

Abstract
The ob/ob mouse is genetically deficient in leptin and exhibits a phenotype that includes obesity and non-insulin-dependent diabetes melitus. This phenotype closely resembles the morbid obesity seen in humans. In this study, we demonstrate that a single intramuscular injection of a recombinant adeno-associated virus (AAV) vector encoding mouse leptin (rAAV-leptin) in ob/ob mice leads to prevention of obesity and diabetes. The treated animals show normalization of metabolic abnormalities including hyperglycemia, insulin resistance, impaired glucose tolerance, and lethargy. The effects of a single injection have lasted through the 6-month course of the study. At all time points measured the circulating levels of leptin in the serum were similar to age-matched control C57 mice. These results demonstrate that maintenance of normal levels of leptin (2–5 ng/ml) in the circulation can prevent both the onset of obesity and associated non-insulin-dependent diabetes. Thus a single injection of a rAAV vector expressing a therapeutic gene can lead to complete and long-term correction of a genetic disorder. Our study demonstrates the long-term correction of a disease caused by a genetic defect and proves the feasibility of using rAAV-based vectors for the treatment of chronic disorders like obesity.
Keywords: gene therapy, adeno-associated virus vectors
Obesity is a complex disorder and often leads to peripheral hyperinsulinemia, hyperglycemia, and insulin resistance (1–3). Obesity is also a major risk factor for hypertension and cardiovascular disease (3). It is clear that there are multiple pathways that control the complex balance of caloric intake and expenditure. The intake of calories is controlled by molecules that include leptin, leptin receptor, melanin concentrating hormone, the melanocortin 4 receptor, urocortin, and neuropeptide Y and its type 5 receptor (4). In rodents several genes responsible for obesity, including obese (ob), fat (fa), agouti (ay), and diabetes (db) have been identified in genetically obese models (3, 5–7). Recently, the human and mouse ob genes have been cloned, and the secreted gene product, termed leptin, has been characterized (3, 8–11).
In ob/ob mice, mutation in the ob gene leads to a marked increase in food consumption that results in an increase in adipose tissue mass and a syndrome that resembles morbid obesity in humans (9). Abnormalities include hypothermia, lethargy, hyperglycemia, glucose intolerance, and hyperinsulinemia resembling non-insulin-dependent diabetes melitus in humans. Leptin, a 16-kDa secreted protein, is produced exclusively in the adipose tissue and is thought to act mainly on the hypothalamus, although leptin receptors are also present in peripheral tissues (3, 12–14). Leptin has been shown to mediate its effect via the appetite-stimulating peptide, neuropeptide Y (15). In ob/ob mice repeated administration of recombinant leptin reduces food intake, increases energy expenditure and leads to loss of body weight and fat mass (8–11). These effects require continuous administration of the recombinant leptin protein; withdrawal results in reversal to the obese phenotype (11). In contrast to recombinant protein therapy, somatic gene therapy offers the potential of sustained delivery of missing proteins in metabolic diseases.
Adeno-associated virus (AAV) vectors represent a promising delivery system. These vectors are nonpathogenic and can infect both dividing and nondividing cells. The vector system consists of AAV terminal repeats that are necessary and sufficient for replication, packaging and possibly integration. The AAV vector lacks virally encoded genes, thus avoiding potential undesirable immune responses. Also, AAV preparations are stable and can be produced at high titers (>1012 particles/ml) (16, 17). There are recent reports demonstrating long-term expression of transgenes following delivery of AAV vectors into lung, liver, muscle, heart, and brain (18–23).
In this report, we have generated a recombinant AAV (rAAV) vector carrying the mouse leptin cDNA and demonstrated the ability of this vector to express leptin in vitro and in vivo. In vivo we demonstrated that a single i.m. injection of rAAV vector carrying leptin into ob/ob mice leads to long-term correction (6 months) of all metabolic abnormalities tested, including obesity and diabetes.
