

Abstract
Valuable information on the active sites of molybdenum enzymes has been provided by MoV electron paramagnetic resonance (EPR) spectroscopy. In recent years, multiple resonance techniques have been extensively used to examine details of the active-site structure, but basic continuous-wave (CW) EPR has not been re-evaluated in several decades. Here, we present a re-examination of the CW EPR spectroscopy of the sulfite oxidase low-pH chloride species and provide evidence for direct coordination of molybdenum by chloride.
Introduction
Sulfite oxidase (SO) is an oxo-transferase enzyme responsible for the physiologically vital oxidation of sulfite to sulfate.1 Residing in the mitochondrial inner-membrane space, the enzyme is dimeric with a subunit mass of about 52 000. Each monomer contains molybdenum associated with a single pterin cofactor, and a cytochrome b5-type heme. The two-electron oxidation of sulfite to sulfate is known to occur at the molybdenum site, which is concomitantly reduced from MoVI to MoIV.
| (1) |
The catalytic cycle is completed with reoxidation of the molybdenum first to MoV, and then to MoVI, by intramolecular electron transfer to the cytochrome b5 site, with cytochrome c serving as the external electron acceptor.2,3
With the exception of nitrogenase,4 all molybdenum enzymes described to date contain a novel pyranopterin-dithiolene cofactor (known as molybdopterin) in which the molybdenum is coordinated by the dithiolene moiety.5,6 In SO, the molybdenum atom is coordinated by one dithiolene, with two terminal oxygen atoms and one additional sulfur ligand from a cysteine (Cys207 in human SO).7 As one of the most intensively studied molybdenum enzymes, SO can be regarded as the prototypical member of the family of molybdenum enzymes possessing a dioxo molybdenum structural motif when in the fully oxidized MoVI form.8
One of the major spectroscopic techniques that has provided information on the active site is MoV electron paramagnetic resonance (EPR). Recently, efforts have been focused on studying SO using advanced pulsed EPR techniques, such as electron–nuclear double resonance (ENDOR) and electron-spin envelope echo modulation (ESEEM),9 but the conventional continuous-wave (CW) EPR has not been re-evaluated in nearly two decades. In this work,10 we revisit the MoV EPR of one of the major signals from this important enzyme,11–13 and using the stable magnetic isotopes 35Cl and 37Cl, in conjunction with difference EPR spectroscopy, and analogous complexes with other halides, we present new evidence for halide molybdenum coordination of the MoV low-pH species.
Materials and Methods
CW EPR spectroscopy
X-band EPR spectra were recorded on Varian E109 or Bruker EMX spectrometers. S-band spectra were recorded on a custom-built loop-gap resonator14 equipped with a 2–4 GHz bridge (Medical Advances, Milwaukee, WI) at the National Biomedical EPR Center. Data acquisition, analysis, and EPR computer simulations were performed as previously described,14–16 except that a modulation amplitude of 0.05 mT was used. For the Bruker EMX system, 4096 field points across the scan were taken, whereas for the Varian spectrometers, 1024 field points were used.
Samples
Purified recombinant human sulfite oxidase was obtained as previously described.17 MoV EPR signals were generated by the addition of 1 mM (final) sodium sulfite solution to concentrated enzyme (approximately 0.25 mM Mo) before freezing in quartz sample tubes of 3 mm internal diameter. A mixed buffer system was used, consisting of 50 mM bis-tris-propane and 50 mM MES. Reagents were obtained from Sigma-Aldrich Chemical Co. and were of the best quality available. Isotopically enriched Na35Cl and Na37Cl were obtained from Oak Ridge National Laboratories at isotopic enrichments of 99.9 and 98.2 atom %, respectively.
Molecular Modeling
Density functional theory (DFT) molecular modeling used the program Dmol3 Materials Studio, version 3.2.18,19 We expected bond-length accuracies of around 0.05 Å, and good estimates of energetic trends between postulated molecular entities. The Becke exchange20 and Perdew correlation functionals21 were used to calculate both the potential during the self-consistent field procedure and the energy. Double numerical basis sets included polarization functions for all atoms. Calculations were spin-unrestricted, and all electron relativistic core potentials were used. No symmetry constraints were applied, and optimized geometries used energy tolerances of 2.0 × 10−5 Hartree.
