

Abstract
A variety of nanocarriers such as bioconjugates, dendrimers, liposomes, and nanoparticles have been widely evaluated as potential targeted drug delivery systems. Passive targeting of nanoscale carriers is based on a size-flow-filtration phenomenon that is usually limited to tumors, the reticular endothelial system, and possibly lymph nodes (LN). In fact, targeting the delivery of drugs to pivotal physiological sites such as the lymph nodes has emerged as a promising strategy in treating HIV disease. Ligands for specific cell surface receptors can be displayed on nanocarriers in order to achieve active targeting. The approach has been extensively used preclinically in cancer where certain receptors are over-expressed at various stages of the disease. Unfortunately, markers of HIV infection are lacking and latently infected cells do not show any signs of infection on their surface. However, the disease naturally targets only a few cell types. The HIV receptor CD4, coreceptors (CCR5 and CXCR4), and some receptors relatively specific for macrophages provide potentially valuable surface targets for drug delivery to all susceptible cells in patients infected by HIV. This review focuses on nanoscale targeting with an emphasis on surface modifications of drug delivery nanocarriers for active targeting. A number of related issues, including HIV biology, targets, pharmacokinetics, and intracellular fate as well as literature-cited examples of emerging surface-modified targeted carrier systems are discussed.
Keywords: HIV/AIDS, anti-HIV drugs, nanocarriers, surface modification, macrophages, intracellular delivery
1. Cell surface targets and sites of infection
Tens of millions of people are currently infected by human immunodeficiency virus-1 (HIV-1) and millions of new infections occur each year [1–6]. HIV-1 infection leads to acquired immunodeficiency syndrome (AIDS) as a result of low levels of CD4+ T cells that are directly killed by HIV and/or by apoptosis of infected CD4+ T cells and indirect killing by CD8 cytotoxic T-lymphocytes. The disease naturally targets only a few cell types including CD4+ T cells, CD4+ monocytes/macrophages (M/M), dendritic cells (DC) [7], FDCs and microglial cells. Certain cells such as macrophages are commonly referred to as reservoir sites since they are not killed by HIV infection allowing the virus to reproduce and supply the body with new virus [8, 9]. As such, killing HIV in cellular and tissue reservoirs represents a major challenge for eradicating HIV infection. The infection of a relatively few cell types makes it desirable to direct drug therapy only to infected cells. Unfortunately, markers of HIV infection are lacking and latently infected cells do not show any signs of infection on their surface. Thus, active targeting of HIV drugs to HIV infected cells has been difficult to achieve.
1.1. Entry into target cells and tropism: HIV cell surface targets
An interesting observation can be made about HIV-1, it is a particle with a diameter of about 100 nm and it has surface molecules with post-translational modifications (i.e., ligands) that allow it to efficiently target and be taken up into cells such as T-cells and macrophages. As such, HIV-1 can be viewed as a natural surface functionalized nanocarrier. Much can be learned from understanding how HIV enters target cells. The first step of HIV infection involves adsorption to the cell surface in order to bring the virus closer to and in an optimal position to interact with the HIV cell surface receptors and coreceptors. In order to achieve this, negatively charged heparan sulfate molecules on the cell surface interact with the positively charged HIV glycoprotein (gp120) [10]. HIV then binds to the CD4 receptor and one of the two chemokine coreceptors, CXCR4 or CCR5 via gp120 [10] (Figure 1). This is followed by the fusion of the viral particle’s envelope with the cell that involves viral gp41 protein and the entry of the virus into the target cell [11]. Susceptible cells include CD4+ T cells, CD4+ M/M, DCs, FDCs, and microglial cells. Additional specific types of cells are promyelocytes, intestinal immunocytes, astrocytes, oligodendrocytes, epidermal Langerhans’ cells, and certain fibroblasts.
Figure 1.

Model for HIV-1 entry in T cells. HIV-1 gp120 first binds to cellular CD4. This results in a conformational change in gp120, allowing it to bind to the chemokine receptors CCR5 or CXCR4, thereby forming a trimolecular complex (CD4-gp120-CCR/CXCR). An excess of the natural ligands for CCR5 or CXCR4 can competitively inhibit this step of infection (as noted). After binding to the chemokine receptor, gp120 is thought to become stripped off of the virion, thereby exposing a hydrophobic domain at the N-terminus of gp41, which mediates fusion of the host cell and virus membranes, thereby allowing the virus core to enter the host cell cytoplasm (redrawn from www.bioscience.org).
Whether HIV interacts with CXCR4 or CCR5 on CD4+ cells depends upon the protein sequence of the V3 loop of gp120. Some HIV-1 strains use both receptors, others use CCR5 or CXCR4 exclusively while others may predominantly use one co-receptor in macrophages and another co-receptor in lymphocytes. There are rare clinical strains of HIV that prefer chemokine coreceptors other than CXCR4 or CCR5. A revised paradigm of tropism shows that macrophages express both CCR5 and CXCR4 and can be infected by M-tropic and dual-tropic strains, but not T-tropic strains [12]. HIV-1 can infect FDCs by targeting the CD4-CCR5 pair of receptors or alternative mannose-specific C-type lectin receptors such as DC-SIGN [13]. It cannot be ruled out that other cellular receptors for HIV-1 exist since CD4-negative cells (e.g., fibroblasts, bowel, and glial cells) can also be infected. For example, an alternative receptor in brain- and bowel-derived cells has been identified as galactosyl ceramide. As our knowledge of the number of receptors and coreceptors involved in HIV cell entry grows, the enormity of the challenge becomes obvious. If the various HIV interactions are highly specific and distinct for each receptor, blocking HIV cell entry will be an enormous challenge.
1.2. Sites of HIV infection and reservoirs
Nearly 99% of all viral replication occurs in activated and productively infected CD4+ T-cells of the blood and lymphoid tissues such as the peripheral secondary lymphoid organs, the spleen, lymph nodes (LNs), and gut-associated lymphoid tissue (GALT) [14, 15]. Lymphoid tissues have a greater extent of infection than the peripheral blood since only 2% of lymphocytes are in the general circulation at any one time and the remainder are distributed among the lymphoid tissues primarily in the LNs [15]. HIV infects several subsets of T-cells including Th-17 cells. Th-17 cells, which exist in the lamina propria of the gut, are responsible for host defense against bacterial infections [16]. As such, the GALT is home to large numbers of activated T-cells, which propagate the infection [15]. The primary lymphoid tissues, bone marrow and thymus, are also found to be reservoir sites, however the extent of T-cell infection for these sites remains unknown [15].
M/M and cells derived from that lineage are other major cellular targets of HIV-1 infection [17]. HIV-1 infection does not lead to M/M depletion as occurs for HIV-1 infected CD4+ T-lymphocytes [18]. Productively infected M/M can fuse with CD4+ T-lymphocytes and transfer the virus to these cells within the context of antigen presentation [19] and trigger apoptosis of T-lymphocytes (either CD4+ or CD8+) [20, 21] as well as astrocytes [22]. A few HIV-infected M/M are sufficient to induce the recruitment and activation of HIV-infected resting CD4+ lymphocytes and infect them [23]. HIV-infected monocytes have been implicated in carrying HIV across the blood brain barrier [24]. The physiological proximity of HIV-infected M/M to the other cell types involved in HIV infection compound their importance in viral infection [25]. For these reasons macrophages play a critical role in HIV persistence.
FDCs are unable to produce HIV particles through replication. However, they play a significant role in viral storage. FDCs also contribute to the expansion of HIV infection by migrating toward T-cells and presenting those cells with the viral particles [26].
Latently infected CD4+ T cells, including resting memory (CD45RO) T-cells and resting naive (CD45RA) T-cells are an important HIV reservoir. M/M become more important as a viral reservoir only when the majority of CD4+ T cells have been lost later in HIV-1 disease. Therefore, a critical step for eradicating HIV infection requires the targeted treatment of infected M/M.
1.3. Non-HIV receptor targets: Macrophages
Targeted drug delivery for treating HIV infection requires specific ligands that interact with specific HIV receptor targets. CD4 is an ideal receptor to target because it is expressed on most HIV susceptible cells. However, antibodies and large peptides are too expensive to be used as targeting agents and so far only a few small chemical CD4 antagonists have been developed [27]. Unfortunately, conjugating these small molecule antagonists to drug delivery carriers destroys their receptor-binding specificity and affinity. In contrast, more entry and fusion inhibitors have been identified that target HIV-1 coreceptors [28], some of which may be developed as targeting agents.
Although there are a variety of cells that become infected by HIV, the role of macrophages has become increasingly recognized over the past few years. As such, this section will only focus on the receptor targets on macrophages. More than a dozen non-viral receptors are expressed on macrophages [29] that may serve as targets for drug delivery. Among them, the formyl peptide receptor 1 (fMLF as the targeting agent), mannose receptor (mannose as the targeting agent), and Fc receptor (Fc as the targeting agent) are the most promising (Figure 2).
Figure 2.

Schematic representation of macrophage active targeting by several different ways such as phagocytic receptors mediated endocytosis via formyl peptide receptor, mannose receptor and Fc receptor.
2. Targeting requirements for treating HIV infection
2.1. Intracellular targeting features and mechanisms
The concept of targeted drugs is not new but dates back to 1906 when Ehrlich first postulated the ‘magic bullet’ theory [30]. Ideally, a carrier should target only HIV-1-infected cells. However, targeting HIV infected cells is difficult due to the very short life of infected and virus-replicating CD4+ T cells (half-life ~1–2 days) and the lack of infection markers on latently infected CD4+ cells. The rationale behind nanocarrier-based HIV drug delivery is that the systemic/cellular distribution pattern should be dictated by the properties of the nanocarriers rather than the anti-HIV drugs. Higher drug concentrations and increased residence time at target cells and enhanced viral load reduction can be achieved [31].
Targeting using surface-modified nanocarriers occurs conceptually at two levels, at the organ/tissue and cell levels. At the organ/tissue level, nanocarrier size and surface properties determine which organ/tissue will be targeted by the nanocarriers (i.e., preferentially retained) and how long they will be retained. The dependence of nanocarrier size-flow-organ/tissue filtration is a hallmark of passive targeting. The enhanced permeability and retention (EPR) effect is a good example in cancer tumor targeting [32]. In HIV therapy, the passive targeting to lymph nodes [33] is another example. The most successful targeted drug therapies will ultimately prove to be a combination of passive and active targeting. Once retained in a tissue or organ, a nanocarrier displaying a targeting moiety on its surface would have a much higher specificity than the non cell-targeted nanocarrier because it has been enriched twice, once at the organ/tissue level (i.e., passive targeting) and once at the cellular level (i.e., active targeting).
Cell delivery refers to both cell types and subcellular targets. In most cases, subcellular targets are within the cytosol/nucleus compartment of a cell. Some of the features to be considered for effective intracellular targeting are [34]:
Clearance from the circulation as discussed above.
Release of drugs at non-targeted sites.
Delivery of drug-nanocarrier to the target site.
Release of drugs at the target site.
Removal of drugs from the target.
Elimination of the nanocarrier from the body.