MATERIALS AND METHODS
Vector Construction.
pKm201CMV is an AAV cloning vector in which an expression cassette, consisting of a cytomegalovirus (CMV) immediate early enhancer, promoter, and intron and a bovine growth hormone polyadenylation site, is flanked by inverted terminal repeat sequences from AAV-2. pKm201CMV, was derived from pKm201, a modified AAV vector plasmid in which the ampicillin resistance gene of pEMBL-AAV-ITR (24) has been replaced with the gene for kanamycin resistance. The expression cassette from pCMVlink, a derivative of pCMV6c (25) in which the bovine growth hormone poly(A) site has been substituted for the simian virus 40 terminator, was inserted between the inverted terminal repeats of pKm201 to generate pKm201 CMV. To construct the AAV leptin expression vector pCMVAAV-m-leptin, a 511-bp fragment, encoding murine leptin cDNA (11) was cloned into the XbaI-BamHI sites of pKm201 CMV. A 579-bp fragment, from the posttranscriptional regulatory element region of hepatitis B (26), was amplified using the primer set 5′-ACATACGCGTGCTTGCGTGGAACCTTTG-3′ and 5′-TTGTGGCGCGCCAGCTTATCGATTTCGAACCCG-3′. The resulting fragment was digested with AscI and MluI and inserted into an MluI site between the leptin coding region and the bovine growth hormone Poly(A) site. AAV helper plasmid pKSrep/cap (encoding rep and cap protein) was constructed by cloning the AAV-2 genome, without the inverted terminal repeats (AAV-2 nucleotides 192-4493) into pBluescript II KS+ (Stratagene).
Preparation and Titering of rAAV Particles.
rAAV vectors were produced by a modified transient plasmid transfection protocol (27). Briefly, human embryonic kidney 293 cells, grown to 60% confluence in a 15-cm dish, were cotransfected with 12.5 μg of helper plasmid pKS rep/cap and 12.5 μg of vector plasmid pCMVAAV-m-leptin or pCMVAAV-lacZ using the calcium phosphate coprecipitation method. After 8 hr, transfection medium was replaced with Iscove’s modified Dulbecco’s medium + 10% fetal bovine serum containing adenovirus type 5 dl312 at a multiplicity of infection of 2. Seventy-two hours postinfection, the cells were harvested in Hepes buffer (2.5 ml per dish) and lysed by three cycles of freezing and thawing. The cell lysate was centrifuged at 2,000 × g for 20 min to remove cell debris. The packaged rAAV virus was purified through two rounds of cesium chloride equilibrium density gradients to remove any contaminating proteins and heated at 56°C for 45 min to inactivate residual adenovirus particles. For estimation of the total number of vector particles, the vector stock was treated with DNase 1, and encapsidated DNA was extracted with phenol-chloroform, precipitated with ethanol. Released DNA was compared with a known standard by dot blot analysis.
In Vitro Analysis of rAAV-Leptin.
rAAV-leptin particles were diluted in 2 ml of IMDM + 10% fetal bovine serum and added to human embryonic kidney 293 cells plated at 50% confluence on a 6-well dish. Virus was left on cells for 24 hr, cells were washed and 2 ml of fresh IMDM + 10% fetal bovine serum was added. Supernatant was collected, for Western blot or RIA analysis (24–48 hr postinfection). For transfections, 2 μg of pCMV-AAV-m-leptin plasmid was incubated with 10 μl of transfection reagent LT1 (Panvera, Madison, WI) and added to 5 × 105 human embryonic kidney 293 cells seeded on 6-well dishes. Complexes were incubated with cells for 4 hours and re-fed with 2 ml of media. Cell supernatant was collected 48 hr posttransfection and analyzed by Western blot or RIA.
Western Blot Analysis.
Supernatant (10 μl) from infected or transfected cells was mixed with 5 μl of 3× Laemmli buffer and boiled to denature proteins. Denatured supernatants were electrophoretically separated on 14% SDS/PAGE (NOVEX, San Diego) and transferred onto nitrocellulose. Blots were probed overnight at 4°C with a 1:5,000 dilution of rabbit anti-leptin antibody (11) in PBS + 0.1% Tween + 0.2% nonfat dry milk. Following extensive washing, a horseradish peroxidase conjugate of goat anti-rabbit IgG (Boehringer Mannheim) was added. Following a 1 hr incubation and further washing, immunoreactive bands were visualized by chemiluminescence (ECL kit, Amersham).
In Vivo Administration of rAAV-Leptin.
Female C57BL/6J-ob/ob mice (4–6 weeks old) were obtained from The Jackson Laboratory. Following anesthesia with a mixture of ketamine and xylazine, 50 μl of normal saline or normal saline containing 5 × 1010 or 1 × 1011 rAAV particles was injected into the tibialis anterior muscle. In some experiments, both legs were injected on two successive days, while in other experiments twice as many particles were injected on one day.
Glucose Tolerance Test.