Results and Discussion
MoV Electron Paramagnetic Resonance Spectroscopy
Figure 1 compares the high-pH signal and low-pH MoV EPR signals of human SO. Both have an exchangeable strongly coupled proton thought to originate from Mo–OH coordination, but a well-resolved splitting is only seen for the low-pH signal. The splitting is not directly observed for the high-pH signal, but its presence is betrayed by formally forbidden ΔmI = ±1 satellites separated by the proton frequency (the so-called proton spin-flip lines).16,22 Two different explanations have been suggested for the lack of observed proton hyperfine splittings in the high-pH signal; both assume a proton coordinated as Mo–OH with a small isotropic coupling and postulate different reasons why the anisotropic splitting is not directly observed. The first hypothesizes that the anisotropic proton hyperfine tensor and the g tensor are oriented close to the so-called trimagic angle, resulting in essentially no anisotropic splittings at each of the principal g values (gx, gy, and gz), whereas the ΔmI = ±1 will be at maximal intensity, and thus readily observed.16 Computer simulations showed that this suggestion could account for the powder line shape of the high-pH signal.16,23 The second suggestion followed confirmation of the presence of proton hyperfine splitting by pulsed EPR spectroscopy24 and conjectured that rotational averaging about the Mo–O bond caused essentially zero anisotropic splittings at each gx, gy, and gz.24 Which of these two suggestions more accurately explains the lack of proton splitting in the SO high-pH EPR signal remains uncertain. Previous work has shown that the addition of chloride promotes the conversion of the high-pH species to the low-pH species.13 Moreover, Bray and co-workers found that increasing the chloride concentration by a factor of 10 shifted the pKa of the interconversion between high- and low-pH signal-giving species by one pH unit.13 In addition, they reported that the low-pH EPR signals were identical in the absence and presence of chloride and attributed this to trace amounts of contaminating chloride in their “chloride-free” buffers.25 Figure 1b and c compare the low-pH spectra obtained in the presence and in the rigorous absence of added chloride. The signal obtained in the presence of chloride is essentially identical to that obtained by Bray et al., but the spectrum in the absence of chloride is clearly different, possessing slightly different g values and much narrower line widths (Table 1).26We will use this EPR signal-giving species as the low-pH halide-free species. The observed pK relating the high-pH and low-pH EPR signalgiving species shifts from 8.1 in the absence of chloride to 6.8 in the presence of 0.1 M chloride (Figure 2), which approximately corresponds to the lowest pK measured by Bray and co-workers13 and might suggest a difference in structure at the active site in the presence and absence of chloride.
Figure 1.
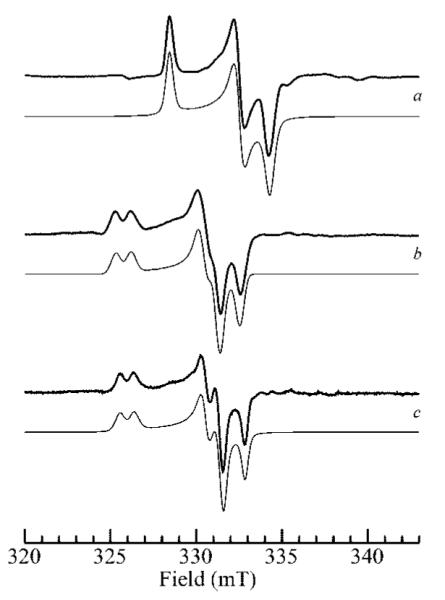
Effects of pH and chloride on MoV EPR spectra of human sulfite oxidase. Part a shows the high-pH species at pH 9.0 in 2H2O; parts b and c show the low-pH species (pH 6.0) in the presence of 0.1 M Cl- (b) and in the absence of chloride (c). Traces shown as bold lines are experimental data, and traces shown as light lines are computer simulations.
Table 1.
