

Abstract
The methodology used to establish tolerable upper intake levels (UL) for nutrients borrows heavily from risk assessment methods used by toxicologists. Empirical data are used to identify intake levels associated with adverse effects, and Uncertainty Factors (UF) are applied to establish ULs, which in turn inform public health decisions and standards. Use of UFs reflects lack of knowledge regarding the biological events that underlie response to the intake of a given nutrient, and also regarding the sources of variability in that response. In this paper, the Key Events Dose-Response Framework (KEDRF) is used to systematically consider the major biological steps that lead from the intake of the preformed vitamin A to excess systemic levels, and subsequently to increased risk of adverse effects. Each step is examined with regard to factors that influence whether there is progression toward the adverse effect of concern. The role of homeostatic mechanisms is discussed, along with the types of research needed to improve understanding of dose-response for vitamin A. This initial analysis illustrates the potential of the KEDRF as a useful analytical tool for integrating current knowledge regarding dose-response, generating questions that will focus future research efforts, and clarifying how improved knowledge and data could be used to reduce reliance on UFs.
Keywords: dose-response, tolerable upper intake levels, vitamin A, retinol, uncertainty factors, homeostatic mechanisms
INTRODUCTION
Nutrients are bioactive substances classified as either macronutrients (energy-producing substances—carbohydrates, fats, and proteins), or micronutrients (vitamins, minerals, and electrolytes). While the various nutrients are required for normal growth, maintenance, and repair of tissue, excess nutrient intake can cause adverse effects. Thus, considerable effort has gone toward identifying “safe” nutrient intake levels. For micronutrients, the methods for setting “tolerable upper intake levels” or “safe upper levels” have borrowed from risk assessment methodologies used by toxicologists for environmental chemicals, in which data from experimental, observational, and clinical studies are used to identify both the nature of effects resulting from excess intake and the intake levels associated with such effects (Food and Nutrition Board, 1998; Institute of Medicine 2006; World Health Organization/Food and Agriculture Organization of the United Nations, 2006; Taylor and Yetley, 2008; FSA, 2003; SCF, 2006).
As is the case with many environmental chemicals, the lowest nutrient intake level associated with an adverse effect is generally considered to represent a “threshold” level, such that any intake at or above that threshold level is expected to pose a health risk (with the specific nature of the risk being dependent on the type of nutrient and the actual intake level). To account for differences, for example, between experimental species and humans or among individual humans, Uncertainty Factors (UF) are applied to the empirically-observed threshold levels to generate reference values of use for various public health purposes. Efforts to establish such values are not as robust or refined as would be desired, due to significant data deficiencies for many substances and also due to various methodological issues, particularly relating to the ability to select appropriate UFs (Renwick and Walker, 2008; Renwick 2006).
Nutrients differ from environmental chemicals in that adverse effects may result from either inadequate intake (of an essential nutrient), or excess intake (of most nutrients). Thus, for many nutrients, two threshold levels are presumed to exist: 1) an intake level that must occur on a regular basis to prevent the adverse effects of deficiency, and 2) an intake level that must be exceeded on a regular basis for a toxic effect to occur. Between these two thresholds, there is a range of safe and sufficient nutrient intake levels.
Nutritional scientists have evaluated, on a nutrient by nutrient basis, available data and have identified values associated with such upper and lower thresholds. For example, in North America, the Institute of Medicine (IOM) is charged with identifying Dietary Reference Intake values (DRIs), which are used for a variety of purposes (e.g., food and nutrition programs and public policies, research design and evaluation, population monitoring, patient counseling, etc.). DRIs are a set of nutrient-based reference values for intakes, established for specified life-stage groups. They include the Estimated Average Requirement, defined as the value at which 50% of the population will have sufficient intake; the Recommended Dietary Allowance (RDA), defined as a level that will cover the needs of essentially all individuals; and the Tolerable Upper Intake Level (UL), the highest level of daily nutrient intake that is likely to pose no risk of adverse health effects for almost all individuals (Institute of Medicine, 2006).
Nutrient intake levels typically vary over time and, on any given day, may fall short of RDAs or go above ULs. But various physiological homeostatic mechanisms exist to maintain systemic and cellular nutrient levels within a safe and adequate range.1 These mechanisms provide stability to the organism, and allow the body to adjust to the intake of a wide variety of foods, differing in nutrient contents, as present in most human diets. Homeostatic mechanisms also allow for adjustments to normal physiological states, such as pregnancy. However, homeostatic mechanisms may be inadequate or overwhelmed. These mechanisms cannot compensate when long-term nutrient intake is very low. Conversely, when long-term nutrient intake is very high, homeostatic mechanisms may become “saturated,” or they may become undermined by altered pathophysiological states (e.g., certain disease states, drug actions etc.). When homeostatic mechanisms fail, adverse effects (nutrient deficiency or toxicity) may ultimately result.
As explained in the introductory paper of this series (Julien et al., 2009), a systematic analysis of individual Key Events along the mode-of-action pathway between intake and effect of concern may contribute new insight with regard to dose-response, biological thresholds, and safe intake levels. The “Key Events Dose-Response Framework” (KEDRF) provides an organizing structure for such an analysis whereby each Key Event is considered with regard to its fundamental biology (including the dose-response relationship for the individual event), and with regard to its influence on the overall dose-response relationship for the effect of concern. Figure 1, reproduced from the first paper in this series, illustrates this conceptual framework.
Figure 1.
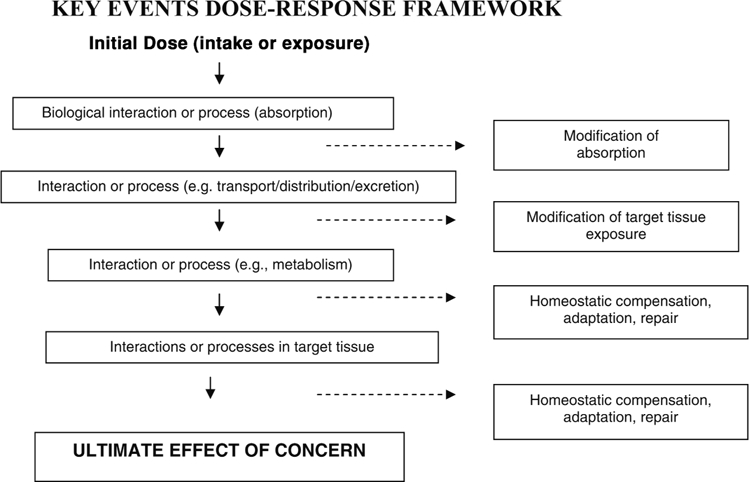
Schematic of the Key Events Dose-Response Framework. The Key Events Dose-Response Framework organizes information on the multiple biological events that occur between an initial dose and the specified effect of concern. Events are indicated generically; but a given pathway may have several kinetic and dynamic events, and may include events that are reversible. In theory, each event may be characterized by its dose and its outcome. Various mechanisms may be available at some events to respond to conditions caused by the agent.
