

Abstract
The effects of oleamide, an amidated lipid isolated from the cerebrospinal fluid of sleep-deprived cats, on serotonin receptor-mediated responses were investigated in cultured mammalian cells. In rat P11 cells, which endogenously express the 5-hydroxytryptamine2A (5HT2A) receptor, oleamide significantly potentiated 5HT-induced phosphoinositide hydrolysis. In HeLa cells expressing the 5HT7 receptor subtype, oleamide caused a concentration-dependent increase in cAMP accumulation but with lower efficacy than that observed by 5HT. This effect was not observed in untransfected HeLa cells. Clozapine did not prevent the increase in cAMP elicited by oleamide, and ketanserin caused an ≈65% decrease. In the presence of 5HT, oleamide had the opposite effect on cAMP, causing insurmountable antagonism of the concentration-effect curve to 5HT, but had no effect on cAMP levels elicited by isoproterenol or forskolin. These results indicate that oleamide can modulate 5HT-mediated signal transduction at different subtypes of mammalian 5HT receptors. Additionally, our data indicate that oleamide acts at an apparent allosteric site on the 5HT7 receptor and elicits functional responses via activation of this site. This represents a unique mechanism of activation for 5HT G protein-coupled receptors and suggests that G protein-coupled neurotransmitter receptors may act like their iontropic counterparts (i.e., γ-aminobutyric acid type A receptors) in that there may be several binding sites on the receptor that regulate functional activity with varying efficacies.
Oleamide, the primary amide of oleic acid, and other long alkane-chained fatty acid amides, including anandamide, the endogenous ligand for the cannabinoid receptor (1), comprise a novel family of amidated lipids that have been found in the cerebrospinal fluid and plasma of mammals, including humans (2, 3). Since its identification and isolation from the cerebrospinal fluid of sleep-deprived cats, oleamide has been implicated in a number of diverse physiological and neuromodulatory functions. In vivo, oleamide induces normal sleep (3) and decreases body temperature (Steven Henriksen, personal communications) when injected into rats. In vitro, oleamide has been shown to antagonize gap junction communication in cultured glial cells (32) and to potentiate 5-hydroxytryptamine2A/2C (5HT2A/2C) receptor-mediated chloride currents in transfected frog oocytes but not those currents elicited by other seven transmembrane domain receptors coupled via the same G protein-linked pathway, such as metabotropic glutamate receptor and muscarinic receptors (4). These latter findings are of great interest because enhancement or disruption of serotonergic neurotransmission can have a range of consequences involving psychiatric disturbances, appetite, alertness, and sleep.
In this study, we examined the effects of oleamide on 5HT receptors and 5HT-mediated neurotransmission by determining whether oleamide could modulate 5HT2 receptor-induced phosphoinositide turnover in mammalian cells, as was implied by studies using the oocyte system (4), and whether oleamide could modulate a 5HT subtype coupled to a different G protein-coupled receptor. We have used rat pituitary P11 cells, which endogenously express the 5HT2A receptor subtype, and HeLa cells that have been transfected with 5HT7 receptor cDNA, as two 5HT systems coupled to different second messenger systems, phosphoinositide hydrolysis and adenylate cyclase, respectively. In the presence of 5HT, oleamide causes a potentiation of 5HT2A receptor-induced phosphoinositide hydrolysis but an inhibition of 5HT7 receptor-mediated stimulation of cAMP levels. Surprising to note, oleamide exhibited a stimulatory effect on cAMP in the absence of agonist. Our results suggest that oleamide interacts at an allosteric site on the 5HT7 receptor and can influence G protein signaling via activation of this site.
METHODS
Cell Culture.
HeLa cells were grown and cultured in OM5 media (5) supplemented with 10% fetal calf serum in a 5% CO2 environment. P11 cells (a subclonal line from rat pituitary tumor 7315a) were supplied generously by Perry Molinoff (Bristol–Myers) and were cultured similarly to those described (6). HeLa cells were transiently transfected with pCMV4REC20 (5HT7 receptor) (7) by using DOTAP {N-[1-(2,3-dioleoyloxy)propyl]-N,N,N,-trimethylammonium methyl sulfate} (Boehringer Mannheim) lipotransfection as described by the vendor.