MoV Electron Paramagnetic Resonance Spin Hamiltonian Parametersa
| g |
A(1H) |
A(35Cl,79Br,127I) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| x | y | z | x | y | z | x’ | y’ | z’ | |
| halide-free | 1.9646 | 1.9723 | 2.0023 | 34.4 | 21.3 | 23.1 | |||
| Cl- | 1.9658 | 1.9724 | 2.0031 | 35.6 | 22.5 | 26.2 | 4.0 | 4.2 | 4.9 |
| Br-b | 1.9655 | 1.9732 | 2.0025 | 42 | 29 | 28 | 10 | 12 | 26 |
| I-c | 1.9670 | 1.9730 | 2.0010 | 38 | 35 | 38 | 21 | 16 | 18 |
Values for couplings are given in MHz. As discussed in the text, the parameters for 79Br and 127I are difficult to unambiguously determine without measurements at a high frequency (35 GHz), because of the large number of spin Hamiltonian parameters, and these values are considered approximate. For “no halide” and Cl-, g values are considered accurate to 2 on the last significant digit, and estimates of A to about 0.5 MHz, as discussed in the text; spin Hamiltonian parameters for the Br- and I- species are considered more approximate because of the number of variables in the simulations.
A(79Br) and g were noncolinear (α,β,γ = 10, 57, −5°). Effects of 81Br were automatically calculated from the ratio of nuclear g values and nuclear electric quadrupole moments using the known natural isotopic abundance. Bromine nuclear electric quadrupole couplings were included and specified by P(x’,y’,z’) = −2.5, +2.9, and −0.3 MHz with the P tensor held collinear with A.
A(127I) and g were noncolinear (α,β,γ = 30, 30, −20°). Iodine nuclear electric quadrupole coupling is included and specified by P(x’,y’,z’) = +4.3, −5.7, and 1.3 MHz with the P tensor held collinear with A.
Figure 2.
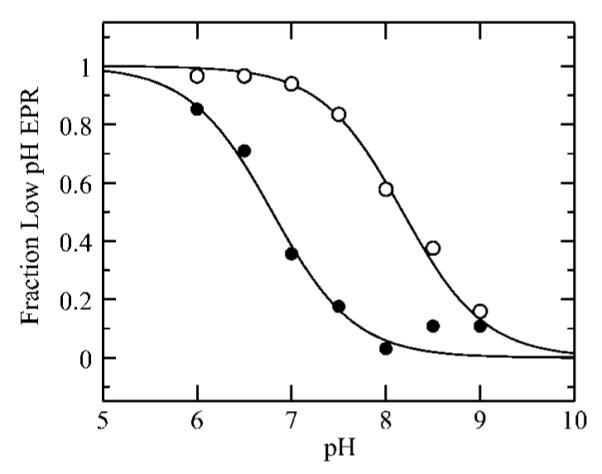
The pH dependency of the high-pH to low-pH conversion in the stringent absence of chloride (◯) and in the presence of 0.1 M NaCl (●). The pKa values determined from these data are 8.1 and 6.8, for no chloride and for 0.1 M NaCl, respectively.
The broad line widths of the low-pH chloride signal have previously been attributed to unresolved hyperfine interaction with I = 3/2 35Cl and 37Cl nuclei.13,28 However, this idea has fallen out of favor because chlorine hyperfine coupling has remained undetected by ENDOR. Thus, the structures currently postulated for the low-pH signal are five-coordinate species, as shown in Figure 3.9 Our finding of a sharper line width for the low-pH halide-free signal supports the original hypothesis of unresolved chlorine hyperfine coupling and encouraged us to perform further experiments using the stable magnetic isotopes 35Cl and 37Cl. Figure 4 shows the low-pH Cl- spectra at X- and S-band microwave frequencies obtained with 35Cl- and 37Cl-enriched sodium chloride. These two isotopes both have I = 3/2, but 35Cl has a slightly larger nuclear g value than 37Cl (gN = 0.548 and 0.456, respectively), and proportionally larger hyperfine couplings are expected. Subtle but fully reproducible differences between 35Cl and 37Cl are observed, which are best visualized by examining the difference spectra (Figure 4). The EPR difference spectra simulations of Figure 4 were computed using the line widths of the low-pH halide-free signal (e.g., Figure 1c), and simulating the broader lines of the 35Cl spectrum using unresolved hyperfine coupling to an I = 3/2 nucleus. Some effects due to nuclear electric quadrupole coupling to the chlorine nuclei are expected to be present. The nuclear electric quadrupole moments of the two nuclei are quite small, at only −0.079 and −0.062 b for 35Cl and 37Cl, respectively, but the nuclear electric quadruple coupling will gain in magnitude due to the significant electric field gradient expected for coordinated chloride. Unless the quadrupole coupling is very large,29 quadrupole effects are usually manifested as additional transitions approximately