As shown in Fig. 1, various mechanisms (e.g., homeostatic mechanisms, repair, adaptive, immune) may engage at individual events to maintain a normal physiological environment, and thereby influence the outcome of the event. Thus, the capacity of such mechanisms to maintain the environment within a normal homeostatic range (HR) will likely influence the overall dose-response relationship. Events with such mechanisms may be considered to be “control points” in the overall pathway. While every event in the pathway must occur in order to achieve the effect of concern, it is conceivable that a particular event could disproportionately influence its likelihood. Such an event could be hypothesized to be a “determining event” in that the outcome of this event tends to determine whether the effect of concern occurs, or not.
It is important to note that the biological interactions that occur at individual key events may be affected by a variety of factors (e.g., genetics, life stage, health status, behavioral patterns); these factors may exert their influence by modifying the effectiveness of homeostatic mechanisms. Understanding the conditions under which homeostatic control can be undermined or lost is fundamental to better characterizing the dose-response for nutrients, and fundamental to characterizing threshold values as well as the variability in their values.
APPLYING THE GENERIC KEY EVENTS DOSE-RESPONSE FRAMEWORK TO VITAMIN A
Overview
Knowledge of specific key events along the pathway from intake to effect of interest is incomplete for most nutrients, and even less is known about homeostatic mechanisms operating at key events. For vitamin A, however, a fair amount is known. Thus, this vitamin was selected for an initial case study to examine the potential value of the KEDRF for nutrients. The present study will be limited to considering adverse effects from excess intake of preformed vitamin A (retinol). Future work may expand consideration to effects resulting from inadequate intake, and may delve deeper into issues and questions raised in this first case study. The remainder of this paper will:
Briefly review the types of adverse effects associated with retinol.
Describe the pathway of Key Events that leads, potentially, from the intake of preformed vitamin A to the effect of concern (specified for this study as inappropriate gene expression).
Discuss what is known about dose-response and homeostatic mechanisms at each Key Event, including implications for the existence of thresholds at the event, and implications regarding the role of the event in protecting against the effect of concern.
Assess lessons learned regarding retinol and future research needs.
Assess the value of the Key Events Dose-Response Frame-work as applied to retinol.
Adverse Effects Associated with Inadequate or Excess Intakes of Vitamin A
Vitamin A is a family of molecules that includes retinol and potent bioactive retinol metabolites that are required for health. As reviewed elsewhere (Institute of Medicine, 2001), inadequate vitamin A status can potentially lead to visual abnormalities, impaired fertility, a decreased immune response, decreased bone growth, and impaired maintenance of the surface lining of the eyes and epithelial lining of the respiratory, urinary, and intestinal tracts. Vitamin A also plays a role in gene regulation and in guiding embryonic development (Clagett-Dame and DeLuca, 2002). In pregnant women, both inadequate intake and excess intake may lead to teratogenic effects (McCaffery et al., 2003). Excess intake of vitamin A or its products has been associated with organ damage and weakened tissues (e.g., bones) in addition to teratogenic effects.
It is important to note that adverse effects resulting from an excess or deficiency of vitamin A are almost certainly a product of both dose level and dose frequency. Acute vitamin A toxicity, resulting even in death, can occur after a single dose or several extremely high doses of vitamin A; acute toxicity can also occur with moderately high intakes of vitamin A during critical physiological periods, such as early pregnancy. Data from observational studies suggest that unhealthful outcomes can also result from more modest elevations in vitamin A intake (via diet or supplements) over a prolonged time period. Similarly, adverse effects from insufficient intake occur only after prolonged or chronic deficiency. Examples of the integrated effects of dose level and frequency are shown in Table 1.
Table 1.
Examples of adverse effects associated with dietary intake of vitamin A. Effects are a product of dose level and dose frequency
| Vitamin A Intake: Level and Frequency | Potential Clinical Effects (Institute of Medicine, 2001) |
| Extremely high acute intake (Excessive consumption of supplements, or foods extremely high in vitamin A) | Severe toxicity or lethality, likely due to rapid changes in membranes (fluid and hemodynamics) and possibly inappropriate gene expression. |
| Very high acute intake | Teratogenicity, likely resulting from inappropriate gene expression. |
| Moderately high chronic intake | Organ damage affecting metabolism (e.g., liver damage), likely as a result of membrane damage and/or inappropriate gene expression and their sequelae – e.g., fibrosis, or altered immune/inflammatory functions. |
| Marginally high chronic intake | Weakened tissues (possible bone fragility). |
| Chronic insufficient intake | Impaired night vision, blindness, reduced reproductive performance, poor antibody response, decreased bone growth, and impaired maintenance of the surface lining (e.g., skin, intestinal tract). |
Toxicity from excess dietary vitamin A intake results from high systemic levels of free retinol (i.e., retinol that is not stored, esterified, or bound to chaperone proteins) and subsequently, increased levels of the metabolite of retinol, retinoic acid (RA). As illustrated in Fig. 2, there are two main etiologies underlying the effects from excess intake:
Figure 2.
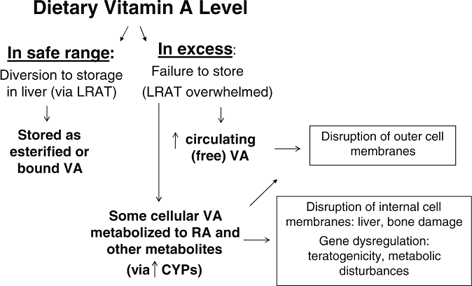
General etiologies leading to adverse effects from excess dietary vitamin A.
Both RA (and possibly its metabolites) as well as free retinol itself (either systemic or cellular) are soluble in cellular lipids, and may intercalate into membranes, destroying membrane integrity and causing cell lysis. Thus, multiple mechanisms are in place to keep retinol in a bound or esterified form.
Excess levels of RA may also lead to increased binding to retinoid receptor proteins and subsequently, increased stimulation of retinoid-responsive genes. Inappropriate gene expression can result in aberrant cell differentiation or induction of apoptosis (Soprano and Soprano, 1995; Williams and Iatropoulos, 2002; Yin et al., 2005).
Overview of Key Events for Retinol
The absorption, fate, and target tissue activity of ingested vitamin A and its metabolites have been reviewed elsewhere (Ross and Harrison, 2007; Harrison, 2005) and will be described only briefly here. Figure 3 summarizes Key Events along the pathway from vitamin A intake to the effect of concern, which in this case is inappropriate gene expression. Each of these Key Events will be systematically examined with regard to its dose-response characteristics, homeostatic mechanisms, and implications for thresholds.