Cyclic AMP Accumulation.
Cyclic AMP accumulation experiments were performed by an adaptation to that described (8). HeLa cells were transfected by the DOTAP method and used 72 h later. Cells were plated into 12-welled culture dishes at a density of 800,000 cells/well in MEM supplemented with 20 μM adenine and 2.5 μCi [3H]adenine/well and incubated for 2–4 h. The cells were washed twice with Krebs–Ringer bicarbonate buffer (124 mM NaCl/5 mM KCl/1.3 mM MgCl2/26 mM NaHCO3/1.2 mM KHPO4/1.8 mM CaCl2/10 mM glucose) and equilibrated with 0.5 mM isobutylmethyl xanthine in 0.5 ml for 15 min at 37°C. After equilibration, the cAMP assay was initiated by the addition of serotonin and/or various fatty acid amide derivatives and incubated for 15 min. The reactions were quenched with the addition of 0.47 ml of trichloroacetic acid (10% wt/vol). After 30 min, the supernatant from each well was applied to a cation exchanges column (1.25 ml of AG 50W-X4, 200–400 mesh) and washed twice with 1.25 ml of water. This eluate was collected, and the radioactivity was measured to determine the incorporation of [3H]adenine into [3H]ATP. The Dowex columns were positioned over neutral alumina columns, and the [3H]cAMP fractions were eluted onto the alumina columns with 5 ml of water. The [3H]cAMP fractions were eluted from the columns with 4 ml of imidazole (0.1 M, pH 7.6). These fractions were collected, and the amount of radioactivity was measured to determine the amount of accumulated [3H]cAMP. The [3H]cAMP values are expressed as a percentage of the amount of incorporation of [3H]ATP to correct for minor variations in the number of cells plated per condition. In some experiments, [32P]cAMP (2,000–4,000) was added to the Dowex column as an internal standard. When used, 5HT antagonists were added 30–45 min before addition of agonists. Only those experiments in which 5HT caused at least a 2-fold increase in cAMP accumulation over basal were included in the calculations. All drugs were purchased from Sigma with the exception of oleamide and its derivatives, which were provided generously by Benjamin Cravatt (The Scripps Research Institute).
Phosphoinositide Hydrolysis.
Measurement of phosphoinositide hydrolysis in cultured cells was performed similarly to that described (6). Cells were plated 700,000 cells/well in OM5 containing myo-[3H]inositol (3 μCi/well). Assay were performed 1–2 days later. Cells were washed twice with Krebs–Ringer bicarbonate buffer and then equilibrated in 1 ml of Krebs–Ringer bicarbonate buffer for 1 h in 10% CO2/90% air. LiCl was added at a final concentration of 20 mM, and cells were incubated for an additional 10 min before the addition of various drugs. After 30 min, reactions were quenched by the addition of 0.125 ml of perchloric acid (10% wt/vol). The supernatants from each well were transferred into glass test tubes and neutralized with 0.35 ml KOH (0.3 M) and 0.35 ml NaHCO3 (5 mM). The entire contents of the test tubes were then applied to 0.5 ml of Bio-Rad AG 1-X8 resin (200–400 mesh) and washed four times with 5 ml of water. Total inositol phosphates were eluted off the columns with 0.5 ml of ammonium formate (1 M)/formic acid (0.1 M). Scintillation fluid (12 ml) was added to each vial, and radioactivity was quantified by a scintillation counter. Cell membranes remaining in each well were solubilized with 1 ml of 0.1 M NaOH, and the radioactivity incorporated into membrane phospholipids was determined. Data are expressed as the percentage of [3H]phospholipids converted to [3H]inositol phosphates.
Calculations.
EC50 values and maximal response values were estimated by using nonlinear regression analysis as described (9).