within the envelope of the hyperfine splitting.30,31 Furthermore, no obvious quadrupole effects are observed in the EPR spectra of other transition metal ions exhibiting chlorine hyperfine coupling.32,33 As we observe only a broadening, our estimates of hyperfine couplings will necessarily be approximate, and quadrupole effects can be safely neglected in our current simulations without substantially changing our conclusions. Difference simulations were generated by subtracting the normalized 37Cl simulation from the normalized 35Cl simulation, with the former being obtained by reducing the chlorine couplings by the ratio of nuclear g values (0.83).34 Our simulations of the observed broadenings yielded an approximate hyperfine coupling (35Cl)A(x,y,z) = 3.9, 4.9, and 3.0 MHz.35 The similarity of the experimental and simulated spectra in Figure 4 provides strong evidence that hyperfine coupling to chlorine in the range of 3–5 MHz is present in the low-pH chloride species. The observation of similar line broadening of the EPR spectra at the two microwave frequencies effectively eliminates g strain as the primary source of broadening in the low-pH chloride species.36 We note that a complete quantitative determination of this hyperfine coupling is not possible with X- and S-band EPR. Hyperfine coupling arises from such mechanisms as electron delocalization, exchange terms such as spin polarization, and direct dipole coupling. Sulfite oxidase contains an arginine-rich pocket ~5 Å from Mo, and Cl- has been observed crystallographically in this pocket in the structure of a mutant sulfite oxidase.37 The dipole coupling at this distance will be only 0.1 MHz, and at such large distances, other mechanisms of coupling are very unlikely. We can thus effectively exclude the 5 Å pocket as the origin of the observed hyperfine coupling. Typical Mo–Cl bond lengths for high-valent Mo species range from 2.38 to 2.65 Å (cis and trans to an Mo=O ligation, respectively).38 Simple dipole hyperfine calculations using these bond lengths yield a maximum 35Cl dipole coupling contribution of 1.2 and 0.8 MHz, respectively, and additional mechanisms will increase these values (vide supra). Thus, the chlorine hyperfine broadening of the MoV EPR signal that we observe provides strong evidence for direct coordination of chloride to Mo. Furthermore, the similarity in spin Hamiltonian parameters for the low-pH Cl- and low-pH halide-free signals (Figure 1) cannot be reconciled with an equatorial coordination of the anion, as this would be expected to influence the g values through bonding interaction with the magnetic Mo 4dxy orbital. Thus, we postulate that the low-pH Cl- species is six-coordinate with the chloride coordinated trans to the Mo=O ligand. In addition, we hypothesize that the structure of the high-pH species is analogous to the five-coordinate Mo site established from protein crystallography and extended X-ray absorption fine structure (EXAFS) spectroscopy (Figure 3). The low-pH signal prepared in the absence of halide (low-pH halide-free, Figure 1) has very similar g values and 1H hyperfine couplings to the low-pH and must have a similar active-site structure, possibly with sulfate or sulfite occupying the axial position instead of chloride. This description of the active site is consistent with the observed shift to higher g values for the low-pH Cl- species as the coordination change will effectively raise the donor orbitals of the dithiolene ligand into the plane of the half-occupied ground-state Mo 4dxy, thus facilitating spin-delocalization onto the dithiolene sulfur ligands through spin–orbit coupling.
Figure 3.
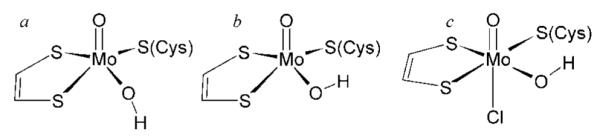
Previously postulated structures for the high-pH (a) and low-pH (b and c) signal-giving species.
Figure 4.