Figure 3.
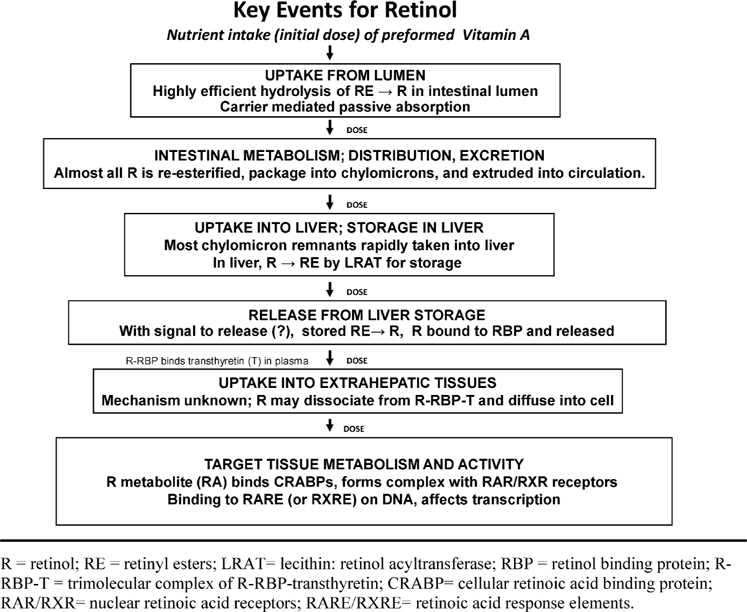
Overview of Key Events between dietary intake of preformed vitamin A and target tissue activity that affects transcription.
In humans, dietary Vitamin A is ingested in two primary forms: i) preformed vitamin A (retinyl esters (RE) in organ meats, fish, whole milk, eggs) and ii) carotenoids, some of which are vitamin A precursors (in vegetable and fruit products), the most prominent being β-carotene. In addition, dietary supplements and fortified foods may contain preformed vitamin A, carotenoids, or a mixture of these (Institute of Medicine, 2001; Ross and Harrison, 2007).
Some carotenoids are converted to retinol in the lumen, via a process that is not very efficient. For example, it is generally assumed that 12 molecules of β-carotene of fruit or vegetable origin will on average convert to 1 molecule of retinol in the human (Institute of Medicine, 2001). However, the conversion of β-carotene to retinol from certain plant sources such as spinach, may be less efficient (∼20:1) (Institute of Medicine, 2001). Only about 15–30% of an oral dose of pure ß-carotene in oil (its most bioavailable form) will be absorbed (Harrison, 2005). The efficiency of carotenoid absorption may be increased somewhat when systemic vitamin A is low, and reduced somewhat when it is high. In general, however, carotenoids are metabolized and absorbed at a low rate, and do not perturb systemic vitamin A homeostasis. Thus, ingestion of preformed vitamin A is the predominant source of systemic retinol, and the present study will focus on retinol obtained via this form.
Analysis of Individual Key Events for Preformed Vitamin A
Uptake from Lumen into Enterocytes
Preformed dietary vitamin A is rapidly and efficiently hydrolyzed to retinol in the intestinal lumen. About 70% of preformed vitamin A is absorbed as retinol into enterocytes, apparently via carrier-mediated passive absorption.
The rates of hydrolysis and absorption are quite constant regardless of the ingested dose, that is, the dose-effect curve for these events is linear, i.e., there is no evidence that either hydrolysis or absorption is “thresholded.”2 There is also no evidence that either RE hydrolysis in the lumen or absorption into enterocytes is significantly regulated in a manner related to an individual's systemic vitamin A status. Thus, there is no evidence that this first key event is an important control point for regulating systemic dose or target tissue levels in response to an excess intake of vitamin A.
Initial (Intestinal) Metabolism and Distribution
Once retinol is absorbed into enterocytes, most of it is reesterified to form RE and packaged into chylomicrons, which are then extruded into the lymph system and then into blood, where they are converted into smaller chylomicron remnants (Ross and Harrison, 2007; Harrison, 2005). A small amount of absorbed retinol in the enterocyte may not be esterified and packaged into chylomicrons, and instead may be passed directly into the blood as oxidized retinoic acid. Thus, the intestine, especially after ß-carotene or vitamin A-containing meals, may contribute to the production of relatively small amounts of acidic retinoids that are present in plasma, and plasma levels may increase in the post-absorptive period (Wang et al., 1992; Wang et al., 1993; Hébuterne et al., 1995).
There is no evidence that this key event—esterification of retinol and packaging into chylomicrons—is regulated based on systemic vitamin A status. In fact, the functional capacity for absorption remains strong across a wide range of vitamin A status, even in cases of vitamin A overload or vitamin A deficiency (Randolph and Ross, 1991; Ross and Harrison, 2007). There may be some homeostatic feedback regulation via circulating RA levels of the enzyme that cleaves some carotenoids (especially ß-carotene) to form retinaldehyde, and then retinol (Bachmann et al., 2002); however, since even in its most bioavailable form, only ∼ 30% of ß-carotene will be absorbed, this feedback mechanism is unlikely to be an important control point for regulating the availability of retinol for packaging and distribution.
Uptake and Storage in the Liver
The liver is the main storage organ for retinol and plays a central role in retinol metabolism. Most (60–80%) of the chylomicron remnants holding RE are taken up by liver receptors and incorporated into hepatocytes, while a lesser portion (20–40%) is taken up into extrahepatic tissues. Chylomicron remnant uptake into liver is both rapid and efficient, and esterified retinol is passively assimilated into hepatocytes as a component of the chylomicron remnant (Ross and Harrison, 2007). Thus, uptake into the liver is not homeostatically regulated. After uptake, esterified retinol in chylomicrons is rapidly hydrolyzed in liver parenchymal cells (hepatocytes). Within a few hours, newly formed RE is found in liver storage cells (stellate cells).
Two enzymes are known to catalyze esterification of retinol [reviewed in Ross and Harrison, 2007; Harrison, 2005]. Lecithin—retinol acyltransferase (LRAT) esterifies retinol in numerous tissues. In the small intestine, LRAT esterifies retinol that is bound to cellular retinol binding protein II (CRBP-II). In the liver (mostly in stellate cells), LRAT esterifies retinol bound to cellular retinol binding protein I (CRBP-I). LRAT is also active in the retina, testis, and other tissues. A second enzyme in liver, acyl-CoA—retinol acyltransferase (ARAT) is capable of esterifying retinol that is not associated with a CRBP (Yost et al., 1988). This activity may be important at higher concentrations of retinol, if CRBPs become saturated. The molecular identity of ARAT is currently unknown; it may be a form of diacylglycerol acyltransferase (DGAT) and its activity may represent side reactions of enzymes involved in the pathway of triglyceride biosynthesis (Orland et al., 2005; Yen et al., 2005). ARAT activity is not as well characterized as LRAT and there is no evidence at present that ARAT is specific for vitamin A, or saturated by high vitamin A. Thus, LRAT is the principal enzyme involved in retinol esterification in both the liver and the enterocyte (Ross and Zolfaghari, 2004; O'Bryne et al., 2005).