Targeted nanocarrier delivery offers three closely interrelated benefits: (a) the recognition of HIV-infectable target cells and tissues; (b) the ability to reach these sites; and (c) the ability to deliver multiple therapeutic agents [35]. The first two aspects comprise the notion of achieving a preferred, substantially higher concentration of a therapeutic agent at site, a phenomenon that is called ‘localization’ as opposed to the term ‘targeting’ that is often used to identify drugs that provide specific action against a target biological pathway [36]. It should also be noted that the term localization is more often employed to denote intracellular, organelle-specific, site delivery [35]. Small nanocarriers can cross the porous fenestrations present in the endothelial lining of some organ/tissues (e.g., the spleen and the liver). LN targeting from the lymph side through porous lymphatics has a wide size range with an optimum at 100 nm. Penetration into most other tissues through fine capillaries is difficult and usually impractical. By modulating polymer characteristics on the nanocarrier surface, one can control the release of an anti-HIV drug from nanocarriers to achieve desired therapeutic level in target tissue. Surface-modified nanocarriers can be delivered to specific sites by means of conjugation or adsorption of a biospecific ligand. Targeted delivery can thus improve the therapeutic index of anti-HIV drugs by minimizing the toxic effects to healthy (non-viral) tissues/cells. The parameter “intracellular delivery index”, the ratio of intracellular delivery to the extracellulary delivered drug (on mass basis), provides a suitable measure for assessing the effectiveness of intracellular drug delivery [35]. It largely depends on the extent of circulation time of the nanocarrier system in the central compartment (e.g., blood-lymph in HIV therapy), release rate, and rate of uptake of nanocarrier system (by internalization). The intracellular delivery of anti-HIV drugs may involve both the extracellular drug release at the interstitium (tissue site) followed by the intracellular delivery upon the internalization of the surface-modified nanocarrier.
A cell-targeted nanocarrier is likely to be taken up by endocytosis (Figure 3). Topologically, the lumen of an endosome is equivalent to the extracellular space. This entails an endosomal escape step. Fortunately, most current HIV drugs are hydrophobic and can diffuse through endosomal membrane. For these drugs, the only trick in nanocarrier design for endosomal escape is luminal release. For those hydrophilic HIV drugs in the pipeline, a more elaborate endosomal escape strategy would be required [37]. In the case of M/M, regardless of cell targeting, nanocarriers may be taken up by phagocytosis if the nanocarriers have a certain size and surface properties.
Figure 3.

Processes leading to intracellular delivery of drug. A. Passive diffusion of free drug. B. Non-specific phagocytosis of a nanocarrier. C. Drug entrapped in fluid and uptake by pinocytosis. D. Receptor-mediated endocytosis. Nanocarriers bypass multidrug-resistant transporters that may efflux drug entering freely through the plasma membrane [38].
Targeted drug delivery to T-cell and macrophages would improve the efficacy of antiviral drugs, limit toxicities/adverse effects, reduce HIV resistance frequency, reduce viral production in macrophages, abort infection in uninfected cells, and abort viral replication in latent cells upon activation. The specificity of intracellular delivery is related to many factors including the type and number of targeting ligands required for optimal cellular uptake. Intracellular disposition and fate are highly dependent on the type of cell surface receptor and may require an additional strategy to promote endosomal escape [39]. These specific interactions result in preferential accumulation of nanocarrier systems into cellular targets [40].
2.2. Benefits of HIV drug delivery to lymph nodes (LNs)
CD4+ T cells continuously circulate between the blood and LNs. At any given moment, only ~2% of CD4+ T cells are present in the blood circulation, while the remainder are in peripheral lymphoid tissues, especially in LNs. LNs concentrate both T cells and antigen-presenting DCs, facilitate T cell activation and differentiation. Thus, in HIV infection, LNs are an important induction site and HIV-1 replication site. HIV drug delivery to this site can have disproportionally larger effect on controlling disease than to other sites. LNs also harbor a large number of macrophages to destroy foreign microbial invaders draining from infected tissues and to clear dead immune cells after an adaptive immune response. Short-live monocytes continuously emigrated from bone marrow and enter various tissues, including LNs, where they differentiate into macrophages. Therefore as described below, four groups have used macrophages to target RES, particularly LNs, all having achieved impressive efficacy in disease models. Three of the four cases discussed in this review delivered HIV drugs. Drug-loaded nanocarriers or cells are engulfed by macrophages; the macrophages then migrate into LNs or additionally to other RES tissues. In all four cases, drugs slowly diffuse out of macrophages in days or weeks, resulting in sustained high drug concentration in LN or other RES tissue. In all cases, administration of nanocarriers or cells is infrequent, total amount of drugs delivered is very low and so is the toxicity, yet the efficacy is high, all due to the important role of LNs in respective diseases. Two of the four cases used cells as delivery carriers, which can lend their surface modification and LN targeting to HIV nanocarrier drug delivery.
A LN has both blood and lymph supplies. One way to deliver HIV drug-encapsulated liposomes to LNs is from the lymph side by subcutaneous injection into an area close to the thoracic duct. Recent data in macaques is shown in Table 1 [33, 41]. This allowed delivery to many LNs throughout the body, avoiding concentrated entrapment in a few local LNs. Without surface targeting modification and purely relying on the optimal 100 nm size for entering lymphatics, the authors achieved up to 23-fold higher IDV concentrations than free IDV in both peripheral and visceral LNs in a non-human primate SIV model. LN delivery resulted in significant CD4+ T cell count rebound and reduction in viral RNA level.
Table 1.
IDV concentrations in selected lymph nodes of HIV-2287-infected juvenile macaques after subcutaneous administration of 10 mg/kg IDV encapsulated liposomes [41]
| Animal ID | Lymph Node | Time (h) | Lymph Node (IDV)(ng/ml) | Plasma (IDV)(ng/ml) | Lymph Node*/Plasma |
|---|---|---|---|---|---|
| Inguinal | 6 | 1004.3 | 44.2 | 22.7 | |
| Inguinal | 28 | 109.2 | 31.8 | 3.4 | |
| M98165 | Mesenteric | 28 | 158.8 | 31.8 | 5.0 |
| Ileocecal | 28 | 1035.2 | 31.8 | 32.5 | |
| Axillary | 28 | 78.3 | 31.8 | 2.5 | |
| Inguinal | 24 | 147.6 | 20.7 | 7.1 | |
| Inguinal | 26 | 130.4 | 20.8 | 6.3 | |
| J98328 | Mesenteric | 26 | 338.3 | 20.8 | 16.3 |
| Ileocecal | 26 | 145.8 | 20.8 | 7.0 | |
| Axillary | 26 | 51.6 | 20.8 | 2.5 |
Ratio of IDV concentration in lymph node/plasma
Another group [42] developed an IDV-loaded NP formulation. After 12 h of in vitro phagocytosis of NP-IDV by mouse bone marrow-derived macrophages (BMMs), IV injection of HP-IDV packed BMMs (NP-IDV-BMMs) into mice resulted robust accumulation in lung, liver and spleen (Figure 4). A single IV injection of 20 × 106 NP-IDV-BMMs to HIV-1 challenged humanized mice, revealed reduced numbers of virus-infected cells in plasma, lymph nodes, spleen, liver, and lung, as well as CD4+ T-cell protection.
Figure 4.

NP-IDV tissue distribution and pharmacokinetics. (A) Sections of spleen, liver, and lung from mice at day 5 after transfer of rDHPE-NP-IDV–labeled BMMs were stained for CD11b and examined by fluorescence microscopy. Higher magnification inserts demonstrate the presence of rDHPE-NP-IDV (red) colocalized in the cell cytoplasm of CD11b cells (green). BMMs (yellow) were abundantly present in spleen but were less in liver and lung. (B-E) IDV distribution in targeted tissues and body fluids was assessed in mice treated with a single intravenous dose of (B) IDV sulfate solution, (C) cell-free NP-IDV, or (D-E) NP-IDV-BMMs. In contrast to IDV concentrations in mice treated with NP-IDV-BMMs, nadirs within 6 hours after treatment in mice treated with IDV sulfate solution or NP-IDV, levels were prolonged in tissues and plasma over 14 days in mice treated with NP-IDV-BMMs. Data represent mean ± SEM for 4 mice/group per time point. Magnifications are (originals) × 100 and (insets) × 400 [42].
A third group [43] used autologous, HIV drug ddCTP-encapsulated red blood cells to target macrophages. The drug-loaded erythrocyte membranes were modified using artificial ageing to enhance macrophage phagocytosis. In a feline immunodeficiency animal model, ddCTP-loaded erythrocytes were able to reduce FIV production by macrophages in naturally or experimentally infected cats. Furthermore, the administration of ddCTP-loaded erythrocytes protected the majority of peritoneal macrophages during a 7-month experimental FIV infection and reduced the percentage of circulating lymphocytes stained by an anti-p24 antibody. Lastly, another strategy uses yeast ghost cells to deliver anti-inflammatory short interfering RNA (siRNA) to macrophages [44]. Yeast ghost cells were made in a way that the cell surface was left with only beta1 3-D-glucan, for which macrophage has a special receptor. The ghost cells can be efficiently absorbed orally through M-cells and, once crossed M-cells, avidly phagocytosed by macrophages in the Peyer’s Patches. Interestingly, macrophages in the Peyer’s Patches migrate into blood circulation and settle at various LNs. Oral gavage of mice with the ghost cells containing as little as 20 μg/kg of siRNA directed against tumour necrosis factor alpha (TNF-α) depleted its messenger RNA in macrophages recovered from the peritoneum, spleen, liver and lung, and lowered serum TNF-α levels.
3. Surface-modified nanocarriers for effective intracellular delivery
Various types of nanocarriers are being developed for anti-HIV drug delivery applications. HIV-1 enters a new host through a mucosal barrier. It is then passed locally from one cell to another through infection in the tissues (notably in lymph nodes and mucosal lamella propria) or spread through blood circulation as free virus or inside infected CD4+ cells. Apart from having a short half-life as a free virus, HIV-1 does not face the challenges a drug nanocarrier faces in transit through the body before reach its target cells. A drug nanocarrier usually is given IV or orally and thus must spend some time in blood circulation. The nanocarrier faces several challenges while in the circulation, including maintaining adequate bioavailabilty and biostability and avoiding clearance by the kidney or the reticular endothelial system (RES, mainly phagocytes in the spleen and the liver). Strategies to meet these challenges have been extensively reviewed [37] including the effect of size and the use of Pegylation [30, 45]. Many anti-HIV drugs can bind to plasma components (principally human serum albumin, HSA) or within other compartments of the tissue, greatly influence the transport and elimination in individual organs and the overall pharmacokinetics. The design of the anti-HIV nanocarrier system needs to eliminate (or minimize) all nonspecific bindings [46].
3.1. Nanocarrier-based deliverables and ‘surface modifiers’
The anti-HIV targeted nanocarriers that have been studied are liposomes, nanoparticles, dendrimers and bioconjugates. The various surface modifiers used in targeted nanocarrier-based delivery systems for HIV therapy are shown in Figure 5 and discussed below:
Figure 5.

Schematic representation of different surface-modified nanocarrier deliverables (liposomes, nanoparticles, dendrimers and bioconjugates – modified and redrawn) [47].
3.1.1. Liposomes
Liposomes as drug delivery vehicles were first proposed by Gregoriadis [47]. They are usually small (80 and 100 nm), encapsulating drugs using a variety of loading methods [48, 49].
Liposome surface can be modified to improve its properties [50, 51]. The most noteworthy modification is the incorporation of the hydrophilic polymer polyethylene glycol (PEG) to prevent interactions with plasma proteins, thus retarding recognition and removal by the RES.
