

Abstract
We report a catalytically promiscuous enzyme able to efficiently promote the hydrolysis of six different substrate classes. Originally assigned as a phosphonate monoester hydrolase (PMH) this enzyme exhibits substantial second-order rate accelerations ((kcat/KM)/kw), ranging from 107 to as high as 1019, for the hydrolyses of phosphate mono-, di-, and triesters, phosphonate monoesters, sulfate monoesters, and sulfonate monoesters. This substrate collection encompasses a range of substrate charges between 0 and -2, transition states of a different nature, and involves attack at two different reaction centers (P and S). Intrinsic reactivities (half-lives) range from 200 days to 105 years under near neutrality. The substantial rate accelerations for a set of relatively difficult reactions suggest that efficient catalysis is not necessarily limited to efficient stabilization of just one transition state. The crystal structure of PMH identifies it as a member of the alkaline phosphatase superfamily. PMH encompasses four of the native activities previously observed in this superfamily and extends its repertoire by two further activities, one of which, sulfonate monoesterase, has not been observed previously for a natural enzyme. PMH is thus one of the most promiscuous hydrolases described to date. The functional links between superfamily activities can be presumed to have played a role in functional evolution by gene duplication.
Keywords: catalytic promiscuity, evolution, mechanism, sulfatase, superfamily
Enzymes are usually seen as highly specific catalysts following the classical rule “one enzyme, one activity.” This view is challenged by an increasing number of enzymes with broad substrate specificities or side activities indicating that enzymes are catalytically more flexible than originally assumed. Some of these promiscuous enzymes can turn over substrates with different structures while catalyzing the same chemical reaction involving the same transition state (substrate promiscuity). Catalytic promiscuity, by contrast, is the ability of an enzyme to catalyze chemically distinct reactions by stabilization of different transition states (TSs) (1). Catalytic efficiencies (kcat/KM) for the promiscuous substrates are often substantially lower (2 to 9 orders of magnitude) than for the native conversions (1–3). The growing number of examples of this phenomenon (1–4) has engendered excitement on a number of fronts. Catalytic promiscuity provides a functional basis for enzyme evolution by gene duplication. The initial head-start activity of the duplicated gene copy could support an immediate selective advantage (1, 2, 5, 6), to be followed by improvement of the initially weak activity (7). Even low kcat/KM values for a promiscuous function can support a significant selective advantage (8). Mimicking this evolutionary shortcut could also provide a more efficient route to changing the function of proteins by directed evolution (5).
We describe multiple and efficient catalytic promiscuity in an enzyme from Burkholderia caryophilli PG2952 originally assigned as a phosphonate monoester hydrolase (BcPMH) when this organism was identified in a screen for growth on glyceryl glyphosate (9). The kcat/KM for glyceryl glyphosate is relatively low (9.5 M-1 s-1) and, because BcPMH also hydrolyzes phosphate diesters with higher efficiency, it has been surmised that its original biological role is to hydrolyze phosphodiesters (9). In a closely related ortholog (RlPMH from Rhizobium leguminosarum; 86% amino acid identity), the unusual formylglycine (fGly) residue, first discovered in type I sulfatases (10), was shown to act as a nucleophile. fGly results from enzymatic posttranslational oxidation of a cysteine embedded in a recognition sequence (CxPxR) to an aldehyde (11). On the basis of structural, mutational, and kinetic studies and in analogy to the structurally related arylsulfatases (12), a double displacement mechanism involving a covalent fGly-substrate intermediate was proposed for RlPMH. PMH is a member of the alkaline phosphatase (AP) superfamily that encompasses structurally related enzymes known to hydrolyze phosphate monoesters and diesters and sulfate monoesters (13). Several members of the AP superfamily show catalytic promiscuity, and in some cases the promiscuous reactions are the native activities of other superfamily members (14–19).
BcPMH catalyzes the hydrolysis of a total of six different substrate classes, four of which correspond to activities seen in the AP superfamily. The collection of substrate classes, for which rate accelerations between 107 and 1019 are observed, encompasses large variations in charge, the reaction center, size, hydrophobicity, reactivity, and nature of the TS of the uncatalyzed reaction. The range and magnitude of promiscuous activities suggest that BcPMH is an enzyme in which high reactivity is combined with a relative lack of specificity, challenging the notion that a very active enzyme must be highly specialized. The kinetic and structural analysis of BcPMH highlights mechanistic and structural features that promote catalytic promiscuity for hydrolytic reactions.
Results
BcPMH: A Highly Promiscuous Hydrolase.
A highly purified sample of BcPMH exhibits 107–1012 second-order rate accelerations ((kcat/KM)/kw) for the hydrolysis of phosphate monoesters 1a/b, phosphate triester 3b, sulfate monoesters 5a/b, and sulfonate monoesters 6a/b, in addition to the previously reported hydrolysis of phosphonate monoesters 4a/b and phosphodiesters 2a/b (Fig. 1 and Table 1). The enzyme performs multiple turnovers with each substrate (Fig. S1A–F), and saturation kinetics are observed for all substrate classes except phosphate triester 3b (Fig. S1G–L). kcat/Kw values for all reactions, including the two fastest conversions, differ by 6 orders of magnitude. Catalysis is efficient with TS binding constants (KTS) ranging from 10-6 to 10-18 M.
Fig. 1.