LRAT expression and activity generally correlates with circulating retinol levels. LRAT activity falls to almost undetectable levels in states of low vitamin A status (Randolph and Ross, 1991). This decrease in esterification and storage of retinol likely serves to preserve retinol for oxidation and/or secretion to plasma. On the other hand, liver LRAT activity remains adequate, and may increase slightly, to deal with increased intake of vitamin A (especially preformed vitamin A)3 (Ross and Harrison, 2007; Randolph and Ross, 1991).
After long-term vitamin A supplementation, however, LRAT expression in liver rises only slightly above control levels (Dawson et al., 2000) This suggests that LRAT is not highly “elastic” in the upwards direction, i.e., there is no extensive compensatory upregulation of LRAT, at least in the liver (Ross and Zolfaghari, 2004), although RE storage increases. Thus, at some level of intake liver LRAT may become saturated acutely with extreme vitamin A intake, or slowly after long-term intakes of high vitamin A. In such situations, the dose-effect relationship for this key event may reach a threshold, i.e., an increase in retinol intake does not lead to an adequate increase in LRAT activity and storage. Instead, it leads to increased circulating levels of retinol and retinyl esters, and increased likelihood of disruptions in cell functions due to unesterified retinol or its metabolite within cells.
The liver's capacity to store vitamin A is considered high when compared to usual daily intakes (Randolph and Ross, 1991), and the liver apparently accommodates moderately elevated intakes over time without adverse effects (Dawson et al., 2000). The amount of stored vitamin A in the liver increases as intake exceeds the amount required for use and the rate of clearance (Green and Green, 1994). The storage capacity of the liver is not inexhaustible, however. Research suggests that when liver vitamin A stores reach a very high level (> 300 µg/g liver), plasma RE begins to accumulate (Olson, 1984); this indicates the liver's lack of capacity to process and store additional vitamin A.
Thus, this key event—esterification of retinol by LRAT to enable storage in the liver—appears to be an important control point in regulating systemic vitamin A levels. There is evidence, however, that this control mechanism can be overwhelmed by very high acute intake of preformed vitamin A, or by high long-term intake.
Release of Stored Retinol into Plasma
Signals for the liver to release stored vitamin A into circulation are not precisely known, but once retinol is freed from its storage form, it is bound to retinol binding protein (RBP) within the hepatocyte and then secreted into circulation as a dimolecular complex (retinol-RBP, also known as holo-RBP) (Soprano and Blaner, 1994). Retinol-RBP then joins with another protein (transthyretin) in the circulation, forming a retinol-RBP-transthyretin (retinol-RBP-TTR) complex. This trimolecular complex prevents retinol from being nonspecifically delivered to tissues in its unbound (toxic) form and also prevents filtration of retinol bound to RBP by the kidney and loss in the urine. Except in cases of hypervitaminosis A,4 the major form of circulating vitamin A is the retinol-RBP-TTR complex. Other minor circulating forms include RE in lipoproteins, a minor amount of free retinol, small amounts of retinoic acid (RA), and glucuronide conjugates of both retinol and RA. Oxidative metabolites of retinol and retinoic acids have also been detected in blood and bile (e.g., 4-oxo-retinol and 13-cis-4-oxo-RA) and rise after vitamin A supplementation (Eckhoff et al., 1991; Hartmann et al., 2001; Arnhold et al., 2002).
Retinol bound to RBP recirculates between liver and extrahepatic tissues multiple times before being degraded (Green and Green, 1994). Except in cases of vitamin A deficiency or excess, the concentration of retinol in plasma, as a retinol-RBP-TTR complex, is kept essentially constant in any given individual via mechanisms that are centered in the liver. Kinetic modeling studies have indicated that the retinol disposal rate (irreversible loss) is only modestly increased when vitamin A intake is elevated chronically or acutely (Green and Green, 1994; Cifelli et al., 2008). The liver, however, appears to detect circulating plasma retinol levels, as liver LRAT activity (Dawson, 2000) and retinol turnover are correlated with plasma retinol concentrations (Kelley and Green, 1998). There may be a feedback loop whereby low circulating levels of RA, a principal metabolite of vitamin A, or other metabolites signal the liver to release vitamin A from its storage form, RE, and convert these stores (via retinyl hydrolase) back into retinol. Over time, liver storage levels may decrease slowly, or increase very slowly to very rapidly, in response to intake levels and corresponding LRAT activity. Across a population, normal liver storage, as inferred from homeostatically controlled plasma retinol concentrations, ranges from ∼20–300 µg retinol/gram of liver (Olson, 1984).
In summary, mechanisms that control retinol storage in the liver appear to be critical “control points” in the mode-of-action pathway whereby excess vitamin A intake can lead to toxicity. These mechanisms serve to regulate circulating vitamin A levels. As circulating vitamin A levels fall, the release of retinol from liver storage increases to compensate; as circulating vitamin A levels rise, the LRAT storage activity in the liver increases and release of retinol from liver storage decreases.
The capacity of these regulatory mechanisms can be overcome, however, by saturation of the storage enzyme LRAT. Long-term exposure to excess vitamin A can also lead to liver fibrosis and compromised ability to store vitamin A (Nollevaux et al., 2006). Either of these conditions is expected to greatly increase the probability of an adverse effect. Saturated LRAT activity could be an early indicator of potential toxicity except that LRAT activity cannot be readily measured in vivo. Thus, circulating biomarkers capable of reflecting liver LRAT activity would be very valuable.
Circulating plasma retinol-RBP-TTR levels are relatively easy to measure but these are not a good indicator of vitamin A status as levels are kept essentially constant (at the expense of fluctuating liver stores).5 An acute intake of vitamin A will not lead to an increase in plasma levels of retinol bound to RBP except in vitamin A deficiency. When there is deficiency, newly absorbed vitamin A will not be stored; rather, it will be bound in the liver to RBP and go directly into circulation. This is made possible by a build up of RBP stores in liver in response to deficiency. This build up ensures that RBP will be readily available to carry retinol to tissues as soon as there is intake. While liver RBP levels increase when there is vitamin A deficiency (Soprano and Blaner, 1994), liver and plasma RBP levels (which correspond to circulating retinol) do not increase in states of vitamin A excess.