RESULTS
Previous studies demonstrated that oleamide potentiated 5HT2A/2C-mediated chloride currents in the frog oocyte expression system (4), so we investigated whether oleamide could modulate this receptor subtype in rat P11 cells, which endogenously express the 5HT2A subtype coupled to phosphoinositide hydrolysis (6). In these cells, 5HT caused a concentration-dependent increase in inositol phosphate formation with an EC50 of 10 μM (Fig. 1B). In the presence of an EC50 concentration of 5HT, oleamide caused a concentration-dependent increase in inositol phosphate turnover, resulting in a substantial potentiation of the 5HT response (Fig. 1A). At a concentration of 100 nM, oleamide caused a significant potentiation of the 5HT concentration–response curve, resulting in an increase in the maximal 5HT response by 228% (Fig. 1B). Oleamide, by itself, had no significant effect of inositol phosphate formation in these cells (data not shown).
Figure 1.
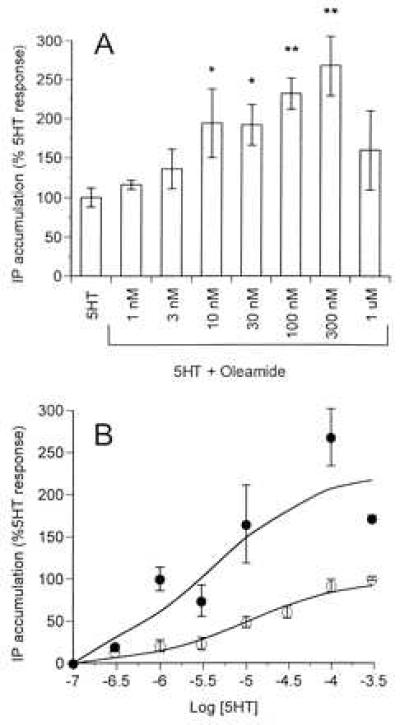
The effects of oleamide on inositol phosphate formation elicited by 5HT in P11 cells. (A) Phosphoinositide hydrolysis in P11 cells was measured in the presence of a single concentration of 5HT (10 μM) in the absence or presence of increasing concentrations of oleamide (1 nM–1 μM). Values plotted are inositol phosphates converted from labeled membrane phospholipids as a percentage of the maximal 5HT-induced response observed. Data are mean ± SEM of four experiments performed in triplicate. Asterisks indicate significant potentiation by oleamide using Student’s t test (two-tailed): ∗, P = 0.010–0.038; ∗∗, P = 0.0002. (B) Phosphoinositide hydrolysis was measured in response to increasing concentrations of 5HT in the absence (○) or presence (•) of oleamide (100 nM). Data points are mean ± SEM of five experiments performed in triplicate.
We next investigated whether oleamide had any modulatory effects on the 5HT7 receptor subtype, which is coupled to a stimulation of adenylate cyclase (7). In HeLa cells transfected with 5HT7 receptor cDNA, 5HT caused a concentration-dependent increase in cAMP accumulation with an EC50 value of 52 nM and a maximal response of 2.56-fold over basal (Fig. 2A, Inset). In the absence of 5HT, oleamide also caused a concentration-dependent increase in cAMP accumulation with an EC50 value of 4.2 nM and a maximal effect of 1.51-fold over basal (Fig. 2A), as determined by nonlinear regression analysis. Neither this effect of oleamide (Fig. 2A) nor that of 5HT (data not shown) was observed in untransfected HeLa cells. We then measured the effect of oleamide in combination with 5HT. Oleamide dose-dependently inhibited the cAMP response elicited by 5HT (100 nM) at a maximally effective concentration of 100 nM (Fig. 2B), which is consistent with the action of a partial agonist. However, oleamide (100 nM) caused insurmountable antagonism of the 5HT concentration–response curve (Fig. 3A), suggesting that oleamide was acting at a site other than the primary binding site for 5HT. To test this hypothesis, two 5HT antagonists, ketanserin and clozapine, were tested for their ability to block oleamide’s effects on cAMP accumulation, as well as 5HT. Clozapine (1 μM) prevented the 5HT-induced cAMP stimulation by 69% but had no significant effect on oleamide-induced increases in cAMP levels (Fig. 4). In contrast, ketanserin (10 μM) blocked both the 5HT- and oleamide-elicited increases in cAMP accumulation (Fig. 4) by ≈65%.