Structures of substrates that are hydrolyzed by BcPMH: 1a: phenyl phosphate, 1b: p-nitrophenyl phosphate, 2a: diphenyl phosphate, 2b: p-nitrophenyl ethyl phosphate, 3b: paraoxon, 4a: phenyl phenylphosphonate, 4b: p-nitrophenyl phenylphosphonate, 5a: phenyl sulfate, 5b: p-nitrophenyl sulfate, 6a: phenyl phenylsulfonate, 6b: p-nitrophenyl phenylsulfonate. Gray lines indicate the bonds that are broken during hydrolysis.
Table 1.
Kinetic parameters for BcPMH wild type at pH 7.5
| Substrate |
kcat (s-1) |
KM (mM) |
kcat/KM (s-1 M-1) |
kcat/kuncat†,‡ |
(kcat/KM)/kuncat†,§ (M-1) |
(kcat/KM)/kw†,¶ |
KTS∥ (M) |
|
| Phosphate monoester | 1a* | (2.1 ± 0.2) ∗ 10-4 | 0.33 ± 0.03 | 0.63 ± 0.08 | 106.2 | 109.7 | 1011.5 | 1.9 ∗ 10-10 |
| 1b | (7.7 ± 0.1) ∗ 10-3 | 0.35 ± 0.02 | 22 ± 1 | 105.2 | 108.7 | 1010.4 | 2.2 ∗ 10-9 | |
| Phosphate diester | 2a | 2.12 ± 0.02 | 0.071 ± 0.004 | (3.0 ± 0.2) ∗ 104 | 1013.7 | 1017.9 | 1019.6 | 1.4 ∗ 10-18 |
| 2b | 5.8 ± 0.1 | 0.63 ± 0.04 | (9.2 ± 0.6) ∗ 103 | 1013.4 | 1016.6 | 1018.3 | 2.8 ∗ 10-17 | |
| Phosphate triester | 3b | > 3.7 ∗ 10-5 | > 2.4 | (1.6 ± 0.1) ∗ 10-2 | > 102.9 | 105.5 | 107.2 | 3.2 ∗ 10-6 |
| Phosphonate monoester | 4a | 1.58 ± 0.04 | 1.23 ± 0.09 | (1.3 ± 0.1) ∗ 103 | 1012.9 | 1015.8 | 1017.6 | 1.4 ∗ 10-16 |
| 4b | 2.73 ± 0.06 | 0.19 ± 0.02 | (1.5 ± 0.1) ∗ 104 | 1011.2 | 1014.9 | 1016.7 | 1.2 ∗ 10-15 | |
| Sulfate monoester | 5a* | (1.0 ± 0.1) ∗ 10-4 | 58 ± 5 | (1.7 ± 0.3) ∗ 10-3 | 108.4 | 109.6 | 1011.3 | 2.5 ∗ 10-10 |
| 5b | (4.0 ± 0.1) ∗ 10-2 | 68 ± 4 | 0.59 ± 0.04 | 107.6 | 108.7 | 1010.5 | 1.9 ∗ 10-9 | |
| Sulfonate monoester | 6a* | (7.0 ± 0.3) ∗ 10-4 | 0.51 ± 0.09 | 1.4 ± 0.2 | 106.7 | 1010.0 | 1011.7 | 1.0 ∗ 10-10 |
| 6b | (1.2 ± 0.1) ∗ 10-2 | 0.24 ± 0.03 | 49 ± 7 | 106.3 | 109.9 | 1011.7 | 1.1 ∗ 10-10 | |
*KM was determined as the competitive inhibition constant (Kic) for the conversion of phosphate diester 2b. By using this Kic as the KM value for that substrate, the kcat could be derived (see SI Text).
†Values for kuncat and kw are listed in Table S1.
‡First-order rate enhancement.
§Catalytic proficiency.
¶Second-order rate enhancement.
∥KTS, the transition state binding constant, was calculated as the inverse of the catalytic proficiency [(kcat/KM)/kuncat].
In each case hydrolysis involves nucleophilic attack at the phosphorus or sulfur center, breaking the ester bond to the leaving group. An alternative addition-elimination (SNAr) mechanism, in which attack occurs at the nitrophenyl ring instead (Fig. S2A), is ruled out by isotope-labeling experiments that were analyzed by mass spectrometry (Fig. S2C). Incubation of substrates 1b–6b in the presence of enzyme and 18O-labeled water showed no incorporation of the label in the p-nitrophenol product.
Thus, all six reactions proceed through an identical reaction sequence involving nucleophilic attack on the various phosphorus and sulfur centers (Fig. S2B). The observed activities involve the making and breaking of bonds to reaction centers with different characteristics via different TSs, so they are genuinely promiscuous.
All Six Reactions Are Catalyzed by BcPMH.
Several lines of evidence suggest that all six reactions are indeed catalyzed by BcPMH and not by contaminant(s) in the purified enzyme preparation.
Copurification of all six different activities.
BcPMH was extensively purified by using anion exchange, hydrophobic interaction, and size-exclusion chromatography (9). Protein-containing fractions that eluted from the three different columns were assayed for activity against substrates 1b–6b. All six activities coincided with the elution of BcPMH during all three purification steps (Fig. S3). Because all activities eluted from the size-exclusion column at the expected molecular weight of tetrameric BcPMH (∼240 kDa) (9), copurifying contaminants interacting specifically with BcPMH are unlikely to be present, because they would have increased the observed mass. Affinity purification of a Strep-tagged version of the enzyme followed by a size-exclusion step yielded pure protein that also catalyzed all six reactions with identical rate parameters as the untagged protein. During size-exclusion chromatography of the tagged protein, all activities coeluted (Fig. S3). When the same expression and purification procedures were followed with an Escherichia coli strain harboring the empty expression vector, no contaminating activity could be detected (Fig. S3). These observations collectively suggest that none of the observed reactions was the result of a contaminant.
pH-rate profiles.