Circulating RE levels are also relatively easy to measure, and do fluctuate with vitamin A intake (i.e., may be higher postprandially if the meal is rich in vitamin A). Several studies have used circulating RE, retinol, or RA metabolites to assess increased potential for adverse effects from intake of vitamin A supplements or foods with high vitamin A content (van Vliet et al., 2001; Hartman, 2001). Measures of circulating RE as a percent of total serum vitamin A may also be useful as an indicator of vitamin A status. Circulating RE is normally present in the range of 2–11% of the total serum vitamin A (Krasinski et al., 1989; Smith and Goodman 1976). If RE in plasma (as percentage of total circulating vitamin A) in a fasting state exceeds this range, this is an indicator of excessive vitamin A status (intake exceeds clearance, and storage capacity of liver has been exceeded). Thus, an increase in proportion of circulating RE may be an indicator of vitamin A overload, but not an early indicator, as increased (fasting) RE levels indicate likely liver damage.
Uptake and Storage in Extrahepatic Tissue
The mechanism for the uptake of retinol into extrahepatic tissues is unknown, but may involve uptake by a specific membrane receptor (Kawaguchi et al., 2007). Alternatively, retinol may dissociate from the retinol-RBP-TTR complex and diffuse into cells where it is then be converted into its storage form, RE, by LRAT. Another source for extrahepatic uptake is circulating chylomicron remnants containing RE; these are also taken up via an unknown mechanism.
Due to lack of knowledge regarding uptake mechanisms, the role of extrahepatic uptake in controlling the consequences of excess vitamin A intake cannot be well characterized. There is no evidence, however, to suggest that uptake mechanisms may control the amount of circulating retinol taken into extrahepatic tissues. But, LRAT activity in extrahepatic tissues, such as lung, may be increased with an increase in circulating vitamin A (Ross et al., 2006). It is not known whether LRAT capacity in extrahepatic tissue becomes saturated in cases of vitamin A overload, or whether there is a limit to upregulation of LRAT expression.
Production and Metabolism of Bioactive Metabolites (Especially RA)
Bioactive retinoids (e.g., RA) in extrahepatic tissue are derived from stored RE, from recently delivered chylomicron remnants, and from retinol taken up from circulating retinol-RBP-TTR. RA plays a critical role in regulating gene expression, especially during development (Underhill et al., 1995; Zile 1998). However, an excess of retinoids can potentially lead to an inappropriate expression of retinoid responsive genes and also damage the cell surface and intracellular membranes. High intakes of RA in experimental animals have been shown to cause teratogenicity, liver abnormalities, altered metabolism, and bone fragility. Less is known about whether metabolites formed from RA are directly toxic, and if so, at what concentrations. RA metabolites probably do not alter gene expression very significantly, as compared to the very potent activity of RA (Perlmann, 2002).
As retinoids are potent bioactive substances, systems have evolved for the control of retinoid levels. Enzymes responsible for RA production (biogenesis from retinol) and RA oxidation have been implicated in controlling intracellular RA levels. There is considerable evidence that at elevated concentrations, RA induces its own metabolism (Perlmann, 2002; Wang et al., 2002). Several CYP genes and proteins, present in the liver and extrahepatic tissues, have been implicated in RA oxidation (formation of hydroxyl and keto metabolites, which are further conjugated and then excreted). Of these, the CYP26 family has been most extensively studied and the mode of regulation of CYP26A1 by RA has been defined at the molecular level (Wang et al., 2002). Metabolic studies in vivo (humans and experimental animals) provide evidence that metabolites similar to those formed in vitro by CYP26A1 are present in plasma after acute vitamin A supplementation (Penniston and Tanumihardjo, 2005) or consumption of high vitamin A foods (Arnhold et al., 1996), and after therapeutic treatment with RA or other related retinoids (van Vliet et al., 2001). Retinoid biokinetic studies in animals and clinical studies in humans have indicated that RA metabolism is induced by RA; however, the dose-response relationship between RA concentration, oxidative activity (CYP26 or other enzymes), and metabolite formation is not well known.
In summary, these data form a strong basis for the hypothesis that RA levels are closely monitored and regulated in vivo, perhaps through several different mechanisms. These mechanisms may include “thresholds” for inducing metabolism. RA metabolite levels may be useful as indicators of excessive RA, as they may mirror the need to catabolize RA.
Retinoid Activity in Cell Nuclei
In the cell nucleus, cellular RA binding proteins (CRABP-I and CRABP-II) bind specific isomers of RA (e.g., all-trans RA) and regulate the activity of retinoid responsive genes.6 The mechanism of action is mediated by two major receptor families, the RAR and the RXR receptors, which exist in alpha, beta, and gamma forms. These receptors subsequently bind to specific DNA response elements, RARE or RXRE, thereby affecting gene transcription. Although RXRs may bind to one another to form homodimers, they most frequently bind to RARs to form heterodimeric complexes. RXRs also bind as heterodimers to several other types of nuclear transcription factors, and thus mediate cross-talk among different signaling pathways that affect the expression of many genes (Rochette-Egly, 2005).
The existence of homeostatic mechanisms within the cell nucleus to control effects from excess vitamin A intake, or RA production, is not clear. Vitamin A deficiency lowers the expression of RAR-ß, which conversely is induced in most cells by RA (DeLuca, 1991), but it is not known whether vitamin A overload affects the expression of any RARs or RXRs. Vitamin A status has no effect on the expression of CRABP-I, although CRABP-II expression can be induced in some tissues by all-trans retinoic acid (Astrom, 1994). Thus, CRABP-II may play a role in controlling free RA by sequestering excess amounts.
DISCUSSION
This initial Key Events Dose-Response Analysis for vitamin A has provided insight at two levels. It has shed light on specific biological events that determine whether high intake of vitamin A will lead to adverse effects. It has also revealed the potential of the KEDRF as a new analytical tool for examining dose-response relationships and for using dose-response data to inform public health decision making.
With regard to vitamin A, this analysis supports the hypothesis that saturation of liver LRAT activity is a “determining event” in the Key Events pathway; such saturation greatly increases the likelihood that high intake levels will lead to excess circulating retinol and RE, and consequently a greatly increased probability of toxicity. The analysis also revealed critical research needs, including: i) characterizing the dose-response relationship for liver LRAT saturation, ii) identifying, and quantitatively characterizing, sources of variability at important key events (e.g., at control points), and iii) characterizing the role of downstream events (e.g., regulation of RA metabolism, extrahepatic storage, or sequestration of retinoids) in protecting target tissues when liver LRAT storage activity is compromised. Because these research needs were generated in the context of a KEDRF, any resulting data will feed directly into a refined understanding of the dose-response relationship for vitamin A. The present analysis also suggested candidate biomarkers that should be evaluated further as potential indicators of vitamin A overload. Additional research is needed to further examine biomarkers for a range of purposes, for example, to assess overall vitamin A status, and to quantify interindividual variability in capacity of homeostatic mechanisms.