3.1.2. Nanoparticles
Nanoparticles are solid colloidal particles ranging in size from 10 to 1000 nm (1 μm), usually containing no lipids as a major component material. Drug is dissolved, entrapped, encapsulated and/or to which the drug is adsorbed or attached [52–55]. They can be internalized by lymphocytes because of their small size, acting as intracellular drug reservoirs [47]. As with liposomes, opsonization (adsorption to plasma proteins) of polymeric nanoparticles can be prevented by providing a surface of the particles with a hydrophilic group. These are known as stealth nanoparticles [56]. Being less than 1 μm, nanoparticles can be administered by intravenous injection (the diameter of the smallest blood capillaries is 4 μm), as well as intramuscular and subcutaneous injection that minimizes any possible embolism [57–59]. As with all colloidal carriers discussed in this review, nanoparticle size must be less than 200 nm to avoid uptake by the RES and its surface should be hydrophilic to avoid clearance by macrophages [60].
3.1.3. Dendrimers
Dendrimers are a class of monodisperse polymers distinguished by their repeated branching structure emanating from a central core [61]. It has been used to carry anti-HIV drugs [62–64]. Narrow molecular weight distribution, small size (less than 100 nm), and easy incorporation of targeting ligand are attractive features.
3.1.4. Bioconjugates
Polymer bioconjugates have also been used for HIV drug delivery [65]. Surface targeting modifiers specific for macrophages include the chemo-attractant peptide, N-formyl-methione-leucine-phenylalanine (fMLF) and mannose. Biodegradation inside the target cells has been conceptually attempted in this type of nanocarrier [66]. Tat cell penetrating peptide has been used as surface modifier to facilitate endosomal escape [39].
3.2. Examples of nanocarrier-based deliverables and 'surface modifiers'
3.2.1. Liposomes
Lipid composition and charge can affect the properties of a drug-encapsulated liposome. The intracellular stability of 2′,3′-dideoxycytidine (ddC) encapsulated in different liposomal formulations has been evaluated as a function of drug concentration in RAW 264.7 macrophages. An enhanced uptake of liposomal-encapsulated ddC by macrophages was observed when the liposomal drug concentration was increased. In addition, the anionic character of liposomes, due to the presence of cholesterol in the lipid composition of liposomes, seemed to be an important factor to obtain a high intracellular uptake of ddC. Liposomes having long saturated fatty acyl phospholipids and containing 50% (molar ratio) of cholesterol displayed the best stability in the intra-macrophagic compartments at all liposomal ddC concentrations used. On the other hand, although the leakage of ddC from liposomes sterically stabilized with polyethylene glycol chains was similar to that of other cholesterol-containing liposomes, the anti-HIV drug was readily released from cells for all concentrations of liposomal ddC tested. These results suggest that some lipid components such as cholesterol can modulate the liposomal stability of drugs such as ddC in response to the conditions of the environment, the properties of the drug used and the nature of interactions between liposomes and cells [67]. In another study, the effect of lipid used, cholesterol concentration and the presence of surface charge on the liposome bilayer, encapsulated with tritium labeled stavudine (d4T), on the cellular uptake by M/M was investigated. d4T uptake was found to be maximum (approximately 950 ng/million cells) in liposomes containing dipalmitoyl phosphatidylcholine (DPPC). The presence of sphingomyelin (which increases bilayer rigidity) decreased cellular uptake of d4T and the presence of negative charge on the bilayer enhanced the uptake of d4T compared to positive charge. There was no apparent difference in uptake when varying amounts of cholesterol was added in liposomal formulations. This study shows that the sensitivity of macrophages to different charge and lipid type can be used to either decrease or increase cellular uptake as desired [68]. Thus, lipid composition has been shown to affect the stability of ddC-encapsulated liposomes [67] and liposome composition and charge to affect uptake of d4T-encapsulated liposomes into macrophage-like cells [68]. These results confirm the generally accepted rule that cargo drugs do not change the fate of nanocarriers in vivo.
Soluble CD4 [69] as well as CD4-derived peptide [70] have been used to surface-modify liposomes to improve cellular delivery to HIV-1-infected cells. In one study, N-butyldeoxynojirimycin (NB-DNJ), an inhibitor of HIV gp 120 folding, is encapsulated into soluble CD4-surface-modified liposome to target PBMCs. The improvement is based on targeting viral gp120/41 complex expressed on HIV-1 infected cells. Low pH sensitivity has also been engineered into the liposomes to facilitate endosomal escape. A 5-fold increase in uptake into HIV-infected cells compared with uninfected cells was observed [69]. In another study, a synthetic peptide from the complementarily determining region 2 (CDR-2)-like domain of CD4 could bind specifically to HIV-infected cells and mediate the binding of peptide-coupled liposomes to these cells. A peptide from the CDR-3 like domain of CD4 inhibited HIV-induced syncytia formation, but failed to target liposomes to infected cells. This apparent discrepancy may be due to the requirement for a conformational change in the CD4 receptor for the CDR-3 region to interact with the HIV envelope protein. These results demonstrate the feasibility of using synthetic peptides to target liposomes containing antiviral drugs to HIV-infected cells [70].
Low density lipoprotein (LDL) has been used both as a targeting moiety and drug carrier to target macrophages [17, 71]. LDL is about 25.5 nm in size in normal people [72]. In these studies, LDL is conjugated with AZT and the drug-loaded LDL may be considered a quasi-drug modified liposome. LDL is recognized by scavenger receptors specifically expressed on macrophages and taken into cells by endocytosis [71]. A 10-fold increase in cellular uptake of AZT delivered by LDL-modified liposome as compared to free AZT was observed [17]. Compared to antibody-modification presented below, the specificity of targeting with these non-antibody agent-modifications is modest.
A mouse antibody has been attached to didanosine triphosphate (ddITP)-loaded liposomes and dideoxycytidine triphosphate (ddCTP)-loaded liposomes to target human macrophages in two different studies [73, 74]. Targeting is based on indirect recognition of the mouse antibody by Fc receptor specifically expressed on human M/M. Uptake of ddCTP -loaded liposomal antibody was 4–6 times greater than that of free liposomes [73]. Uptake of ddITP-loaded liposomal antibody was 80 times higher than free ddITP at 48 h [74].
RES, also known as mononuclear phagocyte system (MPS), includes macrophage-rich organs, such as the spleen, LNs, bone marrow, and the liver (Kupffer cells in liver are macrophage-like phagocytes but lack immune function). All of these organs can be infected by HIV-1, but the LNs are especially important in HIV-1 disease due to the fact that they are a hub in lymphocyte trafficking, antigen-interaction and activation. It has been reported that surface charge-modified, liposome-like emulsomes [75] and ddC-loaded liposome without surface targeting modification [76] can be enriched in rat liver and rapidly phagocytosed by macrophages, confirming the known liposome RES clearance mechanism that is based on macrophage phagocytosis. On the other hand, an attempt was made to exploit the macrophage phagocytosis in RES to deliver HIV drug ddCTP into spleen and bone marrow [77]. Anti-HLA-DR immunoliposomes were evaluated for targeting LNs while avoiding other RES organs/tissues. The HLA-DR determinant of major histocompatibility complex class II (MHC II) is highly expressed on FDC, macrophages, and activated CD4+ T cells. Therefore, HLA-DR appears to be a good HIV drug delivery target. The immunoliposomes were very efficient in delivering high concentrations of indinavir (IDV) to lymphoid tissues for at least 15 days post a single subcutaneous injection to C3H mice. There was by up to 126 times more than free drug in IDV accumulation in lymph nodes with no significant damage to liver and spleen [78]. Another study exploited a different RES marker, the mannose receptor, as a target wherein d4T-encapsulated liposomes were surface modified with mannose. The authors demonstrated targeting specificity and anti HIV-1 activity in vitro using MT2 cells and in vivo in rats. The results showed reduced side effects and higher drug accumulation in the liver, spleen and the lungs compared to free drug or non-mannose-modified liposomes [79]. Similarly, galactose targeting modification on liposomes resulted in preferential liver accumulation [80, 81]. These studies demonstrate the superior targeting specificity of antibodies over non-antibody targeting ligands. However, the use of antibodies presents other technical challenges and much higher cost as compared to simpler ligands.
Table 2 summarizes the various surface modifiers used in liposomes for targeting purposes.
Table 2.
Summary of surface modifications of liposome-based nanocarriers investigated for targeting anti-HIV drugs to different cells/tissues/organs
| Anti-HIV drugs | Surface modifications | Cells/tissues/organs | Reference |
|---|---|---|---|
| ddC | Induction of charge in lipid composition of liposomes | RAW 264.7 macrophage- like cells | [67] |
| d4T | Presence of charge on the liposome bilayer. Stearylamine or dicetylphosphate used for induction of positive or negative charge on the bilayer | U937 M/M cells | [68] |
| NB-DNJ | Soluble recombinant CD4 molecule and IgG antibody conjugated separately to liposomes | PBMCs | [69] |
| No drug | CD4-derived peptide coupled to liposome | CD4+ Sup-T1, H9 and chronically infected H9/111B cells | [70] |
| AZT-prodrug | Acetylation of LDL | J7774.A and U937 macrophage-like cells | [17] |
| FLT and AZT | Covalent coupling of AZT and FLT to LDL | HIV infected human macrophages and Molt 4/8 cells (CD4+ T cell line) | [71] |
| ddCTP | Antibody attached to liposome surface | Human M/M cells | [73] |
| ddITP | Antibody attached to liposome surface | Human M/M cells | [74] |
| AZT | Induction of cations by sterylamine on liposome-like emulsomes | Liver cells | [75] |
| ddCTP | Induction of charge on liposome surface | Cells of MPS | [77] |
| IDV | Fab′ antibody fragment conjugated to immunoliposomes | LNs | [78] |
| d4T | Mannose conjugated to liposome surface | MT-2 (HTLV-1- transformed human lymphoid) cell line and tissue distribution following IV in rats | [79] |
| AZTP | Galactose conjugated to liposome surface | Organs distribution following IV via tail vein in mice | [80] |
| d4T | Galactose conjugated to liposome surface | Liver parenchymal cells harvested from male albino rats | [81] |
3.2.2. Nanoparticles
Nanoparticle (NP) surface modification can affect their properties related to treatment of HIV-1 infection. In a study, poly(ethylene oxide)-modified poly(epsilon-caprolactone) (PEO-PCL) NP system as an intracellular delivery vehicle for saquinavir (SQV) was developed. Cellular uptake and distribution of the NP was examined in THP-1 human M/M cell line. The PEO-PCL NPs had a smooth surface and spherical shape and showed a relatively uniform size distribution with a mean particle diameter of ~200 nm and a zeta potential of ~ −25 mV. The surface size and charge are typical of most reported NPs. A rapid cellular uptake of the fluorescent probe (rhodamine-123) encapsulated in PEO-PCL NPs was observed in THP-1 cells. Intracellular SQV concentrations were significantly higher than free SQV [82]. The effect of polymer type of AZT-loaded polylactic acid (PLA) and PLA-PEG blend NPs on phagocytosis in rat poly-morphonuclear leucocytes (PMNL) was studied. NP size (about 300 nm) and zeta potential (~ −7 to −20 mV) were not important. The PLA nanoparticles were more efficiently phagocytosed than PLA/PEG blends primarily depending on the PEG and its ratio in the blend. The blend with the highest PEG proportion did not prevent phagocytosis, indicating that the steric effect of PEG was concentration dependent [83]. The effect of polymer architecture of Pegylated NPs on NP properties was evaluated in RAW 264.7 macrophage-like cells. NP size (about 185 nm) and zeta potential (~−3 to −23 mV) did not vary significantly between different architecture. The grafted copolymer NPs reduced macrophage uptake as compared to multi block copolymer, although mechanisms different than phagocytosis were involved. Thus, polymer architecture is an important feature for determining the surface properties and hence, protein binding and cellular interactions of NPs [84]. The effect of NP shape/size on cellular uptake was investigated. A spherical and two cylindrical NPs, whose lengths were distinctively varied, were constructed by the selective cross-linking of amphiphilic block copolymer micelles. When the NPs were modified with Tat cell penetrating peptide (CPP, also known as protein transduction domain), the smaller, spherical NPs had a higher rate of cell entry into Chinese hamster ovary (CHO) cells than did the larger, cylindrical NPs. NPs were released after internalization and the rate of cell exit was dependent on both the NP shape and the amount of surface-bound CPP [85].
