

Abstract
We describe a 62-year-old woman with chronic progressive external ophthalmoplegia (CPEO), multiple lipomas, diabetes mellitus, and a novel mitochondrial DNA (mtDNA) mutation at nucleotide 4302 (4302A>G) of the tRNAIle gene (MTTI). This is the first mutation at position 44 in the variable loop (V loop) of any mitochondrial tRNA.
The muscle biopsy revealed 10% ragged-red or ragged-blue fibers and 25% cytochrome c oxidase (COX)-deficient fibers.
No deletions or duplications were detected by Southern blot analysis. The 4302A>G transition was present only in the patient’s muscle and single-fiber analysis revealed significantly higher levels of the mutation in COX-deficient than in normal fibers. Like tRNALeu(UUR), tRNAIle appears to be a “hot spot” for mtDNA mutations causing CPEO.
Keywords: Mitochondrial DNA (mtDNA), Point mutation, tRNAIle, chronic progressive external ophthalmoplegia(CPEO)
1. Introduction
Chronic progressive external ophthalmoplegia (CPEO) is a common clinical feature of mitochondrial diseases [1,2], often presenting as ptosis followed by gradual involvement of the extraocular muscles [3], along with variable degrees of oropharyngeal and limb weakness. As in many other mitochondrial clinical syndromes, the genetic basis of CPEO is heterogeneous. Most cases are caused by large-scale rearrangements - deletions, duplications, or both - of mtDNA [4, 5]. Patients with single deletions (with or without cognate duplications) are sporadic and the mutations are thought to arise in öogenesis or in early embryonic development [6]. In contrast, multiple deletions of mtDNA are often observed in autosomal dominant or recessive forms of CPEO [7]. In some cases, CPEO is maternally inherited and due to mtDNA point mutations, most frequently in tRNA genes (MTT). Together with tRNALeu(UUR)(MTTL), MTTI has emerged as one the hotspot regions for mutations causing CPEO.
Here, we describe a patient with apparently maternally inherited CPEO, who had many ragged-red and even more abundant COX-deficient fibers in her muscle biopsy and a novel mutation in MTTI gene. This is the first polymorphism described at position 44 of the variable loop (V-loop) of mitochondrial tRNA fulfilling canonical criteria for pathogenicity and the first mutation of the V-loop associated with CPEO.
2. Case Report
A 62 year-old Ecuadorian woman presented with a 10 years history of ptosis associated with diplopia. Approximately 4 years after the onset, she developed slowly progressive proximal limb weakness and muscular atrophy. She described trouble climbing stairs and putting dishes on high shelves. After beginning L-carnitine (990mg/day) and coenzyme Q10 (300 mg/day) supplementation, her diplopia improved. She developed diabetes mellitus, trouble swallowing, enlargement of her tongue and neck. General examination was notable for macroglossia and submandibular and upper arm lipomas. Neurological examination showed bilateral ptosis and severe impairment of vertical and right gaze whereas left gaze was almost normal. Fundoscopy was normal. She had mild facial weakness and her palate elevated briskly. She had mild neck flexor and proximal limb weakness. Sensory examination and coordination were normal.
Audiometry showed 10% deficit at high frequencies. Electrocardiograms and echocardiogram were normal. Venous lactate and pyruvate at rest, and thyroid stimulating hormone levels were normal. Her serum CK was 435 U/L (normal 39–238). Brain MRI, nerve conduction studies, and electromyography were normal. Little information about her mother and grandmother was available except both had unilateral ptosis without ophthalmoplegia. Her parents were cousins, as her paternal grandfather and maternal grandfather were brothers. The patient reported that none of her four siblings or two children had neuromuscular symptoms. Histological and histochemical reactions using standard procedures showed 10% of ragged red fibers as well as intense succinate dehydrogenase histochemistry (SDH) stained fibers. Roughly 25% of fibers showed COX deficiency.
Muscle biochemistry showed normal activities of all mitochondrial respiratory chain complexes and citrate synthase (Table 1).
Table 1.
| Enzyme | (Complex) | Activity* | (Controls±SD) |
|---|---|---|---|
| Cytochrome c oxidase | (IV) | 2.4 | (2.80±0.52) |
| Succinate cyt. c reductase | (II + III) | 0.73 | (0.70±0.23) |
| NADH-cyt. c reductase | (I + III) | 0.71 | (1.02±0.38) |
| NADH-dehydrogenase | (I) | 30.16 | (35.48±7.07) |
| Citrate synthase | 9.64 | (9.88±2.55) | |
| Succinate dehydrogenase | 1.22 | (1.00±0.53) |
micromoles/min/gm
DNA was extracted from skeletal muscle, blood, and urinary sediment using Puregene® DNA purification kit (Gentra System).