Animals were fasted for 18 hr and blood was collected from the tail vein to determine fasting glucose levels. Mice then received 1 mg/g body weight of a sterile glucose solution by i.p. injection. Blood samples were collected at 15, 30, 60, 120, and 180 min following the injection and circulating glucose was measured using the Lifescan (Mountain View, CA) One Touch monitor.
Serum Insulin, Glucose, and Leptin levels.
Blood was collected from mice at indicated times by retroorbital bleeds of isoflurane anesthetized mice. Blood glucose levels were determined using a Lifescan One Touch Basic glucose monitor. Insulin levels were measured with the Linco Rat Insulin RIA kit (Linco Research Immunoassay, St. Charles MO). Leptin levels were measured with the Linco mouse Leptin RIA kit.
Statistics.
Statistics were calculated using a Mann–Whitney two sample T test. Calculations were performed on the instat software program.
RESULTS
Vector Construction and in Vitro Analysis.
In addition to the CMV immediate early promoter/enhancer and intron, the AAV vector used in these studies contains a posttranscriptional regulatory element from hepatitis B virus. The posttranscriptional regulatory element, which increases efficiency of mRNA transport, (26) was included to increase the size of the vector genome for more efficient packaging. Inclusion of the coding region for mouse leptin into this construct results in a 3.4-kb packageable vector genome. This vector plasmid was packaged using standard methods, and a purified stock of 1.25 × 1012 particles/ml was obtained. The ability of this vector to direct the production of leptin protein was assessed in vitro after infection of 1 × 106 293 embryonic kidney cells with 109 or 1010 particles of recombinant virus. Supernatants were collected from infected cells 48 hr postinfection and analyzed by Western blot and RIA. As a control, supernatant was harvested from cells transfected with 2 μg of the pCMVAAV-m-leptin packaging plasmid. Western blot analysis, using a rabbit anti-mouse leptin antibody, revealed a single immunoreactive protein in supernatant from transfected cells and cells infected with 1010 particles (Fig. 1). Previous studies have shown that this protein, which migrates slightly faster than the prestained 16-kDa marker, is identical in size to recombinant mouse leptin produced in Eschericha coli (data not shown). No band was visible in supernatant from cells infected with 109 particles or mock transfected cells. The level of protein was quantitated using a sensitive RIA. While mock infected cells released no detectable leptin into media, cells infected with 109 and 1010 particles released 47 and 290 ng of leptin per 24 hr/106 cells, respectively.
Figure 1.
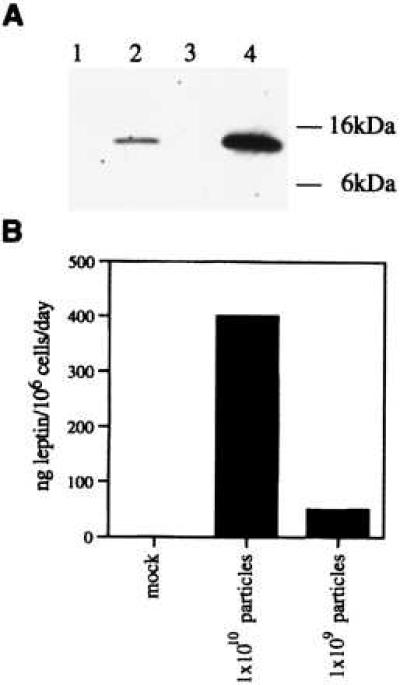
Analysis of leptin expression in vitro. Supernatant was harvested from human 293 cells and analyzed by Western blot (A) or RIA (B). (A) Western blot analysis of supernatant from cells infected with 1 × 109 (lane 1), 1 × 1010 (lane 2) AAV-leptin particles, mock infected cells (lane 3) or cells transfected with 2 μg of pCMVKm201-leptin (lane 4). (B) Quantitation of expression by RIA.
Effect of Intramuscular Injection of AAV Leptin on Weight Gain and Food Intake.