More fundamentally, this vitamin A case study illustrates the value of the KEDRF as an analytical approach for integrating and utilizing available knowledge, and for generating questions that will focus future research efforts. The KEDRF facilitates identification of the most critical events, i.e., determining events within the overall mode-of-action, and also helps tease out the role of specific homeostatic mechanisms in countering the effects of excess intake. A more robust understanding of these events and mechanisms provides the necessary foundation for characterizing and quantifying sources of variability (genetic, behavioral, life stage, etc.) in response to a given dose. In turn, this provides the evidence necessary to reduce reliance on Uncertainty Factors and other assumptions when identifying safe and adequate intake levels for a population or sub-population. Thus the KEDRF approach emerges as a useful new way of thinking about dose-response and the process for setting recommendations for nutrient intakes.
This analytical approach should be further tested by application to other endpoints (including effects from vitamin deficiency) and other vitamins and minerals. The present case example used a vitamin for which there is a relatively good understanding of mode-of-action and Key Events. It might be instructive to test the broader value of this analytical approach on a vitamin or mineral for which much less is known.
In addition to broadening application to other nutrients and endpoints, it would be valuable to “deepen” this KEDRF analysis for vitamin A. This would involve compiling and evaluating current knowledge regarding Key Events for vitamin A as seen from a range of perspectives (molecular, cellular, systemic, organismal, and population). This strategy would leverage data and knowledge from multiple disciplines toward a single goal of refining the characterization of threshold doses (both for safe intake, and for adequate intake) for vitamin A.
ACKNOWLEDGMENTS
This paper is one of the work products of an international group of experts convened by the International Life Sciences Institute (ILSI) Research Foundation. ILSI is a nonprofit, worldwide organization established in 1978 to advance the understanding of scientific issues relating to nutrition, food safety, toxicology, risk assessment, and the environment. The ILSI Research Foundation was established in 1984 to create a vehicle for ILSI to support research. Its risk assessment program sponsors, organizes, and participates in a wide range of activities to develop and disseminate new scientific knowledge, encourage exchange of ideas, and build consensus among scientists from academia, industry, government, and public interest groups in its efforts to improve the scientific basis of risk assessment.
Financial support for this project from the following sources is gratefully acknowledged: ILSI Research Foundation, Health Canada, Ajinomoto, Coca-Cola Company, Groupe Danone, Kellogg Company, Kraft Foods, Inc., Mars, Inc., Mead Johnson Nutritionals, Monsanto Company, Nestlé, PepsiCo, Inc., The Procter & Gamble Company, Syngenta Ltd., Flavor Extract Manufacturers Association, and Grocery Manufacturers Association. Assistance from Ms. Julie Fitzpatrick with coordination of the final preparation of these papers is also gratefully acknowledged.
Declaration of Interests: All authors declare that they have no relevant interests to disclose.
Footnotes
1 Throughout this discussion, the phrase “nutrient levels” is intended to refer not only to the parent nutrient compound, but also to metabolites or other downstream effectors generated by the nutrient.
2 The term “thresholded” is commonly used in the field of chemical risk assessment. It refers to a dose-response relationship that is characterized by a biological threshold and a non-linear or discontinuous dose-response curve. A biological threshold is a transition point between the highest dose that will not elicit a specified biological effect, and the lowest dose that will (see Appendix to Julien et al. 2009 for further discussion).
3 LRAT also increases in some extrahepatic tissues, such as the lungs, when vitamin A or retinoic acid are above usual levels (Ross et al., 2006). In addition, ARAT activity becomes active in liver and intestine when retinol is present in high concentrations.
4 With hypervitaminosis A (acute or chronic), RE may exceed retinol-RBP-TTR as the major form of circulating VA. RE may be released directly from the liver and chylomicron containing RE may not be taken up by liver. Some species are known to secrete RE from the liver in lipoproteins (Wilson et al., 1987) and this could be, in part, the case in humans. Also, some chylomicron may exchange RE with circulating lipoproteins before being cleared.
5 While plasma retinol levels are not a sensitive indicator of status, they have some predictive value, as epidemiological data have correlated very low levels of plasma retinol levels to certain types of disease risk (e.g., impaired growth, eye disease, mortality from infectious disease) (Underwood, 1994).
6 Other specific vitamin A binding proteins are present in the retina's photoreceptor cells and in retinal pigment epithelial cells.
REFERENCES
- Arnhold T., Nau H., Meyer S., Rothkoetter H. J., Lampen A. D. Porcine intestinal metabolism of excess vitamin A differs following vitamin A supplementation and liver consumption. J. Nutr. 2002;132((2)):197–203. doi: 10.1093/jn/132.2.197. [DOI] [PubMed] [Google Scholar]
- Arnhold T., Tzimas G., Wittfoht W., Plonait S., Nau H. Identification of 9-cis-retinoic acid, 9,13-di-cis-retinoic acid, and 14-hydroxy-4,14-retro-retinol in human plasma after liver consumption. Life Sci. 1996;59((12)):169–177. doi: 10.1016/0024-3205(96)00408-0. [DOI] [PubMed] [Google Scholar]
- Aström A., Pettersson U., Chambon P., Voorhees J. J. Retinoic acid induction of human cellular retinoic acid-binding protein-II gene transcription is mediated by retinoic acid receptor-retinoid X receptor heterodimers bound to one far upstream retinoic acid-responsive element with 5-base pair spacing. J. Biol. Chem. 1994;269((35)):22334–22339. [PubMed] [Google Scholar]