Since macrophages efficiently phagocytose foreign particles, this has become a major element of macrophage targeting strategies. For example, human M/M uptake of poloxomer surface-modified (Pluronic F68 and F108) AZT loaded NP was studied. The influence of size, composition, concentration, and surface of the NP as well as macrophage activation state on phagocytosis were tested. NP made of polyhexylcyanoacrylate (PHCA) or HSA with a diameter of about 200 nm were found most useful for specific delivery to macrophages. Macrophages isolated from HIV-infected patients also show good incorporation of NP [86]. Additional targeted moieties have been added to NP to enhance drug delivery to macrophages. Two studies used mannan and mannose as the targeting modifiers [87, 88]. Mannan-coated NPs for macrophage targeting of didanosine (ddI) was investigated. Results of the ex-vivo cellular uptake study indicated 5-fold higher uptake of ddI from the mannan-coated NP formulation by the macrophages in comparison with free drug. Results of the quantitative biodistribution study showed 1.7, 12.6 and 12.4 times higher localization of ddI in the spleen, LNs and brain, respectively, after administration of mannan-coated NPs compared to that after injection of ddI solution in buffer. Thus, mannan-coated NPs appeared to successfully target ddI to macrophages by exploiting mannosyl receptor mediated endocytosis [87]. Mannosylated gelatin NPs (MN-G-NPs) for the selective delivery of ddI to macrophage tissues was investigated. Cellular uptake by MN-G-NPs was 2.7 times higher as compared to the non-targeted G-NPs. Intravenous administration of free-drug solution resulted in a high serum drug concentrations whereas serum concentrations were much lower for G-NPs suggesting targeted delivery and local release of drug. Coupling of the NPs with mannose significantly enhanced the lung, liver, and lymph nodes uptake of drug, which is reflected in the recovery of a higher percentage of the dose from these organs following administration of MN-G-NPs in comparison to non-coupled G-NPs or free drug [88].
NP oral absorption has been studied for years. The mechanism of absorption is not entirely clear but uptake through M-cells seems to play a dominant role. The specific targeting of the anti-HIV drug AZT to rat RES cells by the oral route was investigated [89]. 14C-labelled AZT was bound to hexylcyanoacrylate NPs formed by emulsion/polymerization. The NPs were 230 ± 20 nm in size and had a zeta potential of −51.6 ± 3 mV. The radioactivity in several organs including those containing large numbers of macrophages was measured after peroral administration of the nanoparticle preparation and compared to a 14C-AZT control solution containing the same components without the nanoparticles. The area under the curve (AUC) of 14C-AZT in the liver after oral administration of the NPs was 30% higher than that after oral administration of the free 14C-labelled AZT solution [89]. In the case of IV delivery of AZT to RES, AZT concentrations were up to 18 times higher in rat organs belonging to the RES when the drug was bound to nanoparticles compared with unbound AZT [90]. NP delivery of AZT to the intestinal epithelium and gut-associated lymphoid tissues has been attempted with promising results. Unlike the solution, nanoparticles concentrated very efficiently in rat intestinal mucosa, as well as in the Peyer’s patches. Additionally, the release of free AZT could be controlled. AZT concentration in Peyer’s patches was 4 times higher for nanoparticles, compared to the control solution. The tissue concentration was 30–45 μM, which was much higher than the reported IC50 of AZT (0.06–1.36 μM) and was regularly distributed along the gastrointestinal tract. One disappointing aspect of this study was the very low AZT concentration in the lymph since the draining mesenteric LNs are an important induction site in HIV-1 infection [91].
NPs have also been used to deliver DNA vaccines. This study was aimed to induce immunity to HIV-1 Tat protein. Poly(methylmethacrylate)-based polymeric NPs efficiently adsorbed plasmid DNA encoding HIV-1 Tat, mainly through electrostatic interactions. The NPs showed no in vitro or in vivo toxicity. Two intramuscular immunizations followed by two Tat protein boosts resulted in both antibody and cell-mediated immune response [92].
A separate review in this theme issue surveys HIV drug delivery to the CNS. However, surface modifications of NPs carried out in two studies for brain delivery are mentioned. Thiamine was used as the targeting modifier [93] and Tat CPP was used as a cell penetration modifier [94]. Enhancement across blood brain barrier (BBB) was observed in both cases.
Table 3 summarizes the various surface modifiers used in nanoparticles for targeting purposes.
Table 3.
Summary of surface modifications of nanoparticle (NP)-based nanocarriers investigated for targeting anti-HIV drugs to different cells/tissues/organs
| Anti-HIV drugs | Surface modifications | Cells/tissues/organs | Reference |
|---|---|---|---|
| SQV | PEO coated on PCL- NP surface | M/M cells | [82] |
| AZT | PEG coated on PLA- NP surface | PMNL | [83] |
| No drug | PEG coated on PLA- NP surface | RAW 264.7 Macrophage-like cells | [84] |
| Tat* | HIV Tat PTD | CHO cells | [85] |
| No drug | Poloxamers (Pluronic F68 and Pluronic F108) coated on PBCA-NP surface | M/M cells | [86] |
| ddI | Mannan coated on NP surface | (a)Ex-vivo with heparinized human blood (b)Tissue distribution following SC and oral administration in rats | [87] |
| ddI | Mannose conjugated to NP surface | (a)Macrophage-like cells (b)Organ distribution following IV administration | [88] |
| Plasmid DNA encoding HIV-1 Tat | Covalently linking PEG chains to the NP core | (a)HeLa cells (b)Blood and organ distribution following bilateral intramuscular injection in mice | [92] |
| No drug | Thiamine ligand covalently linked to NP surface via a PEG spacer | brain following in-situ perfusion | [93] |
| RTV | Tat conjugated to NP surface | (a)MDCK-MDRI cells and MDCK-wt cells (b)Biodistribution following injection via tail vein in mice | [94] |
Behaves both as an anti-HIV drug and a cell penetrating peptide
3.2.3. Dendrimers
Surface modified fifth generation poly(propyleneimine) (PPI) dendrimer-based nanocarriers were constructed for targeting lamivudine (3TC) and efavirenz (EFV) to MT-2 cells and M/M cells, respectively. Mannose was used as the surface-targeting modifier. The results of a ligand agglutination assay indicated that even after conjugation with PPI, mannose displayed binding specificity towards Con A (a well-investigated lectin, known to bind specifically with saccharides such as mannose). Significant increase in uptake of 3TC by MT-2 cells was observed when MPPI (mannosylated PPI) was used, which was 21 and 8.3 times higher than that of free drug (p<0.001) and PPI (p<0.001) at 48 h respectively. Significantly higher anti-HIV activity of PPI and MPPI was observed and this was due to the enhanced cellular uptake of 3TC in formulation as compared to that of free drug [95]. The cellular uptake of EFV was found to be both concentration and time dependent. Significant increase in cellular uptake of EFV by M/M cells were observed in case of mannose conjugated dendrimer which is 12 times higher than that of free drug and 5.5 times higher than that of t-Boc-glycine conjugated PPI dendrimer. While mannose conjugated dendrimer was taken up by the lectin receptors of the cells, phagocytosis of the t-Boc-glycine conjugated dendrimer might be responsible for its enhanced uptake. Interestingly, the cytotoxicity of the PPI dendrimer was found negligible when these dendrimers were surface-modified with t-Boc-glycine or mannose. Results suggest that the proposed dendrimer-based nanocarriers hold potential to increase the efficacy and reduce the toxicity of antiretroviral therapy [96].
Table 4 summarizes the various surface modifiers used in dendrimers for targeting purposes.
Table 4.
Summary of surface modifications of dendrimer-based nanocarriers investigated for targeting anti-HIV drugs to different cells/tissues/organs
3.2.4. Bioconjugates
Bioconjugate-based targeted anti-HIV drug delivery nanocarriers have been studied. The drug-polymer bioconjugate-based nanocarriers can significantly influence HIV therapy by specific localization to viral reservoir sites. Bioconjugates prepared from different combinations of the approved drug, SQV, the cell penetrating peptide retro-inverso-cysteine-lysine-Tat nonapeptide (R.I.CK-Tat9), the polymeric carrier, PEG, and the cell uptake enhancer, biotin, were studied. The anti-HIV-1 activity ED50 of the PEG3.4k-based-SQV nanocarriers was evaluated in HIV-1-infected MT-2 cells. The activity of conjugated SQV was reduced (ED50 = 900 nM) when SQV was conjugated to PEG3.4k alone, but restored with the addition of biotin (ED50 = 125 nM), R.I.CK-Tat9 (ED50 = 15 nM), and R.I.CK(stearate)-Tat9 (ED50 = 62 nM) as compared to maximum achievable antiviral efficacy (unconjugated SQV, control, ED50 = 15 nM), suggesting enhanced cellular uptake of conjugates [97]. However, the mechanism of antiviral enhancement for the SQV-PEG3.4k-R.I.CK-Tat9 bioconjugate-based nanocarrier was not clearly established since, in addition to targeting intracellular TAR, Tat possesses cell penetrating properties that may promote conjugate uptake into the cell and/or it may exert anti-HIV-1 activity by means of cell surface binding to CXCR4 receptors [97, 98]. In separate experiments, confocal microscopy studies showed that in cells incubated with R.I.CK(fluorescein)-Tat9 (Tat alone) or PEG3.4k-R.I.CK(fluorescein)-Tat9 (PEG-Tat; 1 copy Tat), there was significantly higher fluorescence intracellularly than on the cell surface. On the other hand, cells incubated with PEG10k-[R.I.CK(fluorescein)-Tat9]8 (PEG-Tat; 8 copy Tat) showed primarily cell surface bound fluorescence. This result is consistent with the results from flow cytometry (Figure 6) which concludes that the majority of R.I.CK-Tat9 is within cells, the majority of PEG10k-(R.I.CK-Tat9)8 is on the cell surface, and PEG3.4k-R.I.CK-Tat9 is roughly equally distributed between cell surface and intracellular locales. Results further indicate that R.I.CK-Tat9 and PEG3.4k-R.I.CK-Tat9 bioconjugates are all predominantly within endosomes. Thus, from different studies, variations in intracellular uptake and intracellular localization, as well as synergistic inhibitory effects of SQV and Tat peptides, contributed to the unexpected and substantial differences in antiviral activity. The experimental results demonstrate that highly potent nanoscale multi-drug conjugates with low non-specific toxicity can be produced by combining moieties with anti-HIV-1 agents for different targets onto macromolecules having improved delivery properties [97, 98].
Figure 6.

Quantitative data from flow cytometry showing the intracellular fluorescence and cell surface bound fluorescence of PEG10k-(fluorescein)8 (no Tat), R.I.CK(fluorescein)-Tat9 (Tat alone), PEG3.4k-R.I.CK(fluorescein)-Tat9 (PEG-Tat, 1 copy Tat) and PEG10k-[R.I.CK(fluorescein)-Tat9]8 (PEG-Tat, 8 copy Tat) after 24 h incubation with MT-2 cells at 37°C. The total cell associated fluorescence was quantified by flow cytometry. The intracellular fluorescence was measured after the cell surface-bound fluorescence was quenched by 0.2 mg/ml trypan blue at pH 5.8. The cell surface bound fluorescence is the difference between the total cell associated fluorescence and the intracellular fluorescence [98].