Injection of rAAV vector into skeletal muscle has been shown to lead to persistent, high level expression of transgenes (20, 21). To determine if the morbid obesity and diabetes characteristic of the ob/ob phenotype could be prevented, young mice (4–6 weeks old and ≈25 g) were treated by administration of rAAV-leptin into the tibialis anterior muscle. The weights of 10 rAAV-leptin-treated mice were compared with 10 mice treated with 0.9% saline vehicle on a weekly basis and the results are summarized in Fig. 2A. The effects of rAAV leptin administration were gradual. Treated mice continued to gain weight for the first 3 weeks, but at a rate that was significantly less than the saline-treated controls (Fig. 2A) (P = 0.004 for week 2 and 0.0005 for week 3). During the fourth week following administration of vector, the rAAV-injected mice began to lose weight while the saline-treated mice continued to gain weight. During weeks 5, 6, and 7, the treated mice continued to lose weight at a relatively constant rate (average of 2.3, 2.4, and 2.0 g per animal per week, respectively). Weight loss in the treated mice continued through week 11 although the rate of loss declined (1.4, 0.59, and 0.35, 1.02 g per animal per week, respectively). From weeks 11–26, the weight remained relatively constant with mice gaining, on average, <3 g during these 15 weeks. The saline-injected mice continued to gain weight throughout the course of the study. By week 8, the average weight of leptin-treated mice was less than half of the control (saline-treated) ob/ob mice (25.9 vs. 52.1 g) (Fig. 3). At week 26 treated mice weighed 27.5 g while untreated mice weighed 77.9 g. From week 4 onward the weight difference between the two groups was statistically significant with P < 0.0005. The treated mice were observed to be much more physically active than saline-treated ob/ob mice.
Figure 2.
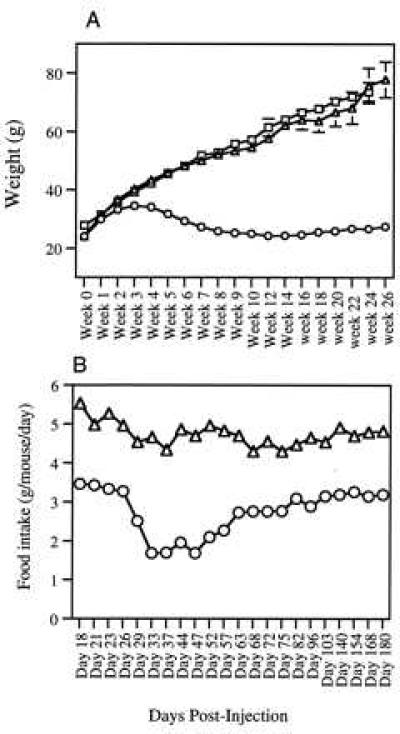
Effect of rAAV-leptin treatment on body weight and food intake. Mice received i.m. injections of either 1011 particles of rAAV-leptin (○), β-galactosidase (□), or saline (▵), and weights were monitored three times weekly. (A) Mean ± SEM of 10 mice in each group. For simplicity, only one time point is shown for each week. (B) Mice were caged in groups of five and food intake over a 24-hr period was measured. The average food consumption per mouse per day is shown. The variation between the two cages in each group was negligible.
Figure 3.
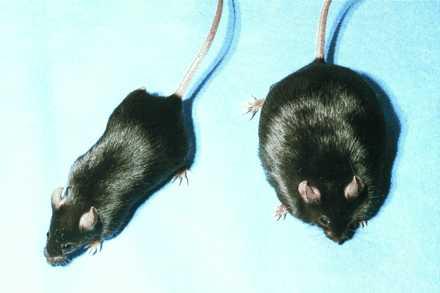
Physical appearance of mice following rAAV-leptin treatment. Mice were photographed 6 weeks after being treated with rAAV-leptin (Left) or saline vehicle (Right).
Monitoring of food intake was begun in the third week following injection. Preweighed standard mouse food was added to cages (containing five mice) each evening and the amount of food consumed was measured the following day. The reduction in food intake of treated mice corresponded with the extent of weight loss. At the earliest time points monitored (day 18–23) rAAV leptin-treated mice ate a daily average of 3.4 g of food per mouse as compared with an average of 5.1 g for saline-treated controls (Fig. 2B). The following week (day 24–29) the mice ate on average 2.9 g of food per day and untreated mice ate 4.6 g per day. From weeks 4–7 (day 33–47), the leptin-treated mice consistently consumed an average 1.9 g of food/day and by week 9 the consumption was ≈2.3 g per day. During weeks 10 and 11, consumption in the leptin-treated mice plateaued at 2.75 g per mouse per day and remained at this level through week 15. Although food intake rose slightly after week 15, treated mice never ate >3.5 g of food per day. Throughout the course of the study, the saline controls ate an average of 4.6 g per mouse per day.
In a study to ensure that the observed weight loss was not due to a side-effect of rAAV injection, ob/ob mice were injected with an 1 × 1011 rAAV-β-galactosidase particles as a negative control. The kinetics of weight gain for the β-galactosidase-treated mice were identical to the saline-treated mice in the initial experiment (Fig. 2A).