Catalysis by BcPMH is dependent on its protonation state between pH 6 and 10 (Fig. 2). The pH-rate profiles of the kcat/KM values for substrates 1b, 2b, 4b, 5b, and 6b largely coincide. For the pH range sampled, in which the protonation states of the substrates do not change, the hydrolysis of these five compounds could be fitted to a two-pKE model with a pKE1 of 7.0–7.2 (5.8 for sulfate monoester 5b) and a pKE2 of 7.5–8.1. The fGly residue that results from the oxidation of Cys57 (Fig. 3D) is likely to contribute to catalysis in its deprotonated state (11), represented by pKE1. The protonated forms of residues His218 and Lys337 (Fig. 3B) have been suggested to act as general acids in its close ortholog RlPMH (11), which could explain the presence of pKE2. The coincidence of the pH-rate profiles for kcat/KM for these five substrates suggests that their conversions are catalyzed by following a similar mechanism as proposed for phosphonate monoester hydrolysis by RlPMH (11). Furthermore, the striking similarity between the pH-rate profiles suggests that these five reactions are catalyzed by the same active site, because a contaminant would be unlikely to have a similar pH-rate profile. The enzyme-catalyzed hydrolysis of phosphotriester 3b shows a pH-rate profile that differs substantially from that of the other five substrates, suggesting that hydrolysis of this substrate is promoted in a different way.
Fig. 2.

pH-rate profiles of kcat/KM for all six substrates: (A) phosphate monoester 1b, (B) phosphate diester 2b, (C) phosphate triester 3b, (D) phosphonate monoester 4b, (E) sulfate monoester 5b, and (F) sulfonate monoester 6b. For details see SI Text.
Fig. 3.

X-ray structure of BcPMH. (A) BcPMH is tetrameric in the crystal and in solution. The surface of the active site residues at the bottom of the active site cavity is orange (Upper Left protomer). The arrow indicates the position of the cut for C. (B) Active site residues as identified by homology to RlPMH (11) and P. aeruginosa arylsulfatase. Hydrogen bonds and metal-binding contacts are indicated by dotted lines. The metal ions identified in the protein were iron and zinc. (C) A cut through the active site illustrates the spacious active site opening. (D) Proposed mechanism for all five different conversions involving the fGly nucleophile [on the basis of the proposed mechanism of RlPMH (11)]. Upon nucleophilic attack of fGly on the central P/S atom (X) the leaving group (ROH = phenolate) is abstracted. For all substrates except 3b, a covalent intermediate formed via fGly is broken down by cleaving the same C-O bond (red, instead of a second attack at X).
Cross-inhibition.
Because the kcat values for the hydrolysis of substrates 1b, 3b, 5b, and 6b are at least 100-fold lower than those of the phosphodiesterase and phosphonate monoesterase reaction, these four weaker substrates will act as inhibitors of the faster reactions. Apparent kcat/KM values for the conversion of phosphate diester 2b were determined from Michaelis–Menten curves recorded at various inhibitor concentrations (Fig. S4B). The Kic was determined by fitting the dependence of 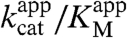 on inhibitor concentration (Fig. S4). Inhibition of native phosphodiesterase activity was shown for substrates 1b, 5b, and 6b (Fig. S4) with competitive inhibition constants (Kic) that were in agreement with the KM for the conversion of these nonnative substrates (Table 2), suggesting that these reactions are catalyzed by the same active site. For phosphotriester 3b, no inhibition was observed within the sampled concentration range of the inhibitor consistent with the assumption that this reaction is catalyzed by following a different pathway or in a different part of the active site than the other five reactions (see below).
on inhibitor concentration (Fig. S4). Inhibition of native phosphodiesterase activity was shown for substrates 1b, 5b, and 6b (Fig. S4) with competitive inhibition constants (Kic) that were in agreement with the KM for the conversion of these nonnative substrates (Table 2), suggesting that these reactions are catalyzed by the same active site. For phosphotriester 3b, no inhibition was observed within the sampled concentration range of the inhibitor consistent with the assumption that this reaction is catalyzed by following a different pathway or in a different part of the active site than the other five reactions (see below).
Table 2.
Inhibition of BcPMH catalyzed phosphate diester 2b hydrolysis by additional substrates
| Substrate |
KM (mM) |
Kic (mM) |
| 1b | 0.35 ± 0.02 | 0.32 ± 0.02 |
| 3b | > 2.4 | > 2.4 |
| 5b | 68 ± 4 | 56 ± 11 |
| 6b | 0.24 ± 0.03 | 0.32 ± 0.05 |
Active site mutation.
Mutation of Cys57 to alanine prevents the generation of the fGly nucleophile and results in an impaired enzyme with reduced activity for all six substrates, suggesting that all reactions are catalyzed by BcPMH in the same active site. kcat values decreased 250–1400-fold whereas KM values were almost unchanged, resulting in reductions of kcat/KM in the range of 7–1200-fold (Table 3). The smallest mutational effect was observed for phosphate triesters, consistent with differences in catalysis for this substrate.
Table 3.