Southern blot revealed no deletions in the patient’s muscle. Due to the parental consanguinity, we screened for autosomally recessive POLG1 mutations. Direct sequencing of tRNAs demonstrated a heteroplasmic transition from adenine to guanosine at nt position 4302 (m.4302A>G) (Fig. 1A). This change is not described in the URLs of MITOMAP and mtDB-Human Mitochondrial Genome Database [see Mitomap: http://mitomap.org and http://www.genpat.uu.se/mtDB/] and it affects a phylogenetically well-conserved site from sea urchin to human (Fig. 1B). This nucleotide change was not found in 100 mtDNA from normal or disease controls.
Fig. 1.
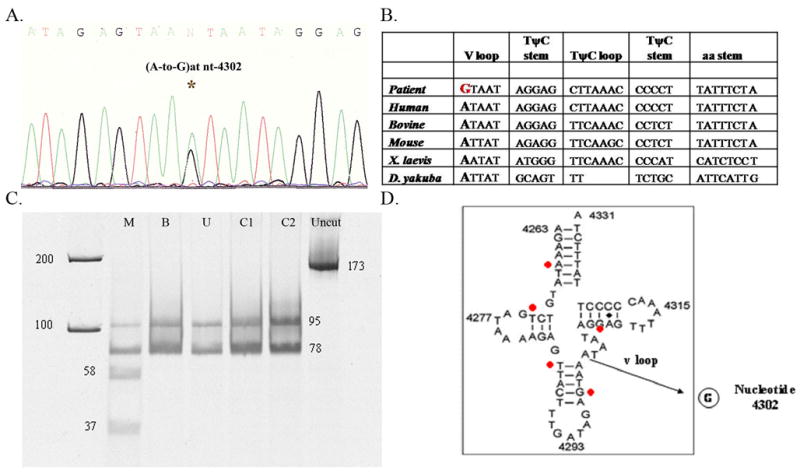
(A) Electropherogram of DNA sequence. Electrophoresed samples were analyzed with Sequence Analysis software (Perkin-Elmer Applied Biosystems). At position 4302 a heteroplasmic adenine to guanine transition is evident (4302A>G).
(B) Nucleotide sequence showing inter-species similarity of the tRNAIle gene and the highly conserved base at nt 4302. The mutation in our patient is highlighted with a bold red letter and compared with human, bovine, mouse, Xenopus (X) laevis, Drosophila (D) yakuba.
(C) PCR-RFLP analysis. Fragments after digestion with DdeI were separated on a 15% non-denaturing acrylamide gel, visualized and quantitated using SYBR Gold® Nucleic Acid Gel Stain (Invitrogen). DdeI cuts the wild type into a 95 and a 78 bp fragment, whereas mutant mtDNA was digested into three (58, 37 and 78 bp). Patient’s tissues: M, muscle; B, blood; U, urine; C1, C2 (controls); uncut PCR product.
(D) Secondary structure of the human wild type tRNAIle showing the nt. 4302 base pair substitution A-to-G at the variable loop (v loop) region. The red points show previously described mutations associated with CPEO phenotype (position 4267, 4274, 4285, 4298, and 4309).
Southern blot analysis of PvuII-digested muscle mtDNA was performed as previously described [9]. We sequenced all 22 mitochondrial tRNAs as described [10]. PCR products were purified with Big Dye® Termination 1.1 V System Kit and analyzed in an Applied Biosystem 3130xl Genetic Analyzer.
To assess quantitatively the level of the m.4302A>G mutation, restriction fragment length polymorphism (RFLP) was performed on DNA extracted from muscle, blood, and urinary sediment. A 173 base-pair (bp) mtDNA fragment spanning nucleotides (nts) 4262–4434 was amplified from total DNA samples using a forward mismatch primer (5′-AAGAAATATGTCTGATAAAAGAGTTACTTTGATAGACTAA-3′) (nts 4262–4301, mismatch nucleotide underlined) and a backward primer (nts 4415–4434). Wild-type mtDNA was cut into two fragments (95 and 78 bp) while mutant mtDNA was digested into three fragments (58, 37, and 78 bp) by the enzyme DdeI. The digestion products were run in a 15% non-denaturing acrylamide gel and subjected to SYBR Gold® Nucleic Acid Gel Stain (Invitrogen) so as to quantitate the percentage of the mutation. Quantitative PCR/RFLP analysis showed that the mutation was heteroplasmic in the patient’s muscle (56%) and undectectable in blood and urinary sediment (Fig. 1C).