- Bachmann H., Desbarats A., Pattison P., Sedgewick M, Riss G., Wyss A., Cardinault N., Duszka C, Goralczyk R., Grolier P. Feedback regulation of beta, beta-carotene 15,15′-monooxygenase by retinoic acid in rats and chickens. J. Nutr. 2002;132((12)):3616–3622. doi: 10.1093/jn/132.12.3616. [DOI] [PubMed] [Google Scholar]
- Cifelli C. J., Green J. B., Wang Z., Yin S., Russell R. M., Tang G., Green M. H. Kinetic analysis shows that vitamin A disposal rate in humans is positively correlated with vitamin A stores. J. Nutr. 2008;138((5)):971–977. doi: 10.1093/jn/138.5.971. [DOI] [PubMed] [Google Scholar]
- Clagett-Dame M., DeLuca H. F. The role of vitamin A in mammalian reproduction and embryonic development. Annu. Rev. Nutr. 2002;22:347–381. doi: 10.1146/annurev.nutr.22.010402.102745E. [DOI] [PubMed] [Google Scholar]
- Dawson H. D., Yamamoto J., Zolfaghari R., Rosales F., Dietz J., Shimada T., Li N-Q., Ross A. C. Regulation of hepatic vitamin A storage in a rat model of controlled vitamin A status during aging. J. Nutr. 2000;130:1280–1286. doi: 10.1093/jn/130.5.1280. [DOI] [PubMed] [Google Scholar]
- De Luca L. M. Retinoids and their receptors in differentiation, embryogenesis, and neoplasia. FASEB J. 1991;5((14)):2924–2933. [PubMed] [Google Scholar]
- Eckhoff C., Collins M. D., Nau H. Human plasma all-trans-, 13-cis- and 13-cis-4-oxoretinoic acid profiles during subchronic vitamin A supplementation: Comparison to retinol and retinyl ester plasma levels. J. Nutr. 1991;121:1016–1025. doi: 10.1093/jn/121.7.1016. [DOI] [PubMed] [Google Scholar]
- Food and Nutrition Board, Institute of Medicine. Dietary Reference Intakes: A Risk Assessment Model for Establishing Upper Intake Levels for Nutrients. Washington, DC: National Academy Press; 1998. [PubMed] [Google Scholar]
- FSA (Food Standards Agency) Safe Upper Levels for Vitamins and Minerals: Report of the Expert Group on Vitamins and Minerals. United Kingdom: Food Standards Agency; 2003. http://www.food.gov.uk/multimedia/pdfs/vitmin2003.pdf. [Google Scholar]
- Green M. H., Green J. B. Vitamin A intake and status influence retinol balance, utilization and dynamics in rats. J. Nutr. 1994;124:2477–2485. doi: 10.1093/jn/124.12.477. [DOI] [PubMed] [Google Scholar]
- Harrison E. H. Mechanisms of digestion and absorption of dietary vitamin A. Annu. Rev. Nutr. 2005;25:87–103. doi: 10.1146/annurev.nutr.25.050304.092614. [DOI] [PubMed] [Google Scholar]
- Hartmann S., Froescheis O., Ringenbach F., Wyss R., Bucheli F., Bischof S., Bausch J., Wiegand U. W. Determination of retinol and retinyl esters in human plasma by high-performance liquid chromatography with automated column switching and ultraviolet detection. J. Chromatogr. B Biomed. Sci. Appl. 2001;751((2)):265–275. doi: 10.1016/s0378-4347(00)00481-3. [DOI] [PubMed] [Google Scholar]
- Hébuterne X., Wang X.-D., Johnson E. J., Krinsky N. I., Russell R. M. Intestinal absorption and metabolism of 9-cis-b-carotene in vivo: Biosynthesis of 9-cis-retinoic acid. J. Lipid Res. 1995;36:1264–1273. [PubMed] [Google Scholar]
- Institute of Medicine. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington DC: The National Academies Press; 2001. [PubMed] [Google Scholar]
- Institute of Medicine. Dietary Reference Intakes (DRI): The Essential Guide to Nutrient Requirements. (2006) Washington DC: The National Academies Press; 2006. [Google Scholar]
- Julien E., Boobis A. R., Olin S. S. the ILSI Research Foundation Threshold Working Group. The Key Events Dose-Response Framework: Across-disciplinary mode-of-action based approach to examining dose-response and thresholds. Crit. Rev. Food Sci. Nutr. 2009;49:682–689. doi: 10.1080/10408390903110692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi R., Yu J., Honda J., Hu J., Whitelegge J., Ping P., Wiita P., Bok D., Sun H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science Feb 9; 2007;315((5813)):820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
- Kelley S. K., Green M. H. Plasma retinol is a major determinant of vitamin A utilization in rats. J. Nutr. 1998;128:1767–1773. doi: 10.1093/jn/128.10.1767. [DOI] [PubMed] [Google Scholar]
- Krasinski S. D., Russell R. M., Otradovec C. L., Sadowski J. A., Hartz S. C., Jacob R. A., McGandy R. B. Relationship of vitamin A and vitamin E intake to fasting plasma retinol, retinol-binding protein, retinyl esters, carotene, alpha-tocopherol, and cholesterol among elderly people and young adults: Increased plasma retinyl esters among vitamin A-supplement users. Am. J. Clin. Nutr. 1989;49((1)):112–120. doi: 10.1093/ajcn/49.1.112. [DOI] [PubMed] [Google Scholar]
- McCaffery P. J., Adams J., Maden M., Rosa-Molinar E. Too much of a good thing: Retinoic acid as an endogenous regulator of neural differentiation and exogenous teratogen. Eur. J. Neurosci. 2003;18:457–472. doi: 10.1046/j.1460-9568.2003.02765.x. [DOI] [PubMed] [Google Scholar]
- Nollevaux M. C., Guiot Y., Horsmans Y., Leclercq I., Rahier J., Geubel A. P., Sempoux C. Hypervitaminosis A-induced liver fibrosis: Stellate cell activation and daily dose consumption. Liver Int. 2006;26((2)):182–186. doi: 10.1111/j.1478-3231.2005.01207.x. [DOI] [PubMed] [Google Scholar]
- O'Byrne S. M., Wongsiriroj N., Libien J., Vogel S., Goldberg I. J., Baehr W., Palczewski K., Blaner W. S. Retinoid absorption and storage is impaired in mice lacking lecithin: Retinol acyltransferase (LRAT). J. Biol. Chem. 2005;280((42)):35647–35657. doi: 10.1074/jbc.M507924200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson J. A. Serum levels of vitamin A and carotenoids as reflectors of nutritional status. J. Natl. Cancer Inst. 1984;73((6)):1439–1444. [PubMed] [Google Scholar]
- Orland M. D., Anwar K., Cromley D., Chu C. H., Chen L., Billheimer J. T., Hussain M. M., Cheng D. Acyl coenzyme A dependent retinol esterification by acyl coenzyme A: Diacylglycerol acyltransferase 1. Biochim. Biophys. Acta. 2005;1737((1)):76–82. doi: 10.1016/j.bbalip.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Penniston K. L., Tanumihardjo S. A. Elevated serum concentrations of beta-glucuronide metabolites and 4-oxoretinol in lactating sows after treatment with vitamin A: A model for evaluating supplementation in lactating women. Am. J. Clin. Nutr. 2005;81((4)):851–858. doi: 10.1093/ajcn/81.4.851. [DOI] [PubMed] [Google Scholar]