Figure 2.
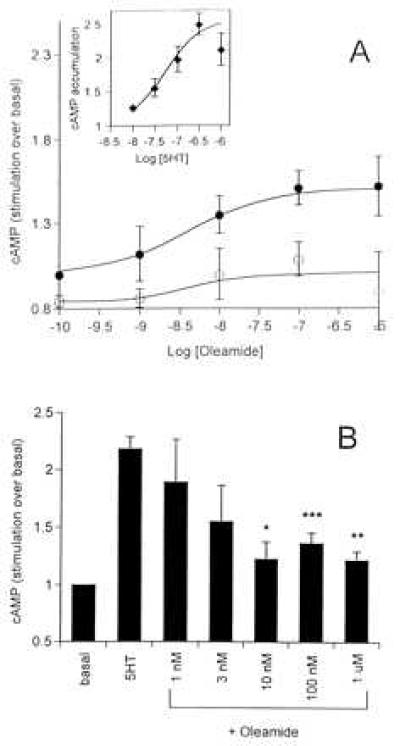
Effects of 5HT and oleamide on cAMP accumulation measured in 5HT7-transfected and untransfected HeLa cells. (A) Synthesis of cAMP in the presence of increasing concentrations of oleamide in 5HT7-transfected HeLa cells (•) and in untransfected HeLa cells (○). (B) cAMP accumulation elicited by 5HT (300 nM) was measured in the presence of increasing concentrations of oleamide (1 nM–1 μM) in 5HT7-transfected HeLa cells. Asterisks indicate significant inhibition by oleamide using Student’s t test (two-tailed): ∗, P = 0.0012; ∗∗, P = 0.0006; ∗∗∗, P = 0.0004. cAMP values are expressed as a multiple of the basal (unstimulated) synthesis.
Figure 3.
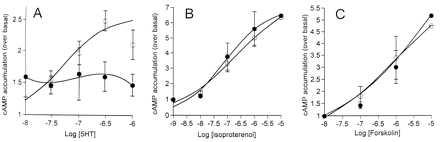
Effect of oleamide on cAMP responses elicited by 5HT (A), isoproterenol (B), and forskolin (C). Synthesis of cAMP was measured in response to increasing concentrations of drug in the absence (○) and presence (•) of oleamide (100 nM). All responses were measured in HeLa cells transfected with 5HT7 receptor cDNA. Data points represent mean ± SEM of at least four experiments performed in triplicate.
Figure 4.
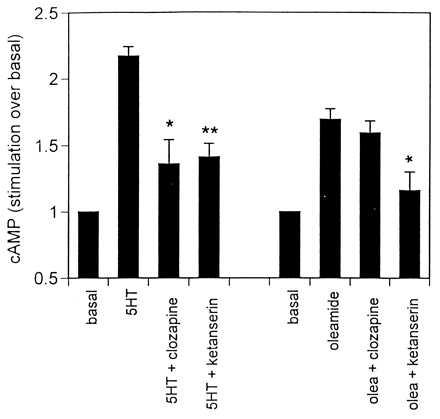
Effects of clozapine and ketanserin on 5HT- and oleamide-elicited cAMP responses in 5HT7-transfected HeLa cells. cAMP values are expressed as stimulation over basal and represent the mean ± SEM of at least four experiments performed in triplicate. Clozapine (1 μM) and ketanserin (10 μM) were incubated with HeLa cells 30–45 min before addition of 5HT or oleamide. Asterisks indicate significant inhibition by clozapine and ketanserin using Student’s t test (one-tailed): ∗, P = 0.024–0.038; ∗∗, P = 0.005.