Kinetic parameters of the BcPMH Cys57Ala mutant for substrates 1b–6b
| C57A mutant |
Ratio (wt:C57A) | |||
| kcat (s-1) | KM (mM) | kcat/KM (M-1 s-1) | kcat/KM | |
| 1b | (3.1 ± 0.1) ∗ 10-5 | 1.16 ± 0.08 | (2.6 ± 0.1) ∗ 10-2 | 828 ± 70 |
| 2b | (5.6 ± 0.1) ∗ 10-3 | 0.71 ± 0.06 | 7.8 ± 0.7 | 1178 ± 131 |
| 3b | > 5.4 ∗ 10-6 | > 2.4 | (2.3 ± 0.1) ∗ 10-3 | 7.0 ± 0.3 |
| 4b | (1.90 ± 0.02) ∗ 10-3 | 0.061 ± 0.005 | 31 ± 3 | 467 ± 55 |
| 5b | (3.1 ± 0.2) ∗ 10-5 | 21 ± 5 | (1.5 ± 0.3) ∗ 10-3 | 405 ± 97 |
| 6b | (1.9 ± 0.2) ∗ 10-5 | 0.14 ± 0.03 | 0.13 ± 0.03 | 361 ± 95 |
BcPMH Has a Formylglycine (fGly) in its Active Site.
BcPMH contains the recognition sequence 57-CXPXR-61 in which Cys57 can potentially be oxidized to the fGly residue. MALDI-TOF spectrometric analysis of a tryptic digest of BcPMH in the presence of an aldehyde-specific labeling agent (20) showed that Cys57 was indeed modified to fGly in this enzyme but that the modification was incomplete (Fig. S5A). Incomplete modification can be explained by saturation of the (as yet unidentified) E. coli fGly-generating machinery under overexpression conditions. Coexpression of BcPMH with the bacterial fGly-generating enzyme from Mycobacterium tuberculosis (MtbFGE) (21) increased the activity of the enzyme 3-fold, but unmodified Cys57 was still detected.
To resolve which form of the enzyme—fGly57 or Cys57—provides the active site nucleophile, BcPMH was expressed under anaerobic conditions to prevent formylglycine formation (22). MALDI-TOF analysis of the tryptic digest of the anaerobically expressed BcPMH confirms an increased fraction of unmodified Cys57 (Fig. S5B/C). Replacement of fGly57 by Cys57 resulted in decreased catalytic efficiency for the hydrolysis of substrates 1b, 2b, 4b, and 6b by 4–60-fold in terms of kcat/KM (Table S2) and activity towards sulfate monoester 5b could not be detected for this “mutant.” These data collectively suggest that the fGly form of BcPMH is mainly responsible for the hydrolysis of these five substrates observed with the partially modified enzyme preparation. In contrast, the activity toward phosphotriester 3b increased 2-fold, suggesting that the unmodified cysteine form of BcPMH is mainly responsible for phosphotriester hydrolysis. The lack of cross-inhibition between phosphotriester 3b and phosphodiester 2b, as well as the differences in the pH-rate profiles in comparison to all other substrates, supports this conclusion. Thus, changing the nucleophile (fGly to Cys) adds the capability to hydrolyze phosphate triester 3b.
Structure of BcPMH.
To obtain insight into the structural features that enable the catalytic variability of BcPMH, we solved its crystal structure (Fig. 3). Strep-tagged BcPMH yielded orthorhombic crystals that diffracted to 2.4 Å. Phases were calculated by molecular replacement using the structure of RlPMH as a search model [Protein Data Bank (PDB): 2vqr] (11). Statistics for the data collection and refinement are listed in Table S3.
The α/β-fold of BcPMH is similar to AP rendering it a member of the AP superfamily. Furthermore, its structure is almost identical to RlPMH (PDB: 2vqr, rmsd 0.5 Å over Cα of 511 residues) and shows high homology to the structure of Pseudomonas aeruginosa arylsulfatase (PDB: 1hdh, rmsd 2.5 Å over Cα of 331 residues). The enzyme crystallizes as a symmetric homotetramer (a dimer of dimers, Fig. 3A) and as the extensive oligomerization interfaces suggest [buried surface area calculated with AreaIMol (23): 5,498 Å2 for the dimer; 17,030 Å2 for the tetramer] is also tetrameric in solution. It elutes from a gel filtration column corresponding to a molecular weight of ∼240 kDa (Mprotomer = 61 kDa) and shows a hydrodynamic radius in dynamic light scattering experiments of 110 Å as predicted for the tetramer by HYDROPRO (24).
The salient feature of the BcPMH structure is the very spacious active site (≈10 × 20 Å2 wide and 15 Å deep) (Fig. 3C), allowing for a range of substrate sizes to be accepted without steric hindrance. BcPMH is a mononuclear metalloenzyme, yet the nature of the native metal ion is uncertain. Metal analysis by microfocused photon-induced x-ray emission (microPIXE) showed the presence of 0.28 ± 0.04 eq iron and 0.26 ± 0.06 eq zinc per protein molecule. Modeling the electron density with Fe and Zn, both at 0.3 occupancy, fitted the electron density well. However, the presence of other metal ions with occupancies < 10% cannot be excluded. Furthermore, Mn2+ ions are known to increase enzyme activity (9), and in RlPMH, Mn2+ has been suggested as the native metal ion (11). Indeed, when BcPMH was coexpressed with MtbFGE to increase the fGly-modification efficiency, 0.19 eq Mn, 0.29 eq Ca, 0.40 eq Fe, and 0.36 eq Zn were detected by microPIXE. When the enzyme was incubated in the presence of excess Mn2+, all six activities increased 1.5–2-fold. Although this result does not exclude that other metal ions can promote activity, Mn2+ enables access to higher efficiency and does so for all reactions, indicating that the promiscuous behavior is not caused by the heterogeneity of the active site metal ion.