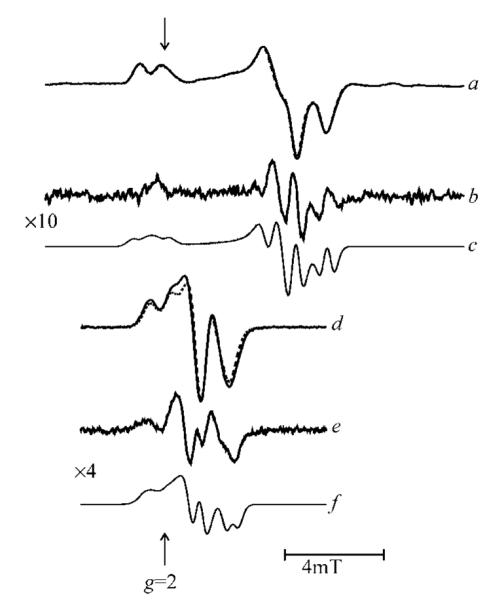
35Cl and 37Cl isotope effects on the low-pH chloride EPR signal at X-band and at S-band microwave frequencies. Traces a and d compare signals from 35Cl (solid line) and 37Cl (broken line) at the X- and S-band, respectively; traces b and e show the corresponding difference spectra, and c and f the corresponding simulations.
In agreement with earlier work, we were unable to detect any X-band ENDOR signals attributable to 35Cl and 37Cl (see the Supporting Information). However, weakly magnetic quadrupolar nuclei such as 35Cl and 37Cl are often difficult to detect by ENDOR unless high microwave frequencies are used to increase the relative size of the nuclear Zeeman term.39 Bray and co-workers also reported a minor species attributed to a low-pH fluoride complex of sulfite oxidase with resolved 19F coupling. This would be expected because the 19F coupling will be inherently larger than the 35,37Cl coupling by approximately a factor of 10. However, we were unable to reproduce their results, possibly because of very subtle differences between the chicken enzyme used by Bray and the human enzyme which is the subject of the present study. Furthermore, we note that Bray’s fluoride spectrum was a minor component and needed to be generated by taking multiple difference spectra. This technique is somewhat prone to artifacts, and whether Bray actually detected a bona fide fluoride species is debatable. Moreover, fluoride is not expected to be a good axial ligand, trans to the Mo=O group, thus our inability to detect a well-defined fluoride complex is not surprising. MoV complexes of bromide and iodide are known,40,41 and these anions should also give larger hyperfine couplings than chloride, and the effects of these anions on the EPR spectrum of SO are thus of relevance to the low-pH chloride signal. Bromine has two naturally abundant stable magnetic isotopes 79Br and 81Br (50.5 and 49.5% natural abundance, respectively), both with I = 3/2 and with similar nuclear g values (1.404 and 1.513, respectively). Iodine has a single stable magnetic isotope 127I with I = 5/2 and a nuclear g value of 1.872. If all other factors are equal, both anisotropic and isotropic hyperfine couplings to bromine and iodine are expected to be similar, and about a factor of approximately 4.5 larger than those to chlorine. Figure 5 compares the signals obtained in the presence of bromide and iodide with those from chloride. Clearly resolved hyperfine splittings can be seen for both bromide and iodide signals. The signals also have well-resolved proton hyperfine splittings, which were demonstrated by development of the signals in 2H2O (not illustrated). Computer simulation was used to quantify the couplings to halide nuclei. We note that bromine and iodine nuclei have significant quadrupole moments (+0.310 and +0.260 b for 79Br and 81Br, respectively, and −0.790 b for 127I). Because of this, we included nuclear electric quadrupole coupling in our simulations. As previously discussed,42 the number of parameters included in simulations such as these is large, and unambiguous determination of the spin Hamiltonian parameters requires spectroscopy at higher microwave frequencies. Nevertheless, the observation that strong hyperfine coupling is present to both bromine and iodine is unambiguous and provides very strong support for the hypothesis of the direct coordination of a halide ligand to Mo in the low-pH halide EPR signal-giving species.
Figure 5.
