, Holmes A B, McCormick F, Hawkins P T. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 40.Alessi D, James S, Downes C, Homes A, Gaffney P, Reese C, Cohen P. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 41.Akimoto K, Takahashi R, Moriyz S, Nishioka N, Takayanagi J, Kimura K, Fukui Y, Osada S, Mizuno K, Hirai S, Kazlauskas A, Ohno S. EMBO J. 1995;15:788–798. [PMC free article] [PubMed] [Google Scholar]
- 42.Palmer R, Dekker L, Woscholski R, LeGood J, Gigg R, Parker P. J Biol Chem. 1995;270:22412–22416. doi: 10.1074/jbc.270.38.22412. [DOI] [PubMed] [Google Scholar]
- 43.Moriya S, Kazlauskas A, Akimoto K, Hirai S, Mizuno K, Takenawa T, Fukui Y, Watanabe Y, Ozaki S, Ohno S. Proc Natl Acad Sci USA. 1996;93:151–155. doi: 10.1073/pnas.93.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herrera-Velit P, Knutson K, Reiner N. J Biol Chem. 1997;272:16445–16452. doi: 10.1074/jbc.272.26.16445. [DOI] [PubMed] [Google Scholar]
- 45.Yao L, Kawakami Y, Kawakami T. Proc Natl Acad Sci USA. 1994;91:9175–9179. doi: 10.1073/pnas.91.19.9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao L, Suzuki H, Ozawa K, Deng J, Lehel C, Fukamachi H, Anderson W, Kawakami Y, Kawakami T. J Biol Chem. 1997;272:13033–13039. doi: 10.1074/jbc.272.20.13033. [DOI] [PubMed] [Google Scholar]
- 47.Arcaro A, Wymann M P. Biochemistry. 1993;296:297–301. doi: 10.1042/bj2960297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu Q, Klippel A, Muslin A J, Fantl W J, Williams L T. Science. 1995;268:100–102. doi: 10.1126/science.7701328. [DOI] [PubMed] [Google Scholar]
- 49.Gold M, Chan V, Turck C, DeFranco A. J Immunol. 1992;148:2012–2022. [PubMed] [Google Scholar]
- 50.Gold M R, Aebersold R. J Immunol. 1994;152:42–50. [PubMed] [Google Scholar]
- 51.Aagaard-Tillery K M, Jelinek D F. J Immunol. 1996;156:4543–4554. [PubMed] [Google Scholar]
- 52.Kitanaka A, Ito C, Nishigaki H, Campana D. Blood. 1996;88:590–598. [PubMed] [Google Scholar]
- 53.Ren C L, Morio T, Fu S M, Geha R S. J Exp Med. 1994;179:673–680. doi: 10.1084/jem.179.2.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wan Y, Bence K, Hata A, Kurosaki T, Veillette A, Huang X-Y. J Biol Chem. 1997;272:17209–17215. doi: 10.1074/jbc.272.27.17209. [DOI] [PubMed] [Google Scholar]
- 55.Takata M, Kurosaki T. J Exp Med. 1996;184:31–40. doi: 10.1084/jem.184.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kiener P A, Lioubin M N, Rohrschneider L R, Ledbetter J A, Nadler S G, Diegel M L. J Biol Chem. 1997;272:3838–3844. doi: 10.1074/jbc.272.6.3838. [DOI] [PubMed] [Google Scholar]
- 57.August A, Sadra A, Dupont B, Hanafusa H. Proc Natl Acad Sci USA. 1997;94:11227–11232. doi: 10.1073/pnas.94.21.11227. [DOI] [PMC free article] [PubMed] [Google Scholar]