Fig. 3B shows the active site configuration of BcPMH. Given the limited modification efficiency in E. coli of Cys57 to fGly57 in the absence of MtbFGE coexpression, modest, but identifiable, electron density was observed for fGly (fitted to an occupancy of 20%). The metal ion is coordinated by five ligands in the form of a distorted pyramid. The coordination of one of the gem-diol hydroxyl groups of the nucleophile fGly57 to the metal ion is set up to increase the concentration of the reactive deprotonated form. Arg61 and Thr107 form hydrogen bonds to Cys57/fGly57, stabilizing the fGly hydrate. Arg61 and Tyr105 hydrogen bond to the metal-coordinating residues. His218, Lys337, and Asn78 are in a position to bind the substrate. On the basis of the proposed mechanism for phosphodiester hydrolysis in RlPMH (11), His218 and Lys337 may provide general acid catalysis for leaving group departure and charge stabilization as the TS is approached, whereas Asn78 is involved in substrate binding.
Discussion
BcPMH Combines High Catalytic Efficiency with Low Specificity.
This promiscuous enzyme is capable of hydrolyzing various phosphate and sulfate esters with high rate accelerations (Table 1). Comparison with the native activities of other enzymes illustrates the high efficiency of BcPMH as a catalyst. The second-order rates of PMH for its phosphonate monoester hydrolase activity and the phosphodiesterase activity, its assumed native function, match the kcat/KM of other phosphodiesterases toward model substrates such as phosphate diester 2b (19, 25, 26).
The small rate constants for the uncatalyzed reactions (kuncat) are evidence that the reactions catalyzed by BcPMH are difficult. The second-order rate enhancement (kcat/KM)/kw is a measure for the extent to which the free energy barrier of the enzymatic reaction is lowered compared to the uncatalyzed process. These rate enhancements for BcPMH’s best reaction (1018), but also for four of its additional substrates (1011–1016), correspond to remarkable decreases in the energy of activation between 14.4 and 27.2 kcal mol-1, falling in the range observed for the native reactions of other enzymes (1011–1027) (27).
Metal ions alone accelerate phosphate transfer reactions by up to 2 orders of magnitude (kcat/kuncat) (28), and in enzyme models one metal ion can provide up to 6 orders of magnitude in second-order rate acceleration [(kcat/KM)/kuncat] (29, 30). The larger accelerations observed here for PMH suggest that the active site provides the potential for additional effects that synergize with the reactivity of the metal ion. Proximity effects, medium effects, and acid base catalysis can provide the additional enhancement of 1010-fold for the two best reactions and 103-fold for three of the other reactions. The weakest catalytic reaction, the hydrolysis of phosphotriester 3b, shows similar activity to that exhibited by metal complexes, suggesting that provision of a metal ion/nucleophile combination is chiefly responsible for this activity.
BcPMH is resilient to the removal of a crucial feature of its catalytic machinery. Mutation of the active site nucleophile (fGly/Cys) to alanine compromises catalysis of all reactions, but rate accelerations are still substantial, as has been noted in mutational studies of a range of enzymes (31). Other catalytic factors seem to substitute for the removed nucleophile: Combined with binding features in the active site, a water molecule may occupy the vacant metal ligand position by acting as the replacement nucleophile. The rate reduction indicates that the metal-coordinated fGly nucleophile contributes up to a factor of 103 to PMH’s efficiency.
Many enzyme models are only able to catalyze the hydrolysis of activated (especially p-nitrophenyl) esters and cannot catalyze more demanding reactions with equal proficiency (32). In contrast, BcPMH hydrolyzes the less activated phosphomonoester 1a with a 10-fold higher rate enhancement than 1b despite a better leaving group and thus higher intrinsic reactivity. The same observation holds for substrate pairs 2a/b, 4a/b, 5a/b, and 6a/b. Furthermore, the enzyme is unable to catalyze reactions that are thermodynamically less demanding, such as the hydrolysis of carboxylic esters, lactones, and epoxides. Additionally, the half-lives of the identified substrates vary between 200 days and 105 years under near neutral conditions. Thus, substrate reactivity is not sufficient to explain catalysis in PMH. The observation that catalysis is substantial for less-activated leaving groups suggests that PMH can confer general acid catalysis facilitating leaving group departure, consistent with the proposed mechanism (Fig. 3).
The five substrate classes that are converted by the fGly form of BcPMH are characterized by large differences in their molecular properties. Ground state charges varying from -2 to 0 are tolerated by BcPMH, as well as large differences in substrate size (160–350 Å3), hydrophobicity (logD -12 to 2.9), bond length around the reaction center, and the reaction center itself (phosphorus vs. sulfur). In solution the substrates are hydrolyzed via different mechanisms, as defined by the charge development in the TS. Phosphate and sulfate monoester hydrolysis proceeds via a loose, dissociative TS, whereas phosphodiesters, phosphotriesters, and phosphonate and sulfonate monoesters are hydrolyzed via tighter, associative TSs (Fig. S6). An enzyme would be assumed to provide complementary molecular recognition to the charge development in the TS. However, this does not seem to apply to PMH: The dissociative sulfatase and phosphatase reactions are catalyzed with the same proficiencies as the associative sulfonate hydrolysis, suggesting tolerance in TS recognition. We conjecture that, similar to AP, the enzymatic TSs are not substantially changed from those of the uncatalyzed reactions as has been suggested as the most straightforward solution to lower the thermodynamic barrier for the promiscuous reaction (33). However, this hypothesis has yet to be tested.