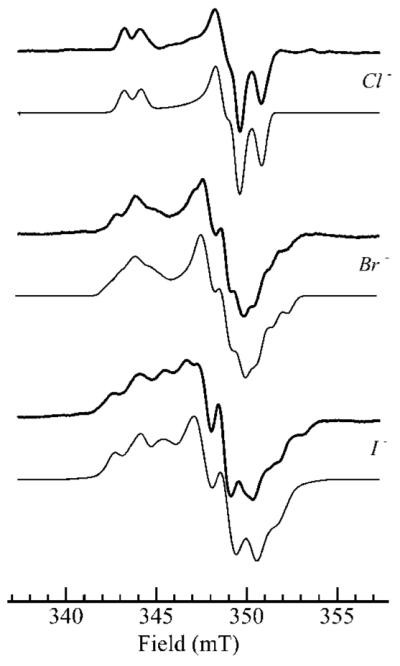
Comparison of the effects of chloride (0.1 M), bromide (0.2 M), and iodide (0.2 M) upon the MoV EPR spectra of human sulfite oxidase reduced with 1 mM sulfite (0.2 M bis-tris-propane, pH 6.0). The bold lines show experimental spectra and the lighter lines computer simulations using the parameters given in Table 1.
This possibility was further tested using density functional calculations, and Figure 6 shows the results of energy minimizations including three of the active-site amino acids (tyrosine 343, arginine 160, and cysteine 207). The α carbons of the amino acids and the pyran ring carbons were constrained to their crystallographic positions, as were the vectors described by the α and β carbons. The results of energy minimizations were found to be very sensitive to the exact starting coordinates; for example, changing the orientation of the –OH group of tyrosine 343 was found to change the result of energy minimization between five-coordinate and six-coordinate, and thus our DFT calculations are of little use as a predictive tool for the active site structure. Nevertheless, the finding of well-defined MoV structures with and without Cl- bound to Mo indicates that our hypothesis of chloride binding to the low-pH species is at least chemically reasonable. Moreover, when tyrosine 343 was omitted from the calculations, chloride typically dissociated from Mo during the energy minimization, suggesting that this amino acid is very important in stabilizing the complex. Figure 6 indicates two different possible Mo–OH orientations, one where the Mo–OH proton is hydrogen-bonded to the tyrosine oxygen (Figure 6a), corresponding to the high-pH species, and the other where the proton of the tyrosine –OH group is hydrogen-bonded to the oxygen of the Mo–OH (Figure 6b), corresponding to the low-pH/chloride species. Conformational differences such as these will significantly affect the orientation of the anisotropic proton hyperfine coupling and may explain the different proton hyperfine splittings observed for the high-pH and low-pH chloride MoV EPR signals; although, as we have discussed above, the exact nature of the proton coupling to the high-pH species is still controversial.
Figure 6.

Postulated structures for the high-pH (a) and low-pH Cl- (b) MoV species. Energy-minimized density functional theory structures are shown, with external atoms approximated by hydrogens (not shown).
Finally, our results are fully consistent with our earlier EXAFS studies of chicken sulfite oxidase which suggested an increase in Mo–S/Cl ligation with reduced conditions of low-pH Cl- formation, relative to those of high-pH formation.43,44 Furthermore, our recent finding that a sulfite oxidase clinical mutant (arginine 160 → glutamine) has a six-coordinate active site in the MoVI state (and in some MoV forms)45 further supports our hypothesis for a six-coordinate structure for the low-pH chloride species. The apparent conformational flexibility of the reduced active site suggested by our new findings may have important implications for the catalytic mechanism. The availability of chloride in the mitochondrial inner-membrane space raises questions about the possibility of in vivo chloride coordination of Mo. Furthermore, the fact that the active site of the native enzyme can apparently adopt both six-coordinate and five-coordinate geometries may be important in the catalytic mechanism, which may involve the binding of anions such as sulfite directly to Mo.
Supplementary Material
Acknowledgment
Work at the University of Saskatchewan was supported by a Canada Research Chair award (G.N.G.), the University of Saskatchewan, the Province of Saskatchewan, the Natural Sciences and Engineering Research Council (Award #283315), the Canadian Institute for Health Research, and the Canada Foundation for Innovation (Award #201742). Work at Duke University was supported by NIH grant GM 00091. The National Biomedical EPR Center is supported by NIH NIBIB EB001980.