Bioconjugates surface-modified with fMLF targeting agent were developed for targeting macrophages in anti-HIV therapy [99–101]. fMLF peptide is a known chemo-attractant and can specifically bind to surface receptors on phagocytic cells, such as macrophages and neutrophils. The enhancement in avidity for the targeted receptor on phagocytic cells by multivalent binding of fMLF was demonstrated. A single copy of fMLF covalently linked to a PEG-based nanocarrier displayed reduced binding avidity (Kd = 190 nM) for neutrophil-like differentiated HL-60 cells relative to free fMLF (Kd = 28 nM). Increasing the number of fMLF residues (up to eight) attached to a single PEG polymer results in enhanced avidity for these cells (Kd = 0.18 nM), which appeared to be independent of whether the polymer backbone was linear or branched. Thus, a concentration of only 0.18 nM B8-PEG-fMLF8 (8-arm branched PEG carrying 8 copies of fMLF) was needed to get 50% occupancy of the differentiated HL-60 cell surface fMLF receptors, as opposed to 190 nM for B8-PEG-fMLF1.1 (8-arm branched PEG carrying 1 copy of fMLF), with only one appended ligand, an increase in avidity of more than 1000-fold (Table 5). Since a single fMLF moiety bound to a branched PEG polymer shows less avidity for differentiated HL-60 cells than does the free peptide, it seems likely that the bulky carrier group sterically interferes with ligand interaction with its cellular receptor, but this interference is overcome by fMLF peptide multivalency on the PEG polymer. In addition, no conjugate showed enhanced activation of phagocytic cells, relative to the free peptide (EC50 = 5 nM), as measured by transient stimulation of release of calcium ions from intracellular stores into the cytoplasm. The results suggest that the PEG nanocarriers with multiple fMLF moieties are capable of binding to and/or entering cells in culture and in vivo. However, this study did not fully define the possible mechanism of internalization and used the reporter compound digoxigenin (DIG) as a surrogate for an HIV-1 drug cargo. A polymer bearing four fMLF and four digoxigenin residues showed specific enhancement in binding to differentiated HL-60 cells and mouse peritoneal macrophages in situ relative to a polymer lacking fMLF; no such enhancement was seen in binding to receptor-negative lymphocytic Jurkat cells. These results suggest that multiple fMLF residues linked to a drug-delivery PEG nanocarrier can be used to target anti-HIV drugs to phagocytic cells with relatively little toxicity due to cellular activation [99].
Table 5.
Binding and cell activation by fMLF-PEG bioconjugate-based nanocarriers [99]
| Competitor | Avidity: Kd (nM) | Cell activation: EC50 (nM) | Relative avidity: Kd × no. of fMLF | Avidity/activation: EC50/Kd |
|---|---|---|---|---|
| fMLF | 28 | 4.7 | 28 | 0.168 |
| B8-PEG-fMLF1.1 | 190 | - | 209 | - |
| B3-PEG-fMLF3 | 1.9 | 9.6 | 5.7 | 5.05 |
| B4-PEG-fMLF4 | 0.8 | 5.0 | 3.2 | 6.25 |
| B8-PEG-fMLF5.5 | 0.5 | 5.3 | 2.75 | 10.6 |
| B8-PEG-fMLF8 | 0.18 | 5.0 | 1.44 | 27.8 |
| PEG-aspartate-fMLF4 | 1.1 | - | 4.4 | - |
| PEG-aspartate-fMLF4-DIG4 | 1.2 | 5.9 | 4.8 | 4.92 |
Multiple fMLF peptide functionalities were also attached through multifunctional commercially available PEGs or to novel peptide-based (PB) PEG bioconjugate-based nanocarriers in which the structures were varied by appending PEGs with average molecular weights of 5, 20 and 40 kDa. The uptake of these targeted bioconjugate-based nanocarriers by macrophage-like differentiated U937cells was evaluated. Studies showed that increasing the number of fMLF residues attached to a single PEG polymer from one to two resulted in significantly enhanced uptake (~ 4-fold, p < 0.05) in U937 cells, while further increasing this number from two to four only resulted in a modest increase in uptake. While the molecular basis of enhanced avidity by multiplicity (i.e., multiple ligands attached to a single polymeric carrier molecule) is not proven, it is likely due to multimeric ligands interacting with multiple cellular receptors. Since typical commercially available multifunctional PEGs limit the numbers of copies of drugs and targeting agents that may be attached simultaneously to a nanocarrier, the alternate PB- PEG carrier (that could potentially carry both drugs and targeting moieties simultaneously) was thereby designed that retained the targeting and uptake properties of non-PB commercial PEGs. The results from these studies indicated that the uptake by these nanocarriers was (a) energy dependent and mediated by a fMLF receptor, (b) appending only 2 copies of the targeting ligand to the multifunctional nanocarrier appears sufficient for binding in vitro, and (c) of the three configurations studied, the nanocarrier with a molecular weight of about 20 kDa, corresponding to a size of 20–60 nm, demonstrated the highest uptake [101] (Figures 7 and 8).
Figure 7.

Uptake of fluorescence-labeled PEG-fMLF nanocarriers of different sizes in differentiated U937 cells after 4 h of incubation at 37°C. The means ± SD for three independent experiments are shown for each value. (*Statistically significant difference between the control PEG5K and PEG-fMLF nanocarriers, p < 0.01) [101].
Figure 8.

Uptake of fluorescence-labeled PEG-fMLF nanocarriers derived from commercial PEGs or novel PB-PEGs in differentiated U937 cells after 4 h of incubation at 37°C. The means ± SD for three independent experiments are shown for each value. (* Statistically significant difference between the control PEG5K and the PEG-fMLF nanocarriers, p < 0.01) [101].
These multivalent fMLF PEG nanocarriers were also assessed for their in vivo macrophage targeting potential. Peritoneal macrophage uptake was evaluated in mice 4 h after IP administration of fluorescence-labeled PEG-fMLF nanocarriers and the preferential receptor-mediated association was examined. In addition, the biodistribution and pharmacokinetic profile of constructs was examined in vivo in Sprague-Dawley rats after IV administration of tritiated PEG-fMLF nanocarriers. Attachment of one, two, or four fMLF copies increased uptake in macrophages by 3.8, 11.3, and 23.6 fold, respectively, compared to PEG without fMLF. Results showed that fMLF-modified PEG nanocarriers (5kDa and 20kDa) accumulated to a greater degree into macrophage-rich organs (liver, kidneys and spleen) than the corresponding unmodified PEG. The enhanced uptake of the peptide-PEG bioconjugates in these organs was attributed to a preferential association with macrophages. However, on a molar basis, penetration was equivalent suggesting nanocarrier size and targeting moieties are important determinants. These results demonstrate the feasibility of targeting macrophages and the suitability of using the synthetically versatile PB-PEG nanocarriers. However, these studies also suggest that balancing peripheral tissue penetration (a size dependent phenomenon) versus target cell uptake specificity remains a challenge to overcome (Figure 9) [100].
Figure 9.
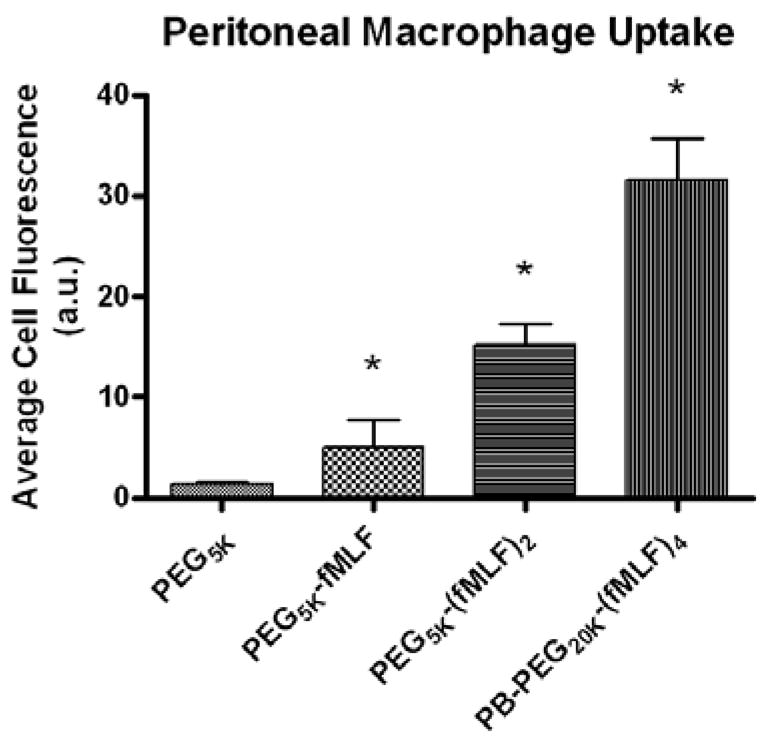
Uptake of fluorescence-labeled PEG-fMLF nanocarriers derived from commercial PEGs or novel PB-PEGs in mice peritoneal macrophages after 4 h of incubation at 37°C. The means ± SD for three independent experiments in mice are shown for each value. (*Statistically significant difference between the control PEG5K and the PEG-fMLF nanocarriers, P<0.05, ** P<0.01) [100].
Recently it was shown that intracellular distribution could be controlled by using a Tat peptide to facilitate endosomal escape [39]. This is particularly important for drugs that are hydrophilic and act in the cytosol or must gain access to the nuclear compartment [66].
A novel polymer bioconjugate scaffold based on the use of a peptide-backbone – PEG scaffold (PB-PEG) was recently reported [66]. The PB-PEG nanocarrier is a polymer bioconjugate type nanocarrier that has multiple attachment sites to which drugs or targeting moieties can be linked. By appending drugs via the bioreversible disulfide bond, which can be designed to varying degrees of susceptibility to cleavage by reducing agents [102], intracellular selective release can be achieved. The plasma circulation time of the carrier can be adjusted, allowing it to reach the targeted cells, before releasing its drug cargo. The multiple attachment sites on the PB-PEG nanocarriers can not only provide a high payload of drug, but also can be used for appending one or more moieties for targeting specific cellular receptors or promoting cell uptake of the conjugate. A biodegradable multimeric PB-PEG nanocarrier (Figure 10) that is relatively stable in the blood circulation, while being selectively degraded inside target cells, was designed in order to exploit the natural extracellular-intracellular gradient of reducing conditions. This multifunctional versatile design is sufficiently flexible so that the component peptidic monomers that build up the multimeric nanocarrier could be used to link PEGs, imaging agents, anti-HIV drugs, targeting ligands or other peptidic cores (Figure 10). Two important design components, the biodegradable bonds between monomeric peptidic core units and using PEGs of the appropriate size in order to promote renal or hepatic elimination, allows for the pre-programming of body elimination properties into the nanocarrier. The nanocarrier biodegradation was evaluated in simulated intracellular and extracellular/blood environments using 3 mM and 10 μM glutathione in buffer, respectively. The multimeric nanocarrier was 9-fold more stable in the extracellular environment. The results suggest selective intracellular degradation of the nanocarrier into components with known body elimination pathways [66].
Figure 10.

Schematic representation of a monodisperse biodegradable dimeric nanocarrier composed of peptidic backbone irreversibly or reversibly conjugated with one or more targeting ligand/drug either directly or through the distal ends of PEG [66].
Table 6 summarizes the various surface modifiers used in bioconjugates for targeting purposes.