Coincident pH-rate profiles and Cys57Ala mutant data suggest that catalysis of the five best reactions follows the same overall reaction pathway. However, a quantitative analysis shows no clear correlation of catalytic proficiency with the type of TS, the amount of negative charge per nonbridging oxygen, substrate hydrophobicity, or bond lengths. This analysis suggests that the specificity of PMH is not dictated by any obvious single characteristic of the substrate and that it is relatively indiscriminate toward the weaker substrates. A closer look reveals a hierarchy of substrate properties that govern the catalytic efficiency of PMH, which are in descending order of importance: Substrates with P centers, negative substrate charge, an associative TS, and smaller substrate size contribute to higher accelerations. These data also suggest that specialization for one compound does not rule out significant catalysis for another.
What Causes the Promiscuous Behavior of BcPMH?
Like many of the promiscuous enzymes described (1–3), BcPMH contains a metal ion (Fig. 3). Metal-coordinated hydroxides or alkoxides are intrinsically highly reactive (3), which may equate to low chemical selectivity. Furthermore, the diameter of the active site is approximately twice that of the largest substrate and therefore allows productive binding of substrates with minimal steric hindrance.
BcPMH is likely to employ covalent catalysis for hydrolysis of all its substrates, confronting it with the problem of different covalent intermediates that have to be broken down to allow multiple turnover. By contrast, imperfect catalysts perform only one turnover: If the covalent intermediate is not broken down, substrates become in effect suicide inhibitors. We hypothesize that the fGly nucleophile provides a solution to this problem: The breakdown of all intermediates is expected to occur by cleavage of the C-O bond in the second step (11, 12) (Fig. 3D, shown in red). This points to a common second step for the collection of promiscuous substrates, where the same hemiacetal-ester cleavage makes different leaving groups depart. Indeed, for all five substrates involving fGly, multiple turnover is observed with 400–600,000 reaction cycles per enzyme molecule (Fig. S1).
Comparison to Other Promiscuous Enzymes.
In comparison to reported cases of catalytic promiscuity, BcPMH is among the most promiscuous hydrolases recorded to date. It can catalyze five chemically distinct reactions with catalytic efficiencies (kcat/KM) that differ by 5 orders of magnitude. By comparison, the five reactions catalyzed by alkaline phosphatase differ by more than 9 orders of magnitude (16, 17, 34). The second-order rate acceleration for the native activity of BcPMH with a (kcat/KM)/kw of 1018 is comparable to that of other known promiscuous enzymes (3). Yet, the simultaneous degrees of freedom of PMH in the identity of the reaction centers, reaction type, and substrate charge distinguish this example of a promiscuous enzyme.
Functional Correlations in the AP Superfamily.
The x-ray structure of BcPMH confirms that it is structurally and mechanistically closely related to arylsulfatases and also, albeit more distantly, to AP and nucleotide phosphodiesterase (11, 13). These three enzymes catalyze at least two reactions besides their native activity that are the native reactions of another family member (14–19). BcPMH promotes all native reactions of these three enzymes, making catalytic promiscuity a widespread feature of this superfamily. Furthermore, various mutants of BcPMH can catalyze the hydrolysis of phosphotriesters and sulfonate monoesters, two activities that have not been observed in the AP superfamily.
Catalytic promiscuity mirrors the chemical potential of the conserved active site arrangement in a superfamily and can be exploited by evolution to modulate specificities leading to differentiated members of a structurally related superfamily. This observation suggests the definition of “catalytic superfamilies” on the basis of reactive active site arrangements. Indeed conservation of chemical functionality has been recognized, e.g., in the enolase superfamily (4). The chemical potential of an active site such as PMH suggests that a targeted search on the basis of promiscuously related chemical reactions may help to identify activities of proteins for which no function has been assigned or where the assignment is computer-generated (and often incorrect). Exploration of other phosphatase superfamilies may reveal promiscuous sulfatase activities indicating the potential of yet unknown sulfatase types in those families. Likewise, this potential could be exploited by directed evolution or enzyme design (35).
Implications of Catalytic Versatility.
Catalytic promiscuity has been suggested to contribute to enzyme evolution, through the mechanism of gene duplication followed by specialization of one of the two copies for the new function (1, 2, 5, 6). The side activities of BcPMH provide a possibly valuable latent functional potential that coexists with a presumed main activity, even if there is no immediate need or even natural substrate for them. The history of BcPMH discovery provides an example of this potential: Even though glyceryl glyphosate is an unnatural compound that the bacterium had never encountered in its original habitat, its ability to hydrolyze this type of phosphonate monoester, thereby enabling the bacterium to use it as the sole source of phosphorus (9), led to the identification of BcPMH. Although BcPMH has a moderate kcat/KM of 9.5 M-1 s-1 against glyceryl glyphosate, this accidental activity conferred survival. Enzymes with kcat/KM values in this region (26, 36), and even as low as 0.3 M-1 s-1, have been shown to confer a selective advantage (8), although in the latter case they were overexpressed from a multiple copy plasmid under control of a strong promotor. The enzyme-catalyzed hydrolysis of sulfonate esters is unprecedented, and there are no examples of sulfonate monoesters in nature, either natural or xenobiotic. However, the observed catalytic efficiencies (kcat/KM = 1.4 and 49 M-1 s-1 for 6a and 6b, respectively) indicate that BcPMH is proficient enough for this unseen substrate that it could be immediately advantageous to an organism.