Single-fiber PCR analysis showed that the proportion of mutant mtDNAs was 77±19% in 12 COX-negative RRFs and 53±27% in 12 COX-positive non-RRFs. This difference was statistically significant (p<0.03, Unpaired t test).
3. Discussion
Both large rearrangements and point mutations in mtDNA have been described in association with sporadic CPEO. We report a patient who presented with CPEO plus limb weakness, diabetes mellitus, macroglossia, and lipomas due to a novel m.4302A>G transition in MTTI. We consider this change “possibly” pathogenic because: 1) it alters a highly conserved base pair within the V-loop region of the isoleucine tRNA and probably disrupts the secondary structure of the molecule 2) it was absent in 100 control DNA samples and public databases; 3) it was present at higher levels of heteroplasmy in COX-negative than in COX-positive fibers.
This case has several interesting features. First, this is the first description of a patient with CPEO associated with macroglossia and lipomas. Second, this is the first reported mutation at position 44 of the V loop in all tRNA’s, except from an unconfirmed similar change in tRNALeu(UUR) associated with LHON phenotype (unpublished data, Dr. Eric Schon). Other mutations have been described in the variable loop, but none have been associated with a pure CPEO phenotype [11]. This position may be structurally important for the proper folding and the 3-dimensional structure of the tRNA, establishing, as it does, a connection with the residue at position 26, which is an essential “linker” between tRNA domains [12, 13]. Although the clinical hallmark of mutations in MTTI appeared to be mitochondrial cardiomyopathy, [14–17] 5 other tRNAIle mutations have been associated with CPEO [18–22]. The fact that none of the mutations affect the anticodon loop illustrates the importance of the overall tRNA structure (Fig. 1D). In our patient and most reported CPEO cases, MTTI mutations were heteroplasmic and restricted to skeletal muscle in contrast to cardiomyopathy-associated tRNAIle mutations, which were nearly homoplasmic and detectable in multiple tissues. These observations suggest that levels of heteroplasmy and tissue distribution of tRNAIle mutations contribute to the variability of clinical presentations. Curiously, the history of unilateral ptosis in the patient’s mother and maternal grandmother suggests maternal inheritance, which would be atypical for muscle-specific mtDNA mutations [1–2]. Unfortunately, matrilineal relatives were not available for mtDNA screening.
In conclusion, this study adds m.4302A>G to growing list of point mutations associated with CPEO and underscores the importance of sequencing all 22 tRNAs in sporadic CPEO patients, after single deletions have being excluded and the necessity, in some cases, of performing DNA analysis in muscle tissue when mutations are not detectable in other tissues.
Acknowledgments
We thank Natalia Carminati for her technical assistance.
This work has been supported by NIH Grant HD32062 and the Marriott Mitochondrial Disorders Clinical Research Fund (MMDCRF).
Footnotes
Conflict of interest
None of the authors has any conflict of interest or financial disclosure to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.DiMauro S, Schon EA. Mitochondrial Disorders in the Nervous System. Ann Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- 2.DiMauro S, Hirano M. Mitochondrial encephalomyopathies: an update. Neuromusc Disord. 2005;15:276–286. doi: 10.1016/j.nmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Hart PE, De Vivo DC, Schapira AH. Clinical features of the mitochondrial encephalomyopathies. In: Shapira AH, DiMauro S, editors. Mitochondrial Disorders in Neurology. Vol. 2. Woburn: Butterworth-Heineman; 2002. pp. 35–68. [Google Scholar]
- 4.Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. doi: 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- 5.Holt IJ, Harding AE, Cooper JM, et al. Mitochondrial myopathies: clinical and biochemical features of 30 patients with major deletions of muscle mitochondrial DNA. Ann Neurol. 1989;26:699–708. doi: 10.1002/ana.410260603. [DOI] [PubMed] [Google Scholar]
- 6.Chinnery PF, DiMauro S, Shanske S, et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet. 2004;364:592–596. doi: 10.1016/S0140-6736(04)16851-7. [DOI] [PubMed] [Google Scholar]