Table 6.
Summary of surface modifications of bioconjugate-based nanocarriers investigated for targeting anti-HIV drugs to different cells/tissues/organs
| Anti-HIV drugs | Surface modifications | Cells/tissues/organs | Reference(s) |
|---|---|---|---|
| SQV, Tat peptide | Conjugation of (a) SQV to PEG, (b)SQV to PEG-biotin, (c) SQV-Tat to PEG, (d) SQV-Tat-stearate to PEG, (e)biotin to Tat, (f)Tat to PEG and (g)Tat-biotin to PEG | HIV-1 infected MT-2 cells | [97, 98] |
| No drug | Conjugation of (a)various copies of fMLF to PEG (different shapes and sizes) and (b)four copy fMLF with four copy digoxigenin to PEG | Differentiated HL-60 cells (expressing formyl peptide receptors) and mouse peritoneal macrophage cells | [99] |
| No drug | Conjugation of (a)various copies of fMLF to PEG (different shapes and sizes) and (b)various copies of fMLF to peptide-based PEG (different sizes) | (a)Differentiated U937 macrophage-like cells (expressing formyl peptide receptors) and (b)tissue distribution following IP administration in mice | [100, 101] |
4. Conclusion
Insufficient concentrations and very short residence time of the anti-HIV drugs at the cellular and anatomical sites are among major factors that contribute to the failure of eradicating HIV from reservoirs and the development of multi-drug resistance against these anti-HIV drugs. The challenges associated with optimizing drug therapy for the effective treatment of HIV/AIDS necessitate research into the field of novel delivery systems. In recent years, some novel nanocarrier-based targeted drug delivery systems have shown remarkable ability to overcome many of the anatomical and physiological barriers and deliver the drugs locally at the HIV-infected sites thereby improving the HIV therapy. Most of the successful approaches have utilized a combination of passive and active targeting.
5. Future perspective
The current focus in pharmaceuticals is shifting to a ‘smart drug’ paradigm, in which increased efficacy and decreased toxicity are motivating factors. In the next few years, research on nanomedicines in targeted anti-HIV drug-delivery systems will lead to breakthroughs that enable their therapeutic application. In the recent past, the simplistic view of HIV infection portrayed that a single receptor or maybe a combination of two receptors was sufficient for HIV infection to occur. However, today we know that there are numerous mechanisms that lead to HIV infection throughout the body. As receptor targets become known and better characterized, nanoscale delivery systems combined with an active targeting approach will allow for the treatment of the numerous variants of HIV disease that are becoming increasingly recognized.
Since current treatment regimens expose the entire body to multiple drugs at high doses, side effects often limit therapy and lead to unsuccessful outcomes. Adherence to drug regimens is crucial in the treatment of HIV infection since it is currently considered an incurable chronic disease. AIDS treatment regimens such as HAART suffer from complexities of frequent administration and high dosages. Development of targeted drug-delivery approaches for AIDS therapy will improve the safety and efficacy of anti-HIV drugs by reducing their dose and adverse effects associated with them. Although nanoscale approaches would add considerable cost per dosage unit, the improvement in therapy resulting from reduced dosages and reduced frequency of administration would result in much higher patient compliance and a greater frequency of successful therapeutic outcomes. This is critical in countries where resources are limited.
Even with breakthroughs in the engineering of novel nanocarriers using various ‘surface modifiers’, there is still the additional challenge of understanding and achieving the dosing that delivers consistent pharmacokinetics. There is no doubt that, with additional understanding of pharmacokinetics and immunity combined with the development of novel biomimetic strategies, these hurdles will be translated into practical therapeutics in the clinic. The toxicity issues of the developed nanocarriers must also be investigated to prove their safe and efficacious clinical use. Short of an effective HIV vaccine, HIV drug therapy enhanced by nanoscale drug delivery carriers will play an important role in devising efficacious regimens over the next decade.
Acknowledgments
Partial financial support from NIH AI51214, NIH CounterACT Program U54AR055073 and the Parke-Davis Endowed Chair in Pharmaceutics and Drug Delivery is gratefully acknowledged. We would like to thank Dr. Howard Gendelman and Dr. Robin Taylor for their help.
List of Abbreviations
- 3TC
lamivudine
- AIDS
acquired immunodeficiency syndrome
- AUC
area under the curve
- AZT
zidovudine
- AZTP
azidothymidine palmitate
- BBB
blood brain barrier
- BMMs
bone marrow-derived macrophages
- CHO
chinese hampster ovary
- CNS
central nervous system
- CPP
cell penetrating peptide
- DC
dendritic cells
- d4T
stavudine
- ddC
2′,3′-dideoxycytidine
- ddCTP
2′,3′-dideoxycytidine-5-triphosphate
- ddI
dideoxyinosine
- ddITP
dideoxyinosine (didanosine) triphosphate
- DIG
digoxigenin
- DPPC
dipalmitoyl phosphatidyl choline
- EC
effective concentration
- ED
effective dose
- EFV
efavirenz
- EPR
enhanced permeability and retention
- Fc
fragment crystallization
- FDA
food and drug administration
- FDCs
follicular dendritic cells
- FIV
feline immunodeficiency virus
- FLT
fluorothymidine
- fMLF
N-formyl-methione-leucine-phenyl alanine
- GALT
gut-associated lymphoid tissue
- gp120
glycoprotein120
- HAART
highly active antiretroviral therapy
- HIV
human immunodeficiency virus
- HSA
human serum albumin
- IDV
indinavir
- IP
intraperitoneal
- IV
intravenous
- LDL
low density lipoprotein
- LNs
lymph nodes
- M/M
monocytes/macrophages
- MN-G
mannosylated gelatin
- MPPI
mannosylated poly(propyleneimine)
- MPS
mononuclear phagocyte system
- NB-DNJ
N-Butyldeoxynojirimycin
- NNRTI
non- nucleoside reverse transcriptase inhibitors
- NP
nanoparticle
- NRTI
nucleoside reverse transcriptase inhibitor
- PB
peptide-based
- PBCA
polybutylcyanoacrylate
- PBMC
peripheral blood mononuclear cells
- PCL
poly(epsilon-caprolactone)
- PEG
polyethylene glycol
- PEO
poly(ethylene oxide)
- PHCA
polyhexacyanoacrylate
- PI
protease inhibitor
- PLA
poly lactic acid
- PMNL
polymorphonuclear leucocytes
- PPI
poly(propyleneimine)
- RES
reticular endothelial system
- R.I.CK-Tat9
retro-inverso-cysteine-lysine-Tat nonapeptide
- RTV
ritonavir
- SC
subcutaneous
- SQV
saquinavir
- Tat
trans-activating transcriptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Büchen-Osmond C, editor. ICTVdB Management Lentivirus. ICTVdB - The Universal Virus Database. Vol. 4. Columbia University; New York, USA: 2006. [Google Scholar]
- 2.Büchen-Osmonda C, editor. ICTVdB Management Spumaretrovirinae. ICTVdB - The Universal Virus Database. Columbia University; New York, USA: 2006. [Google Scholar]
- 3.Lawn SD. AIDS in Africa: the impact of coinfections on the pathogenesis of HIV-1 infection. J Infect Dis. 2004;48:1–12. doi: 10.1016/j.jinf.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Reeves JD, Doms RW. Human Immunodeficiency Virus Type 2. J Gen Virol. 2002;83:1253–1265. doi: 10.1099/0022-1317-83-6-1253. [DOI] [PubMed] [Google Scholar]
- 5.Joint United Nations Programme on HIV/AIDS and World Health Organization. AIDS Epidemic Update. Geneva: 2005. [Google Scholar]
- 6.UNAIDS. AIDS Epidemic Update. Geneva: 2007. [Google Scholar]
- 7.Warr GW, Sljivic VS. Origin and division of liver macrophages during stimulation of the mononuclear phagocyte system. Cell Prolif. 1974;7:559–565. doi: 10.1111/j.1365-2184.1974.tb00439.x. [DOI] [PubMed] [Google Scholar]
- 8.Carter CA, Ehlrich LS. Cell biology of HIV-1 infection of macrophages. Annu Rev Microbiol. 2008;62:425–443. doi: 10.1146/annurev.micro.62.081307.162758. [DOI] [PubMed] [Google Scholar]
- 9.Cassol E, Alfano M, Biswas P, Poli G. Monocyte-derived macrophages and myeloid cell lines as targets of HIV-1 replication and persistence. J Leukoc Biol. 2006;80:1018–1030. doi: 10.1189/jlb.0306150. [DOI] [PubMed] [Google Scholar]
- 10.De Clercq E. Strategies in the design of antiviral drugs. Nature Rev Drug Discov. 2002;1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- 11.Kazmierski WM, Kenakin TP, Gudmundsson KS. Peptide, peptidomimetic and small-molecule drug discovery targeting HIV-1 host-cell attachment and entry through gp120, gp41, CCR5 and CXCR4. Chem Biol Drug Des. 2006;67:13–26. doi: 10.1111/j.1747-0285.2005.00319.x. [DOI] [PubMed] [Google Scholar]
- 12.Collman RG. HIV-1 Env-chemokine receptor interactions in primary human macrophages: entry and beyond. Res Initiat Treat Action. 2003;8:6–9. [PubMed] [Google Scholar]
- 13.Kwon DS, Gregorio G, Bitton N, Hendrickson WA, Littman DR. DC-SIGN-mediated internalization of HIV is required for trans-enhancement of T cell infection. Immunity. 2002;16:135–144. doi: 10.1016/s1074-7613(02)00259-5. [DOI] [PubMed] [Google Scholar]
- 14.Pomerantz RJ. Reservoirs of human immunodeficiency virus type 1: the main obstacles to viral eradication. Clin Infect Dis. 2002;34:91–97. doi: 10.1086/338256. [DOI] [PubMed] [Google Scholar]
- 15.Blankson JN, Persaud D, Siliciano RF. The challenge of viral reservoirs in HIV-1 infection. Annu Rev Med. 2002;53:557–593. doi: 10.1146/annurev.med.53.082901.104024. [DOI] [PubMed] [Google Scholar]
- 16.Reiner SL. Development in motion: helper T cells at work. Cell. 2007;129:33–36. doi: 10.1016/j.cell.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 17.Hu J, Liu H, Wang L. Enhanced delivery of AZT to macrophages via acetylated LDL. J Control Release. 2000;69:327–335. doi: 10.1016/s0168-3659(00)00319-9. [DOI] [PubMed] [Google Scholar]
- 18.Aquaro S, Bagnarelli P, Guenci T, De Luca A, Clementi M, Balestra E, Calio R, Perno CF. Long-term survival and virus production in human primary macrophages infected by human immunodeficiency virus. J Med Virol. 2002;68:479–488. doi: 10.1002/jmv.10245. [DOI] [PubMed] [Google Scholar]