We next tested whether oleamide’s actions were specific to 5HT receptors by measuring the effects of oleamide on the responses elicited by isoproterenol, which acts via the β-adrenergic receptor (10), and forskolin, a direct activator of adenylate cyclase (11). Oleamide (100 nM) had no effect on the EC50 value or maximal response elicited by isoproterenol or forskolin in HeLa cells (Fig. 3 B and C). Hence, oleamide’s actions appear to be specific to 5HT receptors, consistent with the findings of Huidrobro-Toro and Harris (4) in Xenopus oocytes.
Three oleamide-related compounds, which vary in length of alkane chain and number and position of the double bond, as well as oleic acid, which lacks a primary amide group, were tested for their ability to modulate cAMP levels in transfected HeLa cells. Only cis-8,9-octadecenamide had a significant inhibitory effect on the 5HT-induced increase in cAMP accumulation. The compound erucamide, which has a 4-carbon longer backbone, had a small, but nonsignificant, inhibitory effect. Neither oleic acid nor the trans-9,10-octadecenamide had any modulatory effects on 5HT-induced cAMP increases in these cells. Additionally, none of the fatty acid derivatives had any effects on isoproterenol- or forskolin-induced cAMP increases (Table 1).
Table 1.
The effect of various fatty acid amide derivatives and oleic acid on 5HT-, isoproterenol-, and forskolin-mediated stimulation of cAMP in transfected HeLa cells
| Compound | Max cAMP response, %
|
||
|---|---|---|---|
| 5HT (300 nM) | Forskolin (1 μM) | Isoproterenol (100 nM) | |
| Alone | 100 | 100 | 100 |
| Oleic acid | 81.8 ± 10.4 | 104 ± 15 | 101.1 ± 2.8 |
| Oleamide | 45.0 ± 9.3** | 92.0 ± 9.7 | 104.8 ± 3.7 |
| trans-9,10-ODA | 98.5 ± 6.0 | 91.5 ± 20.0 | 101.8 ± 19.6 |
| cis-8,9-ODA | 55.6 ± 8.5* | 104.5 ± 0.25 | 100.2 ± 2.8 |
| Erucamide | 74.8 ± 3.0 | 110.6 ± 16 | 96.8 ± 8.0 |
Values represent cAMP accumulation measured as a percentage of the response elicited by 5HT, forskolin, or isoproterenol. ODA, octadecenamide. Values are mean ± SEM. Asterisks represent values significantly different from the alone value using a Student’s t test (two-tailed). ∗, P = 0.0038; ∗∗, P = 0.0006.
DISCUSSION
We investigated the effects of the fatty acid amide oleamide on 5HT signal transduction in mammalian cells. Previous studies by Huidrobro-Toro and Harris (4) demonstrated that oleamide potentiated 5HT2A/2C-mediated chloride currents in the frog oocyte expression system. In this system, the chloride currents elicited by 5HT result from a signaling cascade involving phosphoinositide hydrolysis and inositol 1,4,5-trisphosphate receptor stimulation (12, 13) although this pathway was not directly measured in their study. We measured the effect of oleamide directly on phosphoinositide hydrolysis, and, in agreement with their studies, our results showed that oleamide can substantially potentiate 5HT-induced phosphoinositide turnover in P11 cells, which endogenously express the 5HT2A receptor. In P11 cells, the Ki value of 5HT at the 5HT2A receptor is ≈33 μM (6). Previous studies using these cells typically have measured submaximal increases in phosphoinositide hydrolysis at 5HT concentrations between 10 and 50 μM (6, 14, 15); however, an EC50 value for 5HT also has been reported to be <1 μM (6). In this study, we measured similar increases in phosphoinositide hydrolysis at a 5HT concentration of 10 μM, but our EC50 value for 5HT was higher than that reported previously (6). This observed lower potency may be accounted for by the slightly different conditions used in culturing the P11 cells. Oleamide caused significant potentiation of the 5HT response at a concentration as low as 10 nM (Fig. 1A), which is in general agreement with the results of Huidrobro-Toro and Harris (4) although those workers observed a small, but significant, effect on 5HT-elicited chloride currents at 1 nM oleamide. This may be explained by the greater sensitivity in measuring ion currents via electrophysiology vs. the column elution method of detecting inositol phosphates or by the fact that the simplified oocyte system is more sensitive to the effects of lipid compounds.