- Perlmann T. Retinoid metabolism: A balancing act. Nat. Genet. 2002;31:7–8. doi: 10.1038/ng877. [DOI] [PubMed] [Google Scholar]
- Randolph R. K., Ross A. C. Vitamin A status regulates hepatic lecithin: Retinol acyltransferase activity in rats. J. Biol. Chem. 1991;266:16453–16457. [PubMed] [Google Scholar]
- Renwick A. G., Walker R. Risk assessment of micronutrients. Toxicol. Lett. 2008;180((2)):123–130. doi: 10.1016/j.toxlet.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Renwick A. G. Toxicology of micronutrients: Adverse effects and uncertainty. J. Nutr. 2006;136((2)):493S–501S. doi: 10.1093/jn/136.2.493S. [DOI] [PubMed] [Google Scholar]
- Rochette-Egly C. Dynamic combinatorial networks in nuclear receptor-mediated transcription. J Biol Chem. 2005;280((38)):32565–32568. doi: 10.1074/jbc.R500008200. [DOI] [PubMed] [Google Scholar]
- Ross A. C., Ambalavanan N., Zolfaghari R., Li N. Vitamin A combined with retinoic acid increases retinol uptake and lung retinyl ester formation in a synergistic manner in neonatal rats. J. Lipid Res. 2006;47:1844–1851. doi: 10.1194/jlr.M600061-JLR200. [DOI] [PubMed] [Google Scholar]
- Ross A. C., Harrison E. H., Rucker R.B., Zempleni J., Suttie J.W., McCormick D.B. Handbook of Vitamins. 4th ed. Boca Raton, FL: Taylor & Francis Group; 2007. Vitamin A: Nutritional Aspects of Retinoids and Carotenoids; pp. 1–40. [Google Scholar]
- Ross A. C., Zolfaghari R. Regulation of hepatic retinol metabolism: perspectives from studies on vitamin A status. J. Nutr. 2004;134((1)):269S–275S. doi: 10.1093/jn/134.1.269S. [DOI] [PubMed] [Google Scholar]
- SCF (Scientific Committee on Food) Tolerable Upper Intake Levels for Vitamins and Minerals. European Food Safety Authority, Scientific Committee on Food, Scientific Panel on Dietetic Products, Nutrition and Allergies; 2006. [Google Scholar]
- Smith F. R., Goodman D. S. Vitamin A transport in human vitamin A toxicity. N. Engl. J. Med. 1976;8((15)):805–808. doi: 10.1056/NEJM197604082941503. 294. [DOI] [PubMed] [Google Scholar]
- Soprano D. R., Blaner W. S., Sporn M.B., Roberts A.B., Goodman D.S. The Retinoids: Biology, Chemistry and Medicine. NY: Raven Press; 1994. Plasma retinol-binding protein; pp. 257–281. [Google Scholar]
- Soprano D. R., Soprano K. J. Retinoids as teratogens. Annu. Rev. Nutr. 1995;15:111–132. doi: 10.1146/annurev.nu.15.070195.000551. [DOI] [PubMed] [Google Scholar]
- Taylor C. L., Yetley E. A. Nutrient risk assessment as a tool for providing scientific assessments to regulators. J. Nutr. 2008;138:1987S–1991S. doi: 10.1093/jn/138.10.1987S. [DOI] [PubMed] [Google Scholar]
- Underhill T. M., Kotch L. E., Linney E. Retinoids and mouse embryonic development. Vitam. Horm. 1995;51:403–457. doi: 10.1016/s0083-6729(08)61046-8. [DOI] [PubMed] [Google Scholar]
- Underwood B. A. Hypovitaminosis A: International programmatic issues. J. Nutr. 1994;124((8 Suppl)):1467S–1472S. doi: 10.1093/jn/124.suppl_8.1467S. [DOI] [PubMed] [Google Scholar]
- van Vliet T., Boelsma E., de Vries A. J., van den Berg H. Retinoic acid metabolites in plasma are higher after intake of liver paste compared with a vitamin A supplement in women. J. Nutr. 2001;131((12)):3197–3203. doi: 10.1093/jn/131.12.3197. [DOI] [PubMed] [Google Scholar]
- Visani G., Manfroi S., Tosi P., Martinelli G. All-trans-retinoic acid and pseudotumor cerebri. Leukemia and Lymphoma. 1996;23:437–442. doi: 10.3109/10428199609054851. [DOI] [PubMed] [Google Scholar]
- Wang X.-D., Krinsky N. I., Tang G., Russell R. M. Retinoic acid can be produced from excentric cleavage of b-carotene in human intestinal mucosa. Arch. Biochem. and Biophys. 1992;293:298–304. doi: 10.1016/0003-9861(92)90399-h. [DOI] [PubMed] [Google Scholar]
- Wang X.-D., Russell R. M., Marini R. P., Tang G., Dolnikowski G. G., Fox J. G., Krinsky N. I. Intestinal perfusion of b-carotene in the ferret raises retinoic acid level in portal blood. Biochim. Biophys. Acta. 1993;1167:159–164. doi: 10.1016/0005-2760(93)90157-5. [DOI] [PubMed] [Google Scholar]
- Wang Y., Zolfaghari R., Ross A. C. Cloning of rat cytochrome P450RAI (CYP26) cDNA and regulation of its gene expression by all-transretinoic acid in vivo. Arch. Biochem and Biophys. 2002;401:235–243. doi: 10.1016/S0003-9861(02)00043-7. [DOI] [PubMed] [Google Scholar]
- Williams G. M., Iatropoulos M. J. Alteration of liver cell function and proliferation: differentiation between adaptation and toxicity. Toxicol. Pathol. 2002;30((1)):41–53. doi: 10.1080/01926230252824699. [DOI] [PubMed] [Google Scholar]
- Wilson D. E., Hejazi J., Elstad N. L., Chan I. F., Gleeson J. M., Iverius P. H. Novel aspects of vitamin Ametabolism in the dog: distribution of lipoprotein retinyl esters in vitamin A-deprived and cholesterol-fed animals. Biochim. Biophys. Acta. 1987;922((3)):247–258. doi: 10.1016/0005-2760(87)90047-6. [DOI] [PubMed] [Google Scholar]
- World Health Organization/Food and Agriculture Organization of the United Nations. A Model for Establishing Upper Levels of Intake for Nutrients and Related Substances. 2006. Internet: http://www.who.int/ipcs/methods/en/
- Yen C. L., Monetti M., Burri B. J., Farese R. V., Jr. Acyl coenzyme A dependent retinol esterification by acyl coenzyme A: diacylglycerol acyltransferase 1. J. Lipid Res. 2005;46((7)):1502–1511. doi: 10.1194/jlr.M500036-JLR200. [DOI] [PubMed] [Google Scholar]
- Yin W. H., Raffelsberger W., Gronemeyer H. Retinoic acid determines life span of leukemic cells by inducing antagonistic apoptosisregulatory programs. Int. J. Biochem. Cell Biol. 2005;37:1696–1708. doi: 10.1016/j.biocel.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Yost R. W., Harrison E. H., Ross A. C. Esterification by rat liver microsomes of retinol bound to cellular retinol-binding protein. J. Biol Chem. 1988;263:18693–18701. [PubMed] [Google Scholar]
- Zile M. H. Vitamin A and embryonic development: An overview. J. Nutr. 1998;128((2 Suppl)):455S–458S. doi: 10.1093/jn/128.2.455S. [DOI] [PubMed] [Google Scholar]