Serum Leptin Levels.
The level of circulating leptin was measured at 5, 7, 9, 11, and 14 weeks after i.m. delivery of rAAV-leptin. At week 5, the serum leptin levels from five AAV-leptin-treated mice and five saline-treated controls were measured. Average leptin concentration for the treated mice was 1.7 ng/ml, with a range of 1.3–2.34 ng/ml (Fig. 4). The saline-treated ob/ob mice averaged 1.19 ng/ml, with a range of 1.01–1.34 ng/ml. This background may be due to reactivity with the truncated leptin protein that is the predicted product of the ob mutation. At week 7, the same groups of mice and five C57 control mice were tested. The average serum leptin level of the rAAV-leptin-treated mice increased to 3.33 ng/ml (range = 2.9–4.56), the saline-treated control mice again measured 1.2 ng/ml and the normal C57 mice had serum leptin levels averaging 3.76 ng/ml. Serum leptin levels in the rAAV-leptin-treated mice decreased to 2.46 ng/ml at week 9. Untreated ob/ob mice had circulating leptin levels of 0.65 ng/ml and the wild-type C57 mice had levels of 4.19 ng/ml at this time point. At the 11 week time point, the leptin concentration was 2.97 ng/ml in treated mice vs. 1.03 ng/ml of reactive protein in the untreated ob/ob mice. This is again in the range of normal C57BL6 mice (2.31 ng/ml). The leptin level at week 14 ranged from 3.5–5 ng/ml with an average of 4.8 ng/ml.
Figure 4.
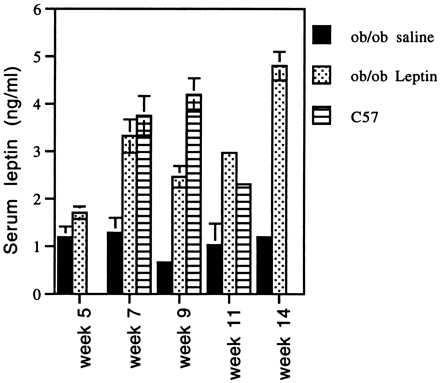
Circulating leptin levels. Serum was collected from mice at the indicated times and leptin levels were measured using the Linco RIA kit. Values are the average of five mice ± SEM. Week 7 mice were fasted for 18 hr prior to serum collection. Mice tested at weeks 5, 9, 11, and 14 were fed ad libitum prior to serum collection. Serum was not collected from C57 mice at weeks 5 and 14. (P values for treated vs. untreated are 0.09, 0.0005, 0.007, 0.0079, and 0.0079 for weeks 5, 7, 9, 11, and 15, respectively.)
Serum Insulin and Glucose Measurements.
The ob/ob phenotype is characterized by insulin-resistant diabetes; ob/ob mice are hyperglycemic, despite elevated levels of circulating insulin. To determine the effects of leptin gene therapy on diabetes, fasting blood glucose and insulin were measured. At week 6, all five saline-treated mice tested were hyperglycemic (Fig. 5A). The fasting glucose levels ranged from 168–355 mg/dl (normal = 91–129 mg/dl) with an average of 259.2. In contrast, all of the rAAV-leptin-treated mice were within, or slightly below, the normal range with a group average of 91.2 mg/dl and a range of 74–125 mg/dl. The insulin levels in serum from these fasted mice, were also measured (Fig. 5b). All mock-treated animals showed marked hyperinsulinemia, with serum insulin levels between 8 and 20 ng/ml. The average serum insulin concentration for rAAV-leptin-treated animals was 0.54 ± 0.2 ng/ml. This is within the normal range of 0.4 ± 0.1 ng/ml.
Figure 5.