- 19.Crowe SM, Mills J, Kirihara J, Boothman J, Marshall JA, McGrath MS. Full-length recombinant CD4 and recombinant gp120 inhibit fusion between HIV infected macrophages and uninfected CD4-expressing T-lymphoblastoid cells. AIDS Res Hum Retroviruses. 1990;6:1031–1037. doi: 10.1089/aid.1990.6.1031. [DOI] [PubMed] [Google Scholar]
- 20.Badley AD, Dockrell D, Simpson M, Schut R, Lynch DH, Leibson P, Paya CV. Macrophage-dependent apoptosis of CD4+ T lymphocytes from HIV-infected individuals is mediated by FasL and tumor necrosis factor. J Exp Med. 1997;185:55–64. doi: 10.1084/jem.185.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herbein G, Mahlknecht U, Batliwalla F, Gregersen P, Pappas T, Butler J, O’Brien WA, Verdin E. Apoptosis of CD8 T cells is mediated by macrophages through interaction of HIV gp120 with chemokine receptor CXCR4. Nature. 1998;395:189–194. doi: 10.1038/26026. [DOI] [PubMed] [Google Scholar]
- 22.Aquaro S, Panti S, Caroleo MC, Balestra E, Cenci A, Forbici F, Ippolito G, Mastino A, Testi R, Mollace V. Primary macrophages infected by human immunodeficiency virus trigger CD95-mediated apoptosis of uninfected astrocytes. J Leukoc Biol. 2000;68:429–435. [PubMed] [Google Scholar]
- 23.Aquaro S, Muscoli C, Ranazzi A, Pollicita M, Granato T, Masuelli L, Modesti A, Perno CF, Mollace V. The contribution of peroxynitrite generation in HIV replication in human primary macrophages. Retrovirology. 2007;4:76–80. doi: 10.1186/1742-4690-4-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahsan F, Rivas IP, Khan MA, Torres Suarez AI. Targeting to macrophages: role of physicochemical properties of particulate carriers—liposomes and microspheres—on the phagocytosis by macrophages. J Control Release. 2002;79:29–40. doi: 10.1016/s0168-3659(01)00549-1. [DOI] [PubMed] [Google Scholar]
- 25.Kedzierska K, Crowe SM, Turville S, Cunningham AL. The influence of cytokines, chemokines and their receptors on HIV-1 replication in monocytes and macrophages. Rev Med Virol. 2003;13:39–56. doi: 10.1002/rmv.369. [DOI] [PubMed] [Google Scholar]
- 26.Burton GF, Keele BF, Estes JD, Thacker TC, Gartner S. Follicular dendritic cell contributions to HIV pathogenesis. Semin Immunol. 2002;14:275–284. doi: 10.1016/s1044-5323(02)00060-x. [DOI] [PubMed] [Google Scholar]
- 27.Rusconi S, Scozzafava A, Mastrolorenzo A, Supuran CT. An update in the development of HIV entry inhibitors. Curr Top Med Chem. 2007;7:1273–1289. doi: 10.2174/156802607781212239. [DOI] [PubMed] [Google Scholar]
- 28.Kuritzkes DR. HIV-1 entry inhibitors: an overview. Curr Opin HIV AIDS. 2009;4:82–87. doi: 10.1097/COH.0b013e328322402e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–944. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 30.Ehrlich PR, Himmelweit F, Marquardt M, Dale HH. The collected papers of Paul Ehrlich. Pergamon Press; London: 1960. [Google Scholar]
- 31.Panyam J, Labhasetwar V. Targeting intracellular targets. Curr Drug Deliv. 2004;1:235–247. doi: 10.2174/1567201043334768. [DOI] [PubMed] [Google Scholar]
- 32.Singh Y, Palombo M, Sinko PJ. Recent trends in targeted anticancer prodrug and conjugate design. Curr Med Chem. 2008;15:1802–1826. doi: 10.2174/092986708785132997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinman L, Bui T, Larsen K, Tsai CC, Anderson D, Morton WR, Hu SL, Ho RJY. Optimization of Lipid-Indinavir Complexes for Localization in Lymphoid Tissues of HIV-Infected Macaques. J Acquir Immune Defic Syndr. 2006;42:155–161. doi: 10.1097/01.qai.0000214822.33905.87. [DOI] [PubMed] [Google Scholar]
- 34.Huang C, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prokop A, Davidson JM. Nanovehicular intracellular delivery systems. J Pharm Sci. 2008;97:3518–3590. doi: 10.1002/jps.21270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 37.El-Sayed A, Futaki S, Harashima H. Delivery of Macromolecules Using Arginine-Rich Cell-Penetrating Peptides: Ways to Overcome Endosomal Entrapment. AAPS J. 2009;11:13–22. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fahmy TM, Fong PM, Goyal A, Saltzman WM. Targeted for drug delivery. Nano Today. 2005;8:18–26. [Google Scholar]
- 39.Zhang X, Jin Y, Plummer M, Pooyan S, Gunaseelan S, Sinko P. Endocytosis and membrane potential are required for HeLa cell uptake of RI-CKTat9, a retro inverso Tat cell penetrating peptide. Mol Pharm. 2009;6:836–848. doi: 10.1021/mp800121f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker JR, Banaszak Holl MM. The binding avidity of a nanoparticle-based multivalent targeted drug delivery platform. Chem Biol. 2007;14:107–115. doi: 10.1016/j.chembiol.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 41.Kinman L, Brodie SJ, Tsai CC, Bui T, Larsen K, Schmidt A, Anderson D, Morton WR, Hu SL, Ho RJY. Lipid-drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissues and viral load reduction: a proof of concept study in HIV-2287-infected macaques. J Acquir Immune Defic Syndr. 2003;34:387–397. doi: 10.1097/00126334-200312010-00005. [DOI] [PubMed] [Google Scholar]
- 42.Dou H, Destache CJ, Morehead JR, Lee Mosley R, Boska MD, Kingsky J, Gorantla S, Poluektova L, Nelson JA, Chaubal M, Werling J, Kipp J, Rabinov BE, Gendelman HE. Development of a macrophage-based nanoparticle platform for antiretroviral drug delivery. Blood. 2006;108:2827–2835. doi: 10.1182/blood-2006-03-012534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magnani M, Rossi L, Fraternale A, Silrotti L, Quintavalla F, Piedimonte G, Matteucci D, Baldinotti F, Bendinelli M. Feline Immunodeficiency Virus Infection of Macrophages: In Vitro and in Vivo Inhibition by Dideoxycytidine-5′-triphosphate-loaded Erythrocytes. AIDS Res Hum Retrovir. 1994;10:1179–1186. doi: 10.1089/aid.1994.10.1179. [DOI] [PubMed] [Google Scholar]
- 44.Aouadi M, Tesz GI, Nicoloro SM, Wang M, Chouinard M, Soto E, Ostrpff GR, CMP Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;458:1180–1184. doi: 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarker DK. Engineering of nanoemulsions for drug delivery. Curr Drug Deliv. 2005;2:297–310. doi: 10.2174/156720105774370267. [DOI] [PubMed] [Google Scholar]
- 46.Opanasopit P, Nishikawa M, Hashida M. Factors affecting drug and gene delivery: effects of interaction with blood components. Crit Rev Ther Drug Carrier Syst. 2002;19:191–233. doi: 10.1615/critrevtherdrugcarriersyst.v19.i3.10. [DOI] [PubMed] [Google Scholar]
- 47.Fahmy TM, Fong PM, Park J, Constable T, Saltzman WM. Nanosystems for simultaneous imaging and drug delivery to T cells. AAPS J. 2007;9:E171–E180. doi: 10.1208/aapsj0902019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waterhouse DN, Madden TD, Cullis PR, Bally MB, Mayer LD, Webb MS. Preparation, characterization, and biological analysis of liposomal formulations of vincristine. Methods Enzymol. 2005;391:40–57. doi: 10.1016/S0076-6879(05)91002-1. [DOI] [PubMed] [Google Scholar]
- 49.Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151:201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 50.Sapra P, Allen TM. Ligand-targeted liposomal anticancer drugs. Prog Lipid Res. 2003;42:439–462. doi: 10.1016/s0163-7827(03)00032-8. [DOI] [PubMed] [Google Scholar]
- 51.Allen TM, Sapra P, Moase E, Moreira J, Iden D. Adventures in targeting. J Liposome Res. 2002;12:5–12. doi: 10.1081/lpr-120004771. [DOI] [PubMed] [Google Scholar]
- 52.Kreuter J. Nanoparticles as drug delivery systems. Encyclopedia of nanoscience and nanotechnology. 2004;7:161–180. [Google Scholar]
- 53.Shive MS, Anderson JM. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 54.Langer R, Folkman J. Polymers for the sustained release of proteins and other macromolecules. Nature. 1976;263:797–800. doi: 10.1038/263797a0. [DOI] [PubMed] [Google Scholar]
- 55.Visscher GE, Robison RL, Maulding HV, Fong JW, Pearson JE, Argentieri GJ. Biodegradation of and tissue reaction to 50: 50 poly (DL-lactide-co-glycolide) microcapsules. J Biomed Mater Res. 1986;20:667–676. doi: 10.1002/jbm.820190315. [DOI] [PubMed] [Google Scholar]
- 56.Muthu MS, Singh S. Targeted nanomedicines: effective treatment modalities for cancer, AIDS and brain disorders. Nanomed. 2009;4:105–118. doi: 10.2217/17435889.4.1.105. [DOI] [PubMed] [Google Scholar]
- 57.Mainardes RM, Evangelista RC. PLGA nanoparticles containing praziquantel: effect of formulation variables on size distribution. Int J Pharm. 2005;290:137–144. doi: 10.1016/j.ijpharm.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 58.Muthu MS, Singh S. Studies on biodegradable polymeric nanoparticles of risperidone: in vitro and in vivo evaluation. Nanomed. 2008;3:305–319. doi: 10.2217/17435889.3.3.305. [DOI] [PubMed] [Google Scholar]
- 59.Singh S, Muthu MS. Preparation and characterization of nanoparticles containing an atypical antipsychotic agent. Nanomed. 2007;2:233–240. doi: 10.2217/17435889.2.2.233. [DOI] [PubMed] [Google Scholar]
- 60.Jain RK. Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliv Rev. 2001;46:149–168. doi: 10.1016/s0169-409x(00)00131-9. [DOI] [PubMed] [Google Scholar]
- 61.Wiener EC, Brechbiel MW, Brothers H, Magin RL, Gansow OA, Tomalia DA, Lauterbur PC. Dendrimer-based metal chelates: a new class of magnetic resonance imaging contrast agents. Magn Reson Med. 1994;31:1–8. doi: 10.1002/mrm.1910310102. [DOI] [PubMed] [Google Scholar]
- 62.Jansen J, de Brabander-van den Berg EM, Meijer EW. Encapsulation of guest molecules into a dendritic box. Science. 1994;266:1226–1229. doi: 10.1126/science.266.5188.1226. [DOI] [PubMed] [Google Scholar]
- 63.D’Emanuele A, Attwood D. Dendrimerñdrug interactions. Adv Drug Deliv Rev. 2005;57:2147–2162. doi: 10.1016/j.addr.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 64.Kolhe P, Misra E, Kannan RM, Kannan S, Lieh-Lai M. Drug complexation, in vitro release and cellular entry of dendrimers and hyperbranched polymers. Int J Pharm. 2003;259:143–160. doi: 10.1016/s0378-5173(03)00225-4. [DOI] [PubMed] [Google Scholar]
- 65.Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther. 2008;83:761–769. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]
