

Abstract
We investigated the ability of pyrroloquinoline quinone (PQQ) to confer resistance to acute oxidative stress in freshly isolated adult male rat cardiomyocytes. Fluorescence microscopy was used to detect generation of reactive oxygen species (ROS) and mitochondrial membrane potential (Δψm) depolarization induced by hydrogen peroxide. H2O2 caused substantial cell death, which was significantly reduced by preincubation with PQQ. H2O2 also caused an increase in cellular ROS levels as detected by the fluorescent indicators CM-H2XRos and dihydroethidium. ROS levels were significantly reduced by a superoxide dismutase mimetic Mn (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP) or by PQQ treatment. Cyclosporine-A, which inhibits mitochondrial permeability transition, prevented H2O2-induced Δψm depolarization, as did PQQ and MnTBAP. Our results provide direct evidence that PQQ reduces oxidative stress, mitochondrial dysfunction, and cell death in isolated adult rat cardiomyocytes. These findings provide new insight into the mechanisms of PQQ action in the heart.
Keywords: mitochondria, rat, ischemia, oxidative stress
Mechanisms of cell injury during myocardial infarction are incompletely understood. Restoration of blood flow as a treatment for acute ischemia is a common and effective therapeutic approach. Although reperfusion after a transient ischemic episode permits tissue recovery, it can also cause further irreparable damage. Ischemia and reperfusion of the heart induce profound cellular damage and tissue dysfunction. It is well-established that a burst of reactive oxygen species (ROS) accompanies reperfusion after an ischemic event and that antioxidants can protect against this injury [1; 2].
It is also well-recognized that mitochondrial dysfunction contributes to impaired cardiac performance in ischemia/reperfusion (I/R) injury. For example, excessive ROS generation by damaged mitochondria has been implicated as one of the mechanisms by which cardiac myocyte death occurs during ischemic injury [3]. This has led to the concept that mitochondria are a rational target for anti-ischemic drugs [4], and to the experimental use of antioxidants to prevent ischemic injury to the heart [5].
Pyrroloquinoline quinone (PQQ) is a nutrient widely distributed in nature and serves as a non-covalently bound redox cofactor in a series of bacterial quinoprotein dehydrogenases [6]. In the presence of reducants, PQQ scavenges ROS in bacteria [7]. In vitro studies show that PQQ protected isolated liver mitochondria from damage after oxidative stress [8] and scavenged superoxide radicals [9; 10]. PQQ was neuroprotective in a rodent stroke model [11; 12] presumably by scavenging peroxynitrite [13]. Our previous in vivo rat studies of I/R injury showed that PQQ reduced myocardial infarct size, improved cardiac function [14; 15], and also reduced lipid peroxidation as measured by malondialdehyde levels [14]. Based on this study, we suggested that PQQ protected the heart from I/R injury via its action as a free radical scavenger.
To date however, there have been no studies characterizing the mechanism of PQQ effects in isolated cardiac myocytes or on ROS generation in these cells. Therefore, the aim of this study was to investigate the effect of PQQ on oxidant-induced mitochondrial dysfunction and cell death.
Materials and Methods
Cardiac myocytes
Adult cardiac myocytes were isolated and cultured from 250-300 g male rats (Charles River) as previously described in our laboratory for murine myocytes [16]. The animal protocol was approved by the San Francisco Veterans Affairs Medical Center Animal Studies Committee. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Chemicals and reagents for experiments were purchased from Sigma (St. Louis, MO), except where otherwise noted.
Myocytes were suspended in minimum essential medium (MEM) with Hanks Buffered Salt Solution (HBSS), 10 μg/ml penicillin, 1.5 μM vitamin B12, and 10 mM BDM. For cell viability assays, myocytes were plated on 35 mm tissue culture dishes coated with laminin (10μg/ml) for a 1.5h attachment period in 2% CO2/air at 37°C. For fluorescence imaging, myocytes were plated overnight on laminin-coated 15 mm glass cover slips for a 1.5h attachment period in 2% CO2/air at 37°C. After this period of attachment, the medium was changed to MEM/HBSS containing 10 μg/ml penicillin, 1.5 μM vitamin B12, and 1 mM BDM, and incubated overnight. Experiments were performed the day following isolation and culture.
Cell Viability
Myocyte survival was determined by counting live cells in the experimental cultures compared to the number of live cells in the control cultures as determined by a trypan blue exclusion assay as previously described in our laboratory [16], and shown in Figure 1.