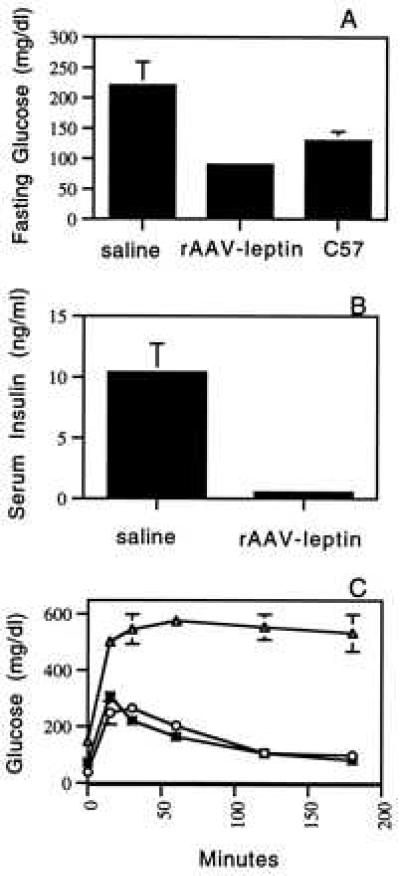
Effects of rAAV-leptin on glucose metabolism and insulin secretion. Untreated C57 mice or ob/ob mice treated with rAAV-leptin or saline were fasted for 18 hr and bled for determination of fasting glucose (A) and insulin (B), 6 weeks postinjection. The values presented here are the mean ± SEM of five mice. (C) Glucose tolerance was determined in saline (▵), rAAV-leptin-treated (○), or C57 (∗) mice by measuring blood glucose levels at indicated times after i.p. injection of glucose. Values are the mean ± SEM of three mice in each group. Test were performed on fasted mice, 8 weeks postinjection.
Glucose Tolerance Test.
Glucose tolerance tests were performed to measure the ability of rAAV-leptin-treated mice to clear glucose from circulation. At 8 weeks after vector administration, a bolus of 1 mg/g glucose was injected i.p. into fasted mice and blood glucose was monitored over time. In control C57 mice and leptin-treated ob mice, the level of circulating glucose peaked at 30 min and returned to normal within 120 min (Fig. 5c). In mock-treated ob/ob mice, the level of glucose was at least 2-fold greater than the leptin-treated mice at all time points. The glucose levels in these mice did not normalize within the 3 hr time course of the study.
DISCUSSION
We have generated a rAAV vector encoding mouse leptin and characterized expression of leptin both in vitro and in vivo. As shown in Fig. 1, rAAV vector encoding mouse leptin can infect 293 cells and the secreted leptin migrates at the expected size of 16 kDa. We have also quantified the amount of leptin secreted by these cells by RIA and the levels range from 50–300 ng/106 cells per 24 hr depending on the multiplicity of infection. Interestingly, we noticed that the packaging capability of the rAAV vector is sensitive to the size of the vector genome packaged. Inclusion of PRE sequence to increase the size of the vector from 2,840–3,430 bp helped in improving the functional titer (data not shown). This result is consistent with the findings of Dong et al. (29), who have demonstrated a direct correlation between genome size and titer of recombinant AAV vectors.
To demonstrate the effectiveness of rAAV mediated mouse leptin delivery, we chose young (4–6 week old) ob/ob mice that gain weight rapidly. Our objective was to evaluate the efficacy of somatic gene therapy in preventing the onset of obesity and diabetes in these mice. Earlier reports using recombinant leptin protein have demonstrated that the protein can be delivered by either i.p. or i.v routes of administration. Reports that demonstrated the delivery of rodent leptin by gene therapy utilized adenoviral based delivery (i.v) and the expression of the transgene presumably occurred in the liver (30, 31). We administered the rAAV vector by the i.m. route and monitored food intake and weight gain over a period of 6 months. In comparison to saline-treated mice, rAAV-leptin-treated mice gained significantly less weight starting from week 1 until the end of the observation period. Treated animals began losing weight by week 4 and continued to lose weight until week 8, at which time weight began to stabilize. By week 8, the average weight of these animals is much closer to the age-matched C57 control mice than to untreated ob/ob mice. Remarkably, this effect on weight lasts for at least six months.
Serum leptin levels were monitored by RIA at five time points during the experiment (5, 7, 9, 11, and 14 weeks). The level of serum leptin in AAV-leptin-treated mice reaches a maximum between week 5 and 7, a time period during which weight loss is also at a maximum. The expression persists for at least 14 weeks, during which time the mice continue to eat less food than their untreated peers and maintain low body weight. Leptin levels in rAAV-leptin-treated ob mice are comparable to the levels in C57BL mice (1.7–3.3 ng/ml vs. 2.3–4.19 ng/ml). Thus, ectopic expression of physiologic levels of leptin can prevent onset of obesity. Interestingly, the RIA employed in this study also detects some activity in untreated ob/ob mice serum. This might be due to the presence of endogenous inactive leptin secreted in this strain of mice (the ob defect is due to premature termination codon in the leptin coding sequence).