The chemical flexibility provided by promiscuous enzymes explains how new activities can be picked up rapidly and enhanced or even introduced by single mutations (35). Our data illustrate this potential evolutionary versatility: By acquiring the fGly modification (instead of Cys), the overall activity of PMH is increased, the sulfatase activity is introduced, and the phosphotriesterase activity is lost (Table S2).
BcPMH efficiently catalyzes difficult reactions with different characteristics by using essentially the same catalytic machinery. The large rate accelerations bring relatively difficult transformations (albeit with activated leaving groups) into a range where they become detectable and potentially evolvable. BcPMH is a striking example of how an active site can be adapted for six chemical tasks, encompassing and extending the chemical repertoire of its superfamily.
Materials and Methods
Details of cloning, mutant construction, protein expression, purification, biophysical characterization, protein crystallization, and x-ray structure determination as well as details on the activity assays are listed in SI Text.
Supplementary Material
Acknowledgments.
We thank A. J. Kirby, A. Aharoni, and O. Khersonsky for useful comments on the manuscript. We thank C. Bertozzi for the MtbFGE gene and Monsanto for the kind gift of plasmid pMON9428. This research was funded by the Biotechnology and Biological Sciences Research Council (BBSRC), the Medical Research Council (MRC), the EU networks ENDIRPRO, and ProSA, and studentships to S.J. (German National Academic Foundation) and A.B. (BBSRC CASE/GlaxoSmithKline). F.H. is an ERC starting investigator.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.H. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0903951107/DCSupplemental.
References
- 1.O’Brien PJ, Herschlag D. Catalytic promiscuity and the evolution of new enzymatic activities. Chem Biol. 1999;6:R91–R105. doi: 10.1016/S1074-5521(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 2.Khersonsky O, Roodveldt C, Tawfik DS. Enzyme promiscuity: evolutionary and mechanistic aspects. Curr Opin Chem Biol. 2006;10:498–508. doi: 10.1016/j.cbpa.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 3.Jonas S, Hollfelder F. Mechanism and Catalytic Promiscuity: Emerging Mechanistic Principles for Identification and Manipulation of Catalytically Promiscuous Enzymes. In: Bornscheuer U, Lutz S, editors. Handbook of Protein Engineering. Vol 1. Weinheim, Germany: Wiley VCH; 2008. pp. 47–79. [Google Scholar]
- 4.Glasner ME, Gerlt JA, Babbitt PC. Evolution of enzyme superfamilies. Curr Opin Chem Biol. 2006;10:492–497. doi: 10.1016/j.cbpa.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Aharoni A, et al. The ‘evolvability’ of promiscuous protein functions. Nat Genet. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 6.Jensen RA. Enzyme recruitment in evolution of new function. Annu Rev Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- 7.Hughes AL. The evolution of functionally novel proteins after gene duplication. Proc Biol Sci. 1994;256:119–124. doi: 10.1098/rspb.1994.0058. [DOI] [PubMed] [Google Scholar]
- 8.Patrick WM, Matsumura I. A study in molecular contingency: Glutamine phosphoribosylpyrophosphate amidotransferase is a promiscuous and evolvable phosphoribosylanthranilate isomerase. J Mol Biol. 2008;377:323–336. doi: 10.1016/j.jmb.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dotson SB, Smith CE, Ling CS, Barry GF, Kishore GM. Identification, characterization, and cloning of a phosphonate monoester hydrolase from Burkholderia caryophilli PG2982. J Biol Chem. 1996;271:25754–25761. doi: 10.1074/jbc.271.42.25754. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt B, Selmer T, Ingendoh A, von Figura K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell. 1995;82:271–278. doi: 10.1016/0092-8674(95)90314-3. [DOI] [PubMed] [Google Scholar]
- 11.Jonas S, van Loo B, Hyvonen M, Hollfelder F. A new member of the alkaline phosphatase superfamily with a formylglycine nucleophile: Structural and kinetic characterisation of a phosphonate monoester hydrolase/phosphodiesterase from Rhizobium leguminosarum. J Mol Biol. 2008;384:120–136. doi: 10.1016/j.jmb.2008.08.072. [DOI] [PubMed] [Google Scholar]
- 12.Hanson SR, Best MD, Wong C-H. Sulfatases: Structure, mechanism, biological activity, inhibition, and synthetic utility. Angew Chem Int Ed. 2004;43:5736–5763. doi: 10.1002/anie.200300632. [DOI] [PubMed] [Google Scholar]
- 13.Galperin MY, Bairoch A, Koonin EV. A superfamily of metalloenzymes unifies phosphopentomutase and cofactor-independent phosphoglycerate mutase with alkaline phosphatases and sulfatases. Protein Sci. 1998;7:1829–1835. doi: 10.1002/pro.5560070819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Babtie AC, Bandyopadhyay S, Olguin LF, Hollfelder F. Efficient catalytic promiscuity for chemically distinct reactions. Angew Chem Int Ed. 2009;48:3692–3694. doi: 10.1002/anie.200805843. [DOI] [PubMed] [Google Scholar]