- 7.Negro R, Zoccolella S, Dell’aglio R, et al. Molecular analysis in a family presenting with a mild form of late-onset autosomal dominant chronic progressive external ophthalmoplegia. Neuromuscul Disord. 2009;19:423–426. doi: 10.1016/j.nmd.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 8.Bonilla E, Sciacco M, Tanji K, Sparaco M, Petruzzella V, Moraes CT. New morphological approaches for the study of mitochondrial encephalopathies. Brain Pathol. 1992;2:113–119. doi: 10.1111/j.1750-3639.1992.tb00679.x. [DOI] [PubMed] [Google Scholar]
- 9.Servidei S, Zeviani M, Manfredi G, et al. Dominantly inherited mitochondrial myopathy with multiple deletions of mitochondrial DNA: clinical, morphologic, and biochemical studies. Neurology. 1991;41:1053–1059. doi: 10.1212/wnl.41.7.1053. [DOI] [PubMed] [Google Scholar]
- 10.Tanji K, Kaufmann P, Naini A, et al. A novel tRNAVal mitochondrial DNA mutation causing MELAS. J Neurol Sci. 2008;270:23–27. doi: 10.1016/j.jns.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanaker PS, Nakkestad HL, Downham E, et al. A novel mutation in the mitochondrial tRNA for tryptophan causing a late-onset mitochondrial encephalomyopathy. Acta Neurol Scand. 2009 Sep 10; doi: 10.1111/j.1600-0404.2009.01243.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 12.Dirheimer G, Keith G, Dumas P, Westhof E. Primary, secondary and tertiary structures of tRNAs. In: Söll D, Bhandary Raj, editors. tRNA: Structure, Biosynthesis, and Function. American Society of Microbiology Press; Washington, DC: 1995. pp. 93–126. [Google Scholar]
- 13.Giegé R, Puglisi J, Florentz C. tRNA structure and aminoacylation efficiency. Prog Nucleic Acid Res Mol Bio. 1993;45:129–206. doi: 10.1016/s0079-6603(08)60869-7. [DOI] [PubMed] [Google Scholar]
- 14.Merante F, Myint T, Tein I, Benson L, Robinson B. An additional mitochondrial tRNA(Ile) point mutation (A-to-G at nucleotide 4295) causing hypertrophic cardiomyopathy. Human Mutation. 1996;8:216–222. doi: 10.1002/(SICI)1098-1004(1996)8:3<216::AID-HUMU4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 15.Casali C, Santorelli FM, D’Amati G, Bernucci P, DeBiase L, DiMauro S. A novel mtDNA point mutation in maternally inherited cardiomyopathy. Biochemical and Biophysical Research Communications. 1995;213:588–593. doi: 10.1006/bbrc.1995.2172. [DOI] [PubMed] [Google Scholar]
- 16.Taniike M, Fukushima H, Yanagihara I, et al. Mitochondrial tRNAIle mutation in fatal cardiomyopathy. Biochemical and Biophysical Research Communications. 1992;186:47–53. doi: 10.1016/s0006-291x(05)80773-9. [DOI] [PubMed] [Google Scholar]
- 17.Ito T, Hattori K, Obayashi T, Tanaka M, Sugiyama S, Ozawa T. Mitochondrial DNA mutations in cardiomyopathy. Japanese Circulation Journal. 1992;56:1045–1053. doi: 10.1253/jcj.56.1045. [DOI] [PubMed] [Google Scholar]
- 18.Smits BW, Hol FA, van den Heuvel LP, Drost G, Rodenburg RJ, Ter Laak HJ, van Engelen BG. Chronic progressive external ophthalmoplegia caused by an m.4267A > G mutation in the mitochondrial tRNAIle. J Neurol. 2007;254:1614–1615. doi: 10.1007/s00415-007-0608-6. [DOI] [PubMed] [Google Scholar]
- 19.Silvestri G, Servidei S, Rana M, et al. A novel mitochondrial DNA point mutation in the tRNA(Ile) gene is associated with progressive external ophtalmoplegia. Biochemical and Biophysical Research Communications. 1996;220:623–627. doi: 10.1006/bbrc.1996.0453. [DOI] [PubMed] [Google Scholar]
- 20.Chinnery P, Johnson M, Taylor R, Durward W, Turnbull D. A novel mitochondrial tRNA isoleucine gene mutation causing chronic progressive external ophthalmoplegia. Neurology. 1997;49:1166–1168. doi: 10.1212/wnl.49.4.1166. [DOI] [PubMed] [Google Scholar]
- 21.Taylor R, Chinnery P, Bates M, Jackson M, Johnson M, Andrews RM, et al. A novel mitochondrial DNA point mutation in the tRNA(Ile) gene: studies in a patient presenting with chronic progressive external ophthalmoplegia and multiple sclerosis. Biochemical and Biophysical Research Communications. 1998;243:47–51. doi: 10.1006/bbrc.1997.8055. [DOI] [PubMed] [Google Scholar]
- 22.Franceschina L, Salani S, Bordoni, et al. A novel mitochondrial tRNA(Ile) point mutation in chronic progressive external ophthalmoplegia. Journal of Neurology. 1998;245:755–758. doi: 10.1007/s004150050283. [DOI] [PubMed] [Google Scholar]