- 66.Gunaseelan S, Pooyan S, Chen P, Samizadeh M, Palombo MS, Stein S, Zhang X, Sinko PJ. Multimeric Peptide-based PEG Nanocarriers with Programmable Elimination Properties. Biomaterials. 2009;30:5649–5659. doi: 10.1016/j.biomaterials.2009.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Makabi-Panzu B, Gourde P, Desormeaux A, Bergeron MG. Intracellular and serum stability of liposomal 2′, 3′-dideoxycytidine. Effect of lipid composition. Cell Mol Biol. 1998;44:277–284. [PubMed] [Google Scholar]
- 68.Katragadda A, Bridgman R, Betageri G. Effect of liposome composition and cholesterol on the cellular uptake of stavudine by human monocyte/macrophages. Cell Mol Biol Lett. 2000;5:483–494. [Google Scholar]
- 69.Pollock S, Dwek RA, Burton DR, Zitzmann N. N-Butyldeoxynojirimycin is a broadly effective anti-HIV therapy significantly enhanced by targeted liposome delivery. AIDS. 2008;22:1961–1969. doi: 10.1097/QAD.0b013e32830efd96. [DOI] [PubMed] [Google Scholar]
- 70.Slepushkin VA, Salem, Andreev SM, Dazin P, Duzgunes N. Targeting of liposomes to HIV-1-infected cells by peptides derived from the CD4 receptor. Biochem Biophy Res Comm. 1996;227:827–833. doi: 10.1006/bbrc.1996.1592. [DOI] [PubMed] [Google Scholar]
- 71.Mankertz J, Matthes E, Rokos K, von Baeyer H, Pauli G, Riedel E. Selective endocytosis of fluorothymidine and azidothymidine coupled to LDL into HIV infected mononuclear cells. BBA-Mol Basis of Dis. 1996;1317:233–237. doi: 10.1016/s0925-4439(96)00059-2. [DOI] [PubMed] [Google Scholar]
- 72.Kondo A, Muranaka Y, Ohta I, Notsu K, Manabe M, Kotani K, Saito K, Maekawa M, Kanno T. Relationship between triglyceride concentrations and LDL size evaluated by malondialdehyde modified LDL. Clin Chem. 2001;47:893–900. [PubMed] [Google Scholar]
- 73.Betageri GV, Black CD, Szebeni J, Wahl LM, Weinstein JN. Fc-receptor-mediated targeting of antibody-bearing liposomes containing dideoxycytidine triphosphate to human monocyte/macrophages. J Pharm Pharmacol. 1993;45:48–53. doi: 10.1111/j.2042-7158.1993.tb03678.x. [DOI] [PubMed] [Google Scholar]
- 74.Betageri GV, Jenkins SA, Ravis WR. Drug delivery using antibody-liposome conjugates. Drug Dev Ind Pharm. 1993;19:2109–2116. [Google Scholar]
- 75.Vyas SP, Subhedar R, Jain S. Development and characterization of emulsomes for sustained and targeted delivery of an antiviral agent to liver. Int J Clin Pharmacol Biopharm. 2006;58:321–326. doi: 10.1211/jpp.58.3.0005. [DOI] [PubMed] [Google Scholar]
- 76.Makabi-Panzu B, Lessard C, Beauchamp D, Désormeaux A, Poulin L, Tremblay M, Bergeron MG. Uptake and binding of liposomal 2′, 3′-dideoxycytidine by RAW 264.7 cells: a three-step process. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;8:227–235. doi: 10.1097/00042560-199503010-00002. [DOI] [PubMed] [Google Scholar]
- 77.Oussoren C, Magnani M, Fraternale A, Casabianca A, Chiarantini L, Ingebrigsten R, Underberg WJM, Storm G. Liposomes as carriers of the antiretroviral agent dideoxycytidine-5 -triphosphate. Int J Pharm. 1999;180:261–270. doi: 10.1016/s0378-5173(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 78.Gagné JF, Désormeaux A, Perron S, Tremblay MJ, Bergeron MG. Targeted delivery of indinavir to HIV-1 primary reservoirs with immunoliposomes. BBA-Biomembranes. 2002;1558:198–210. doi: 10.1016/s0005-2736(01)00432-1. [DOI] [PubMed] [Google Scholar]
- 79.Garg M, Asthana A, Agashe HB, Agrawal GP, Jain NK. Stavudine-loaded mannosylated liposomes: in-vitro anti-HIV-I activity, tissue distribution and pharmacokinetics. The J Pharm Pharmacol. 2006;58:605–616. doi: 10.1211/jpp.58.5.0005. [DOI] [PubMed] [Google Scholar]
- 80.Wu HB, Deng YH, Wang SN, Zhou XY, Wang N, Shi L. The distribution of azidothymidine palmitate galactosylated liposomes in mice. Yao Xue Xue Bao. 2007;42:538–544. [PubMed] [Google Scholar]
- 81.Garg M, Dutta T, Jain NK. Reduced hepatic toxicity, enhanced cellular uptake and altered pharmacokinetics of stavudine loaded galactosylated liposomes. Eur J Pharm Biopharm. 2007;67:76–85. doi: 10.1016/j.ejpb.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 82.Shah LK, Amiji MM. Intracellular delivery of saquinavir in biodegradable polymeric nanoparticles for HIV/AIDS. Pharm Res. 2006;23:2638–2645. doi: 10.1007/s11095-006-9101-7. [DOI] [PubMed] [Google Scholar]
- 83.Mainardes RM, Gremiao MPD, Brunetti IL, da Fonseca LM, Khalil NM. Zidovudine-loaded PLA and PLA-PEG blend nanoparticles: Influence of polymer type on phagocytic uptake by polymorphonuclear cells. J Pharm Sci. 2009;98:257–267. doi: 10.1002/jps.21406. [DOI] [PubMed] [Google Scholar]
- 84.Sant S, Poulin S, Hildgen P. Effect of polymer architecture on surface properties, plasma protein adsorption, and cellular interactions of pegylated nanoparticles. J Biomed Mater Res A. 2008;87:885–895. doi: 10.1002/jbm.a.31800. [DOI] [PubMed] [Google Scholar]
- 85.Zhang K, Fang H, Chen Z, Taylor JSA, Wooley KL. Shape Effects of Nanoparticles Conjugated with Cell-Penetrating Peptides (HIV Tat PTD) on CHO Cell Uptake. Bioconj Chem. 2008;19:1880–1887. doi: 10.1021/bc800160b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schäfer V, von Briesen H, Andreesen R, Steffan AM, Royer C, Tröster S, Kreuter J, Rübsamen-Waigmann H. Phagocytosis of Nanoparticles by Human Immunodeficiency Virus (HlV)-Infected Macrophages: A Possibility for Antiviral Drug Targeting. Pharm Res. 1992;9:541–546. doi: 10.1023/a:1015852732512. [DOI] [PubMed] [Google Scholar]
- 87.Kaur A, Jain S, Tiwary AK. Mannan-coated gelatin nanoparticles for sustained and targeted delivery of didanosine: In vitro and in vivo evaluation. Acta Pharm. 2008;58:61–74. doi: 10.2478/v10007-007-0045-1. [DOI] [PubMed] [Google Scholar]
- 88.Jain SK, Gupta Y, Jain A, Saxena AR, Khare P, Jain A. Mannosylated gelatin nanoparticles bearing an anti-HIV drug didanosine for site-specific delivery. Nanomed. 2008;4:41–48. doi: 10.1016/j.nano.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 89.Löbenberg R, Araujo L, Kreuter J. Body distribution of azidothymidine bound to nanoparticles after oral administration. Eur J Pharm Biopharm. 1997;44:127–132. [Google Scholar]
- 90.Löbenberg R, Araujo L, von Briesen H, Rodgers E, Kreuter J. Body distribution of azidothymidine bound to hexyl-cyanoacrylate nanoparticles after iv injection to rats. J Control Release. 1998;50:21–30. doi: 10.1016/s0168-3659(97)00105-3. [DOI] [PubMed] [Google Scholar]
- 91.Dembri A, Montisci MJ, Gantier JC, Chacun H, Ponchel G. Targeting of 3 -azido 3 -deoxythymidine (AZT)-loaded poly (isohexylcyanoacrylate) nanospheres to the gastrointestinal mucosa and associated lymphoid tissues. Pharm Res. 2001;18:467–473. doi: 10.1023/a:1011050209986. [DOI] [PubMed] [Google Scholar]
- 92.Castaldello A, Brocca-Cofano E, Voltan R, Triulzi C, Altavilla G, Laus M, Sparnacci K, Ballestri M, Tondelli L, Fortini C, Gavioli R, Ensoli B, Caputo A. DNA prime and protein boost immunization with innovative polymeric cationic core-shell nanoparticles elicits broad immune responses and strongly enhance cellular responses of HIV-1 tat DNA vaccination. Vaccine. 2006;24:5655–5669. doi: 10.1016/j.vaccine.2006.05.058. [DOI] [PubMed] [Google Scholar]
- 93.Lockman PR, Oyewumi MO, Koziara JM, Roder KE, Mumper RJ, Allen DD. Brain uptake of thiamine-coated nanoparticles. J Control Release. 2003;93:271–282. doi: 10.1016/j.jconrel.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 94.Rao KS, Reddy MK, Horning JL, Labhasetwar V. TAT-conjugated nanoparticles for the CNS delivery of anti-HIV drugs. Biomaterials. 2008;29:4429–4438. doi: 10.1016/j.biomaterials.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dutta T, Jain NK. Targeting potential and anti-HIV activity of lamivudine loaded mannosylated poly (propyleneimine) dendrimer. Biochim Biophys Acta. 2007;1770:681–686. doi: 10.1016/j.bbagen.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 96.Dutta T, Agashe HB, Garg M, Balakrishnan P, Kabra M, Jain NK. Poly (propyleneimine) dendrimer based nanocontainers for targeting of efavirenz to human monocytes/macrophages in vitro. J Drug Target. 2007;15:89–98. doi: 10.1080/10611860600965914. [DOI] [PubMed] [Google Scholar]
- 97.Gunaseelan S, Debrah O, Wan L, Leibowitz MJ, Rabson AB, Stein S, Sinko PJ. Synthesis of Poly(ethylene glycol)-Based Saquinavir Prodrug Conjugate and Assessment of Release and Anti-HIV-1 Bioactivity Using a Novel Protease Inhibition Assay. Bioconj Chem. 2004;15:1322–1333. doi: 10.1021/bc0498875. [DOI] [PubMed] [Google Scholar]
- 98.Wan L, Zhang X, Gunaseelan S, Pooyan S, Debrah O, Leibowitz MJ, Rabson AB, Stein S, Sinko PJ. Novel multi-component nanopharmaceuticals derived from poly (ethylene) glycol, retro-inverso-Tat nonapeptide and saquinavir demonstrate combined anti-HIV effects. AIDS Res Ther. 2006;3:12–26. doi: 10.1186/1742-6405-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pooyan S, Qiu B, Chan MM, Fong D, Sinko PJ, Leibowitz MJ, Stein S. Conjugates Bearing Multiple Formyl-Methionyl Peptides Display Enhanced Binding to but Not Activation of Phagocytic Cells. Bioconjug Chem. 2002;13:216–223. doi: 10.1021/bc0100657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wan L, Pooyan S, Hu P, Leibowitz MJ, Stein S, Sinko PJ. Peritoneal Macrophage Uptake, Pharmacokinetics and Biodistribution of Macrophage-Targeted PEG-fMLF (N-Formyl-Methionyl-Leucyl-Phenylalanine) Nanocarriers for Improving HIV Drug Delivery. Pharm Res. 2007;24:2110–2119. doi: 10.1007/s11095-007-9402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wan L, Zhang X, Pooyan S, Palombo MS, Leibowitz MJ, Stein S, Sinko PJ. Optimizing Size and Copy Number For PEG-fMLF (N-Formyl-methionyl-leucyl-phenylalanine) Nanocarrier Uptake by Macrophages. Bioconjug Chem. 2008;19:28–38. doi: 10.1021/bc070066k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huang SY, Pooyan S, Wang J, Choudhury I, Leibowitz MJ, Stein S. A poly(ethylene glycol) copolymer for carrying and releasing multiple copies of cystein-containing peptides. Bioconj Chem. 1998;9:612–617. doi: 10.1021/bc980038p. [DOI] [PubMed] [Google Scholar]