In contrast to its augmentation of 5HT2 receptor signal transduction, oleamide demonstrated inhibitory effects on cAMP accumulation elicited by 5HT7 receptor subtype activation. This result indicates that oleamide’s actions are not selective for the 5HT2A receptor subtype per se and are not limited to potentiating effects. Oleamide had no effect on β-adrenergic receptor-stimulated, or forskolin-induced, increases in cAMP synthesis; hence, consistent with the findings of Huidrobro-Toro and Harris (4) in Xenopus oocytes, oleamide’s actions appear to be specific to 5HT receptors albeit not a single subtype.
Oleamide caused a 50% increase in cAMP production in HeLa cells expressing the 5HT7 receptor. In untransfected cells, oleamide did not exhibit a similar increase in cAMP but did appear to have a slight, nonspecific stimulatory effect, which is perhaps because of the lipid nature of the compound. The fact that oleamide, by itself, exhibited a small stimulatory effect on cAMP accumulation in transfected HeLa cells, but inhibited the effects of 5HT, suggests that oleamide acts as a partial agonist. However, our data do not support the notion of oleamide acting at the primary binding site for 5HT, a traditional criterion for a partial agonist. First, clozapine, which has high affinity for the 5HT7 receptor (<50 nM) (7), blocked the 5HT-elicited cAMP response but had no effect on the oleamide-induced increase. This result indicates that oleamide and 5HT do not act at the same recognition site on the receptor. Second, oleamide (100 nM) caused insurmountable blockade of the 5HT concentration–effect curve. If 5HT and oleamide were binding to the same site, then the inhibition by oleamide should have been overcome by high (1 μM) concentrations of 5HT. In addition to clozapine, we used ketanserin, a 5HT2 selective antagonist that also has some affinity for the 5HT7 receptor (≈1 μM) (16). In addition to binding to the primary 5HT binding site on the 5HT2 receptor, ketanserin also has been shown to interact with a well defined allosteric site on this subtype (17, 18). In the present study, ketanserin (10 μM) antagonized the cAMP responses elicited by both 5HT and oleamide. It is possible that ketanserin interacts with an allosteric site on the 5HT7 receptor to prevent the responses induced by oleamide although such sites on the 5HT7 receptor have not yet been reported.
It is well known that ionotropic neurotransmitter receptors, such as the N-methyl-d-aspartate and γ-aminobutyric acid type A receptors, have multiple sites on the receptor that allow for complex modulation of channel opening (19–21). Ligands, which bind to these allosteric sites, typically modify the affinity of the receptor for classical neurotransmitters, such as glutamate and γ-aminobutyric acid, but also can regulate channel function to various extents by themselves (21, 22). Certain G protein-coupled neurotransmitter receptors, such as the muscarinic acetylcholine, α-adrenergic, and serotonin 5HT2 receptors, also exhibit allosteric binding sites, likely located on the extracellular surface of the receptor (22–26). Ligands that bind to these allosteric sites alter the conformation of the classical binding site, located deep within the pocket formed by the seven transmembrane domains, and regulate binding of classical agonists or antagonists (22–25). An independent phenomenon of allosteric antagonism operates by means of uncoupling the G protein from the receptor. For example, compounds such as heparin and trypan blue have been shown to interfere with G protein coupling of muscarinic and α- and β-adrenergic receptors (27–29).