Footnotes
Supporting Information Available: Figure showing X-band ENDOR spectra of low-pH Cl- species. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).McLeod RM, Farkas W, Fridovitch I, Handler P. J. Biol. Chem. 1961;236:1841–1852. [PubMed] [Google Scholar]
- (2).Cohen HL, Betcher-Lange S, Kessler DL, Rajagopalan KV. J. Biol. Chem. 1972;247:7759–7766. [PubMed] [Google Scholar]
- (3).Johnson JL, Rajagopalan KV. J. Biol. Chem. 1977;252:2017–2025. [PubMed] [Google Scholar]
- (4).Rees DC, Tezcan A, Haynes CA, Walton MY, Andrade S, Einsle O, Howard JB. Philos. Trans. R. Soc. London, Ser. A. 2005;363:971–984. doi: 10.1098/rsta.2004.1539. [DOI] [PubMed] [Google Scholar]
- (5).Elliot SJ, McElhaney AE, Feng C, Enemark JH, Armstrong FJ. J. Am. Chem. Soc. 2002;124:11612–11613. doi: 10.1021/ja027776f. [DOI] [PubMed] [Google Scholar]
- (6)(a).Rajagopalan KV. Adv. Enzymol. Relat. Areas Mol. Biol. 1991;64:215–290. doi: 10.1002/9780470123102.ch5. [DOI] [PubMed] [Google Scholar]; (b) Rajagopalan KV, Johnson JL. J. Biol. Chem. 1992;267:10199–10202. [PubMed] [Google Scholar]
- (7).Kisker C, Schindelin H, Pacheco A, Wehbi WA, Garrett RM, Rajagopalan KV, Enemark JH, Rees DC. Cell. 1997;91:1–20. doi: 10.1016/s0092-8674(00)80488-2. [DOI] [PubMed] [Google Scholar]
- (8).Hille R. Chem. Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- (9).Enemark JH, Astashkin AV, Raitsimring AM. J. Chem. Soc., Dalton Trans. 2006:3501–3514. doi: 10.1039/b602919a. [DOI] [PubMed] [Google Scholar]
- (10).The portion of work describing the coupling of 35Cl and 37Cl was originally submitted as a communication in August 2006. Our data thus predate later suggestions of chlorine coupling by others.
- (11).Lamy MT, Gutteridge S, Bray RC. Biochem. J. 1980;185:397–403. doi: 10.1042/bj1850397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cramer SP, Johnson JL, Rajagopalan KV, Sorrell TN. Biochem. Biophys. Res. Commun. 1979;91:434–439. doi: 10.1016/0006-291x(79)91540-7. [DOI] [PubMed] [Google Scholar]
- (13).Bray RC, Gutteridge S, Lamy MT, Wilkinson T. Biochem. J. 1983;211:227–236. doi: 10.1042/bj2110227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Frocisz W, Hyde JS. J. Magn. Reson. 1982;47:515–521. [Google Scholar]
- (15).George GN, Prince RC, Kipke CA, Sunde RA, Enemark JE. Biochem. J. 1988;256:307–309. doi: 10.1042/bj2560307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).George GN. J. Magn. Reson. 1985;64:384–394. [Google Scholar]
- (17).Temple CA, Graf TN, Rajagopalan KV. Arch. Biochem. Biophys. 2000;383:281–287. doi: 10.1006/abbi.2000.2089. [DOI] [PubMed] [Google Scholar]
- (18).Delley B. J. Chem. Phys. 1990;92:508–517. [Google Scholar]
- (19).Delley B. J. Chem. Phys. 2000;113:7756–7764. [Google Scholar]
- (20).Becke AD. J. Chem. Phys. 1988;88:2547–2553. [Google Scholar]
- (21).Perdew JP, Wang Y. Phys. Rev. B: Condens. Matter Mater. Phys. 1992;45:13244–13249. doi: 10.1103/physrevb.45.13244. [DOI] [PubMed] [Google Scholar]
- (22).Proton spin-lip lines are formally forbidden ΔmI = ±1 transitions that are often observed in the EPR of free-radical systems when weakly coupled protons are present. Both proton spin-flip and double spin-flip lines (the latter involving two coupled protons) were the subject of an earlier study on the MoV EPR of molybdenum enzymes.16 The sulfite oxidase high-pH signal was alone among all the signals studied in having a strongly coupled exchangeable proton with no observed splittings, the presence of which was betrayed by the spin-flip lines.