Our data also demonstrates a correlation between weight loss and amount of food intake in the treated mice. In our experiment the food intake in treated mice gradually decreased over a period of 3–6 weeks and stabilized around week 8. By week 10, the average consumption of food in leptin-treated mice was slightly less than in age-matched control C57 mice (2.76 g/day vs. 3.2 g/day).
Hyperinsulinemia and insulin resistance could be corrected in leptin-treated mice. As demonstrated in Fig. 5, at week 6 there was a complete reversal of hyperinsulinemia and hyperglycemia in treated animals. The levels of circulating insulin in treated animals were similar to levels reported for C57 mice (.54 ng/ml vs. 0.40 ng/ml). rAAV-leptin-treated mice had a normal response to a glucose challenge. At week 8, control ob/ob mice failed to correct the exaggerated hyperglycemic state after a postfast injection of glucose. In contrast, leptin-treated and age-matched C57BL mice corrected their hyperglycemia. This demonstrates that insulin resistance has been corrected and that these mice are able to properly regulate insulin secretion in response to glucose challenge.
Our report demonstrates that delivery of leptin by the i.m. route can lead to correction of the ob defect. It is also interesting to note that the weight loss in the treated animals is much more gradual than noticed in experiments where a bolus of recombinant protein is administered. This is reflective of the kinetics of gene expression by rAAV vectors. Expression of a marker gene from rAAV vectors injected into the mouse muscle gradually increases over a period of 4–6 weeks before stabilizing (unpublished data). In contrast, adenoviral gene delivery results in rapid onset of protein expression that is extinguished within 2 weeks, presumably by immune response to adenoviral proteins (30).
Thus, somatic gene therapy using a rAAV vector encoding mouse leptin can lead to total correction of the obese phenotype in ob/ob mice. Preliminary experiments demonstrate that i.m. rAAV-m-leptin administration leads to correction of ob phenotype in older ob/ob mice (12–13 weeks weighing 53 g, data not shown). More importantly, our data point out that long-term correction of genetic defect by somatic gene therapy is possible by rAAV-based vectors.
In summary, we have demonstrated that rAAV can be used as an efficient in vivo gene therapy vector. Direct injection of rAAV vectors can transduce muscle efficiently and lead to expression, secretion and correction of metabolic disorders. Finally, stable persistence of gene expression by a single injection may be useful in treatment of numerous metabolic disorders.
Acknowledgments
We acknowledge Tanya Young and Charles Vitt for expert assistance with all animal procedures. We also thank James Stephans and Wendy Fantl for reagents; Janice Hamer and Michelle Stampien for technical assistance; and Giulia Kennedy for comments on the manuscript.
ABBREVIATIONS
- AAV
adeno-associated virus
- rAAV
recombinant AAV
- CMV
cytomegalovirus
References
- 1.Olefsky J M, Kolterman O G, Scarlett J A. Am J Physiol. 1982;243:E15–E30. doi: 10.1152/ajpendo.1982.243.1.E15. [DOI] [PubMed] [Google Scholar]
- 2.Jahr H, Ratzman K P, Beckert R, Besch W, Hahn H J. Metabolism. 1983;32:1101–1106. doi: 10.1016/0026-0495(83)90055-0. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman J M. Nature (London) 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 4.Spiegelman B M, Flier J S. Cell. 1996;87:377–389. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- 5.Naggert J K, Fricker L D, Varlamov O, Nishina P M, Rouille Y, Steiner D F, Carroll R J, Paigen B J, Leiter E H. Nat Genet. 1995;10:135–142. doi: 10.1038/ng0695-135. [DOI] [PubMed] [Google Scholar]
- 6.Miller M M, Duhl D M J, Vrieling H, Cordes S P, Ollmann M M, Winkes B M, Barsh G S. Genes Dev. 1993;7:454–467. doi: 10.1101/gad.7.3.454. [DOI] [PubMed] [Google Scholar]