That receptors can be activated via their allosteric sites is a notion that only recently has been realized (30). Jakubik and colleagues (30) demonstrated that gallamine, alcuronium, and strychnine, typical allosteric agents, could elicit functional responses of muscarinic receptors (m1–m3) in the absence of a muscarinic agonist. We suggest that oleamide interacts with the 5HT7 receptor in a similar manner, i.e., oleamide binds to an allosteric site on the 5HT7 receptor to stimulate cAMP accumulation, albeit at a lesser efficacy than 5HT, whereas the presence of bound oleamide on the receptor may alter the conformation in a manner that changes its affinity for 5HT or sterically hinders 5HT from binding to its primary binding site, resulting in an inhibition of the 5HT-induced response. Because oleamide had no significant effect on the 5HT2A receptors by itself, we were not able to test the nature of the interaction of oleamide with this subtype; however, it is likely that oleamide can interact with other 5HT subtypes in an allosteric manner. Oleamide’s ability to modulate at least two different subtypes in opposites directions has interesting implications. In cells expressing both subtypes, oleamide could enhance signaling at one receptor while antagonizing the other. This may represent a mechanism for fine tuning complex signaling pathways under certain conditions.
There appears to be some structural specificity for the site at which oleamide interacts, such as amidation of the carboxyl group of the fatty acid and a requirement for a cis conformation. Oleic acid, which lacks the primary amide group, did not exhibit effects on 5HT neurotransmission similar to those of oleamide. It has been shown that some neuropeptides require amidation for activity (31), suggesting that it might be a cellular mechanism that allows for functional activity of certain molecules. Only one other compound, cis-8,9-octadecenamide, which shares a similar double bond configuration as oleamide, exhibited a significant effect on 5HT-induced cAMP accumulation, indicating that the cis configuration is an important structural requirement for activity.
Our data indicate that oleamide acts via an allosteric site on the 5HT7 receptor to influence G protein signaling. This is a unique mechanism of activation for 5HT G protein-coupled receptors, and it suggests that oleamide may influence behavioral responses by interaction with 5HT receptors in this manner. These findings provide evidence that G protein-coupled neurotransmitter receptors may act more like their iontropic counterparts (i.e., γ-aminobutyric acid type A receptors) in that there may be several binding sites on the receptors that regulate functional responses to different ligands with varying efficacies.
Acknowledgments
We thank Perry Molinoff for generously supplying rat P11 cells and Benjamin Cravatt for supplying the oleamide and related compounds. This work was supported in part by National Institutes of Health Grants GM32355, MH47680, and AFOSR F49620-95-1-0247.
ABBREVIATION
- 5HT
5-hydroxytryptamine
References
- 1.Devane W A, Hanus L, Breuer A, Pertwee R G, Stevenson L A, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 2.Arafat E S, Trimble J W, Andersen R N, Dass C, Desiderio D M. Life Sci. 1989;45:1679–1687. doi: 10.1016/0024-3205(89)90278-6. [DOI] [PubMed] [Google Scholar]
- 3.Cravatt B F, Pospero-Garcia O, Siuzdak G, Gilula N B, Hendrikson S J, Boger D L, Lerner R A. Science. 1995;268:1506–1509. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]