- (23).Doonan CJ, Kappler U, George GN. Inorg. Chem. 2006;45:7488–7492. doi: 10.1021/ic0607944. [DOI] [PubMed] [Google Scholar]
- (24).Astashkin AV, Mader ML, Pacheco A, Enemark JH, Raitsimring AM. J. Am. Chem. Soc. 2000;122:5294–5302. [Google Scholar]
- (25).Chloride is rather difficult to eliminate from solution, as there are many sources that are not usually accounted for, such as the KCl contained within the electrode used to measure the pH of samples, some of which will diffuse into the solution. Bray and co-workers13 estimated a contaminating level of chloride of 0.1 mM, and we expect our levels to be similar.
- (26).The lower concentrations of enzyme available to Bray et al. meant that these workers used a comparatively large modulation amplitude of 0.25 mT, which would have broadened the chloride-free low-pH signal so that it looked very similar to the signal obtained in the presence of chloride.
- (27).We note in passing that developing the signals in 2H2O causes a broadening of the MoV EPR signal due to unresolved 2H hyperfine splitting, which makes the difference in broadening caused by 35Cl and 37Cl much harder to discern.
- (28).We note that the high ionic strengths caused by the use of high chloride concentrations might induce strain distributions in spin Hamiltonian parameters (e.g., g-strain), resulting in the observed broadening. This is not likely because low-pH Cl- spectra developed at low pH using low concentrations of chloride (2 mM) are identical within the noise to those obtained at higher (100 mM) or very high (1 M) chloride concentrations.
- (29).George GN, Bray RC. Biochemistry. 1983;22:5443–5432. doi: 10.1021/bi00274a003. [DOI] [PubMed] [Google Scholar]
- (30).George GN, Bray RC. Biochemistry. 1988;27:3603–3609. doi: 10.1021/bi00410a011. [DOI] [PubMed] [Google Scholar]
- (31).Connelly NG, Emslie DJH, Klangsinsirikul P, Rieger PH. J. Phys. Chem. A. 2002;106:12214–12220. [Google Scholar]
- (32).Petersen JL, Egan JW., Jr Inorg. Chem. 1983;22:3571–3575. [Google Scholar]
- (33).Kon H, Sharpless NE. J. Chem. Phys. 1965;43:1081–1082. [Google Scholar]
- (34).Simulations and spectra were in all cases normalized to their maximum amplitude. An alternative might be to normalize to the integrated intensity, but for experimental spectra, this is subject to an approximately 10% error (depending on a number of factors, such as baseline noise etc.), and because of this, normalizing to the amplitude is preferred.
- (35).We note that we estimate the sign of the hyperfine couplings.
- (36).Hagen WR. J. Magn. Reson. 1981;44:447–469. [Google Scholar]
- (37).Karakas E, Wilson HL, Graf TN, Xiang S, Jaramillo-Busquets S, Rajagopalan KV, Kisker C. J. Biol. Chem. 2005;280:33506–33515. doi: 10.1074/jbc.M505035200. [DOI] [PubMed] [Google Scholar]
- (38).This value is an average of all high-valent Mo entries with Mo–Cl coordination in the Cambridge Structure Database: Allen FH, Kennard O, Watson DG. Struct. Correl. 1994;1:71–110.
- (39).We note that ESEEM features attributable to chloride have recently been reported: Astashkin AV, Klein EL, Enemark JH. J. Inorg. Biochem. 2007;101:1623–1629. doi: 10.1016/j.jinorgbio.2007.05.015.
- (40).Cotton FA, Luck RL, Miertschin CS. Inorg. Chem. 1991;30:548–553. [Google Scholar]
- (41).Baird DM, Rheingold AL, Croll SD, DiCenso AT. Inorg. Chem. 1986;25:3458–3461. [Google Scholar]
- (42).George GN, Garrett RM, Graf T, Prince RC, Rajagopalan KV. J. Am. Chem. Soc. 1998;120:4522–4523. [Google Scholar]
- (43).George GN, Kipke CA, Prince RC, Sunde RA, Enemark JH, Cramer SP. Biochemistry. 1989;28:5075–5080. doi: 10.1021/bi00438a026. [DOI] [PubMed] [Google Scholar]
- (44).Note that chlorine and sulfur backscatterers cannot be distinguished by EXAFS; thus, coordination of a chloride would appear as an effective increase in the Mo–S coordination.
- (45).Doonan CJ, Wilson HL, Rajagopalan KV, Garrett RM, Bennett B, Prince RC, George GN. J. Am. Chem. Soc. 2007;129:9421–9428. doi: 10.1021/ja071402a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.