- 7.Tartaglia L A, Dembrski M, Weng X, Deng N, Culpepper J, Devos R, Richards G J, Campfield L A, Clark F T, Deeds J, Muir C, Sanker S, Moriarty A, Moore K J, Smutko J S, Mays G G, Woolf E A, Monroe C A, Tepper R I. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 8.Pelleymounter M A, Cullen M J, Baker M B, Hecht R, Winter D, Boone T, Collins F. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 9.Halaas J L, Gajiwala K S, Maffei M, Cohen S L, Chait B T, Rabinowitz D, Lallone R L, Burley S K, Friedman J M. Science. 1995;269:546–549. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 10.Campfield L A, Smith F J, Guisez Y, Devos R, Burn P. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 11.Giese K, Fantl W J, Vitt C, Stephans J C, Cousens L, Wachowicz M, Williams L T. Mol Med. 1996;2:50–58. [PMC free article] [PubMed] [Google Scholar]
- 12.Maffei M, Halaas J, Ravussin E, Pratley R E, Lee G H, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, Kern P A, Friedman J M. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 13.Emilsson V, Liu Y-Y, Cawthorne M A, Morton M N, Davenport M. Diabetes. 1997;46:313–316. doi: 10.2337/diab.46.2.313. [DOI] [PubMed] [Google Scholar]
- 14.Shimabukuro M, Koyama K, Chen G, Wang M-Y, Trieu F, Lee Y, Newgard N B, Unger R H. Proc Natl Acad Sci USA. 1997;94:4637–4641. doi: 10.1073/pnas.94.9.4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erickson J C, Hollopeter G, Palmiter R D. Science. 1996;274:1704–1707. doi: 10.1126/science.274.5293.1704. [DOI] [PubMed] [Google Scholar]
- 16.Flotte T R, Carter B J. Gene Ther. 1995;2:357–362. [PubMed] [Google Scholar]
- 17.Samulski R, Chang L, Shenk T. J Virol. 1989;63:3822–3828. doi: 10.1128/jvi.63.9.3822-3828.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flotte T R, Afione S A, Conrad C, McGrath S A, Solow R, Oka H, Zeitlin P L, Guggino W B, Carter B J. Proc Natl Acad Sci USA. 1993;90:10613–10617. doi: 10.1073/pnas.90.22.10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koeberl D D, Alexander I E, Halbert C L, Russell D W, Miller A D. Proc Natl Acad Sci USA. 1997;94:1426–1431. doi: 10.1073/pnas.94.4.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher K J, Jooss K, Alston J, Yang Y, Haecker S E, High K, Pathak R, Raper S E, Wilson J M. Nat Med. 1997;3:306–312. doi: 10.1038/nm0397-306. [DOI] [PubMed] [Google Scholar]
- 21.Xiao X, Li J, Samulski R J. J Virol. 1996;70:8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplitt M G, Leone P, Samulski R J, Xiao X, Pfaff D W, O’Malley K L, During M J. Nat Genet. 1994;8:148–154. doi: 10.1038/ng1094-148. [DOI] [PubMed] [Google Scholar]
- 23.Kaplitt M G, Xiao X, Samulski R J, Li K, Ojamma I, Klein, Makimura H, Kaplitt M J, Strumpf R K, Breslow J L, Diethrich E B. Ann Thorac Surg. 1996;62:1669–1676. doi: 10.1016/s0003-4975(96)00946-0. [DOI] [PubMed] [Google Scholar]
- 24.Srivastava C H, Samulski R J, Lu L, Larsen S H, Srivastava A. Proc Natl Acad Sci USA. 1989;86:8078–8082. doi: 10.1073/pnas.86.20.8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chapman B S, Thayer R M, Vincent K A, Haigwood N L. Nucleic Acids Res. 1991;19:193–198. doi: 10.1093/nar/19.14.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Z M, Yen T S. Mol Cell Biol. 1995;15:3864–3869. doi: 10.1128/mcb.15.7.3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou S Z, Cooper S, Kang L Y, Ruggieri L, Heimfeld S, Srivastava A, Broxmeyer H E. J Exp Med. 1994;179:1867–1875. doi: 10.1084/jem.179.6.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X S, Yoder M C, Zhou S Z, Srivastava A. Proc Natl Acad Sci USA. 1995;92:12416–12420. doi: 10.1073/pnas.92.26.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong J Y, Fan P D, Frizzell R A. Human Gene Ther. 1996;7:2101–2112. doi: 10.1089/hum.1996.7.17-2101. [DOI] [PubMed] [Google Scholar]
- 30.Muzzin P, Eisensmith, Copeland K C, Woo S L C. Proc Natl Acad Sci USA. 1996;93:14804–14808. doi: 10.1073/pnas.93.25.14804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen G, Koyama K, Yuan X, Lee Y, Zhou Y-T, O’Doherty R, Newgard C B, Unger R H. Proc Natl Acad Sci USA. 1996;93:14878–14882. doi: 10.1073/pnas.93.25.14795. [DOI] [PMC free article] [PubMed] [Google Scholar]