- 4.Huidobro-Toro J P, Harris R A. Proc Natl Acad Sci USA. 1996;93:8078–8082. doi: 10.1073/pnas.93.15.8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raible D W, McMorris F A. J Neurosci Res. 1990;27:43–46. doi: 10.1002/jnr.490270107. [DOI] [PubMed] [Google Scholar]
- 6.Ivins K J, Molinoff P B. Mol Pharmacol. 1990;37:622–630. [PubMed] [Google Scholar]
- 7.Lovenberg T W, Baron B M, de Lecea L, Miller J D, Prosser R A, Rhea M A, Foye P E, Racke M, Slone A L, Siegel B W, Danielson P E, Sutcliffe J G, Erlander M G. Neuron. 1993;11:449–458. doi: 10.1016/0896-6273(93)90149-l. [DOI] [PubMed] [Google Scholar]
- 8.Thomas E A, Baker S B, Ehlert F J. Mol Pharmacol. 1993;44:102–110. [PubMed] [Google Scholar]
- 9.Candell L M, Yun S H, Tran L L P, Ehlert F J. Mol Pharmacol. 1990;38:689–697. [PubMed] [Google Scholar]
- 10.Koschel K. Eur J Biochem. 1980;108:163–169. doi: 10.1111/j.1432-1033.1980.tb04708.x. [DOI] [PubMed] [Google Scholar]
- 11.Shi Q H, Ruiz J A, Ho R J. Arch Biochem Biophys. 1986;251:156–165. doi: 10.1016/0003-9861(86)90062-7. [DOI] [PubMed] [Google Scholar]
- 12.Manzoni O, Fagni L, Pin J P, Rassendren F, Poulat F, Sladecsek F, Bockaert J. Mol Pharmacol. 1990;38:1–6. [PubMed] [Google Scholar]
- 13.Saxena P R. Pharmacol Ther. 1995;66:339–368. doi: 10.1016/0163-7258(94)00005-n. [DOI] [PubMed] [Google Scholar]
- 14.Ivins K J, Molinoff P B. J Pharmacol Exp Ther. 1991;259:423–429. [PubMed] [Google Scholar]
- 15.Ferry R C, Unsworth C D, Molinoff P B. Mol Pharmacol. 1993;43:726–733. [PubMed] [Google Scholar]
- 16.Bard J A, Zgombick J, Adham N, Vaysse P, Branchek T A, Weinshank R L. J Biol Chem. 1993;268:23422–23426. [PubMed] [Google Scholar]
- 17.Frenken M, Kaumann A, J. Naunyn-Schmiedebergs Arch Pharamcol. 1987;335:359–366. doi: 10.1007/BF00165548. [DOI] [PubMed] [Google Scholar]
- 18.Wang C D, Gallaher T K, Shih J C. Mol Pharmacol. 1993;43:931–940. [PubMed] [Google Scholar]
- 19.Reynolds I J, Miller R J. Adv Pharmacol. 1990;21:101–126. doi: 10.1016/s1054-3589(08)60340-3. [DOI] [PubMed] [Google Scholar]
- 20.Wisden W, Seeburg P H. Curr Opin Neurobiol. 1992;2:263–269. doi: 10.1016/0959-4388(92)90113-y. [DOI] [PubMed] [Google Scholar]
- 21.Deutsch S I, Mastropaolo J, Hitri A. Clin Neuropharmacol. 1992;15:352–364. [PubMed] [Google Scholar]
- 22.Costa E. Neuropharmacology. 1989;2:167–174. [Google Scholar]
- 23.Jakubik J, Tucek S. J Neurochem. 1994;63:1932–1940. doi: 10.1046/j.1471-4159.1994.63051932.x. [DOI] [PubMed] [Google Scholar]
- 24.Lee N H, el-Fakahany E E. Biochem Pharmacol. 1991;42:199–205. doi: 10.1016/0006-2952(91)90703-8. [DOI] [PubMed] [Google Scholar]
- 25.Xu Z, Purdy R E. J Pharmacol Exp Ther. 1989;248:1091–1095. [PubMed] [Google Scholar]
- 26.Howard M J, Hughes R J, Motulsky H J, Mullen M D, Insel P A. Mol Pharmacol. 1987;32:53–58. [PubMed] [Google Scholar]
- 27.Huang R C, Dehaven R N, Cheung A H, Diehl A H, Dixon R A F, Strader C D. Mol Pharmacol. 1989;37:304–310. [PubMed] [Google Scholar]
- 28.Willuweit B, Aktories K. Biochem J. 1988;249:857–863. doi: 10.1042/bj2490857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerstin E H, Luong T, Ehlert F J. J Pharmacol Exp Ther. 1992;263:910–917. [PubMed] [Google Scholar]
- 30.Jakubik J, Bacakove L, Lisa V, El-Fakahany E E, Tucek S. Proc Natl Acad Sci USA. 1996;93:8705–8709. doi: 10.1073/pnas.93.16.8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kizer J S, Busby W H, Jr, Cottle C, Youngblood W W. Proc Natl Acad Sci USA. 1984;81:3228–3232. doi: 10.1073/pnas.81.10.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guan, X., Cravatt, B. F., Ehring, G. R., Hall, J. E., Boger, D. L., Lerner, R. A. & Gilula, N. B. (1997) J. Cell Biol., in press. [DOI] [PMC free article] [PubMed]