- 15.Lassila JK, Herschlag D. Promiscuous sulfatase activity and thio-effects in a phosphodiesterase of the alkaline phosphatase superfamily. Biochemistry. 2008;47:12853–12859. doi: 10.1021/bi801488c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Brien PJ, Herschlag D. Sulfatase activity of E. coli alkaline phosphatase demonstrates a functional link to arylsulfatases, an evolutionary related enzyme family. J Am Chem Soc. 1998;120:12369–12370. [Google Scholar]
- 17.O’Brien PJ, Herschlag D. Functional interrelationships in the alkaline phosphatase superfamily: Phosphodiesterase activity of Escherichia coli alkaline phosphatase. Biochemistry. 2001;40:5691–5699. doi: 10.1021/bi0028892. [DOI] [PubMed] [Google Scholar]
- 18.Olguin LF, Askew SE, O’Donoghue AC, Hollfelder F. Efficient catalytic promiscuity in an enzyme superfamily: An arylsulfatase shows a rate acceleration of 1013 for phosphate monoester hydrolysis. J Am Chem Soc. 2008;130:16547–16555. doi: 10.1021/ja8047943. [DOI] [PubMed] [Google Scholar]
- 19.Zalatan JG, Fenn TD, Brunger AT, Herschlag D. Structural and functional comparisons of nucleotide pyrophosphatase/phosphodiesterase and alkaline phosphatase: Implications for mechanism and evolution. Biochemistry. 2006;45:9788–9803. doi: 10.1021/bi060847t. [DOI] [PubMed] [Google Scholar]
- 20.Peng J, Schmidt B, von Figura K, Dierks T. Identification of formylglycine in sulfatases by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J Mass Spectrom. 2002;38:80–86. doi: 10.1002/jms.404. [DOI] [PubMed] [Google Scholar]
- 21.Carrico IS, Carlson BL, Bertozzi CR. Introducing genetically encoded aldehydes into proteins. Nat Chem Biol. 2007;3:321–322. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]
- 22.Benjdia A, Deho G, Rabot S, Berteau O. First evidences for a third sulfatase maturation system in prokaryotes from E. coli aslB and ydeM deletion mutants. FEBS Lett. 2007;581:1009–1014. doi: 10.1016/j.febslet.2007.01.076. [DOI] [PubMed] [Google Scholar]
- 23.Lee B, Richards FM. The interpretation of protein structures: Estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 24.Garcia De La Torre J, Huertas ML, Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys J . 2000;78:719–730. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shenoy AR, Sreenath N, Podobnik M, Kovacevic M, Visweswariah SS. The Rv0805 gene from Mycobacterium tuberculosis encodes a 3′, 5′-cyclic nucleotide phosphodiesterase: biochemical and mutational analysis. Biochemistry. 2005;44:15695–15704. doi: 10.1021/bi0512391. [DOI] [PubMed] [Google Scholar]
- 26.Ghanem E, Li Y, Xu C, Raushel FM. Characterization of a phosphodiesterase capable of hydrolyzing EA 2192, the most toxic degradation product of the nerve agent VX. Biochemistry. 2007;46:9032–9040. doi: 10.1021/bi700561k. [DOI] [PubMed] [Google Scholar]
- 27.Wolfenden R. Degrees of difficulty of water-consuming reactions in the absence of enzymes. Chem Rev. 2006;106:3379–3396. doi: 10.1021/cr050311y. [DOI] [PubMed] [Google Scholar]
- 28.Kuusela S, Lönnberg H. Metal ions that promote the hydrolysis of nucleoside phosphoesters do not enhance intramolecular phosphate migration. J Phys Org Chem. 1993;6:347–356. [Google Scholar]
- 29.Morrow JR, Iranzo O. Synthetic metallonucleases for RNA cleavage. Curr Opin Chem Biol. 2004;8:192–200. doi: 10.1016/j.cbpa.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 30.Feng G, Mareque-Rivas JC, Torres Martin de Rosales R, Williams NH. A highly reactive mononuclear Zn(II) complex for phosphodiester cleavage. J Am Chem Soc. 2005;127:13470–13471. doi: 10.1021/ja054003t. [DOI] [PubMed] [Google Scholar]
- 31.Peracchi A. Enzyme catalysis: Removing chemically ‘essential’ residues by site-directed mutagenesis. Trends Biochem Sci. 2001;26:497–503. doi: 10.1016/s0968-0004(01)01911-9. [DOI] [PubMed] [Google Scholar]
- 32.Menger FM, Ladika M. Origin of rate accelerations in an enzyme model: The p-nitrophenyl ester syndrome. J Am Chem Soc. 1987;109:3145–3146. [Google Scholar]
- 33.Zalatan JG, Herschlag D. Alkaline phosphatase mono- and diesterase reactions: Comparative transition state analysis. J Am Chem Soc. 2006;128:1293–1303. doi: 10.1021/ja056528r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang K, Metcalf WW. A new activity for an old enzyme: Escherichia coli bacterial alkaline phosphatase is a phosphite-dependent hydrogenase. Proc Natl Acad Sci USA. 2004;101:7919–7924. doi: 10.1073/pnas.0400664101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toscano MD, Woycechowsky KJ, Hilvert D. Minimalist active-site redesign: Teaching old enzymes new tricks. Angew Chem, Int Ed. 2007;46:3212–3236. doi: 10.1002/anie.200604205. [DOI] [PubMed] [Google Scholar]
- 36.McLoughlin SY, Jackson C, Liu JW, Ollis DL. Growth of Escherichia coli coexpressing phosphotriesterase and glycerophosphodiester phosphodiesterase, using paraoxon as the sole phosphorus source. Appl Environ Microbiol. 2004;70:404–412. doi: 10.1128/AEM.70.1.404-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.