Figure 1.
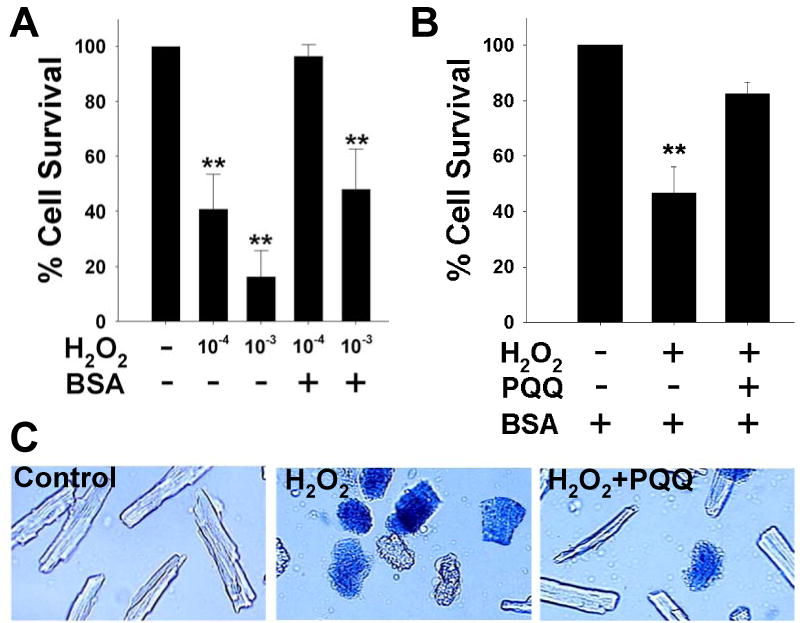
PQQ protects against H2O2 in rat cardiac myocytes. Acutely isolated adult rat cardiac myocytes grown on laminin-coated glass coverslips were exposed to control (vehicle-treated), H2O2, or H2O2 + PQQ. Control – vehicle treatment; H2O2 (1 mM) for 10 min; PQQ (10 μM) pretreatment for at least 30 minutes prior to experiment.
(A) Acutely isolated rat cardiac myocytes treated with from vehicle (control), H2O2 (1 mM and 100 μM), ± bovine serum albumin (BSA).
(B) Rat cardiac myocytes incubated with 0.1% BSA were assayed for cell viability, and mean values (± SEM) from control, H2O2, and H2O2+PQQ experimental groups are represented. Approximately 400-600 cells were counted in a total of 10 fields per dish for each experimental group. Three rat hearts were used for each of the conditions shown in the bargraph.
(C) Representative images of acutely isolated rat cardiac myocytes from control, H2O2, and H2O2+PQQ experimental groups. Live cells were rod-shaped and excluded the vital dye trypan blue, indicating an intact plasma membrane. Dead cells were contracted and stained positive for trypan blue, indicating a compromised plasma membrane. ** p<0.01 compared to control.
Fluorescence microscopy
For fluorescence imaging, experiments were initiated by transferring the coverslips to an open-bath imaging chamber (Warner Instruments, Hamden, CT), and replacing the culture medium with recording buffer containing (in mM): NaCl (120.4), CaCl2 (1.0), KCl (4.7), MgSO4 (1.2), Na2HPO4 (0.6), K2HPO4 (0.6), NaHCO3 (4.6), glucose (5.6), HEPES (10.0), taurine (5.0), and 0.1% bovine serum albumin (BSA), and filtered through a 0.2 μm sterile filter before use. The imaging chamber was placed onto an Axiovert 200M inverted microscope equipped with a 40× oil objective, fast excitation and emission shutters, high-speed filter wheel, and a mechanized stage equipped for holding the imaging chamber. Pharmacological compounds were added at least 30 minutes prior to the addition of H2O2 from concentrated stocks prepared in recording buffer.
Fluorescence was then collected and analyzed using a CCD digital camera (ORCA-ER; Hamamatsu Corporation, Bridgewater, NJ), operated with Openlab software (ImproVision, Boston, MA) on a Mac G4. Fluorescence values were collected as a field average for each excitation / emission pair, with 1-3 cells assessed per coverslip. These values were averaged separately for each experiment and normalized to the baseline values obtained prior to H2O2 or vehicle (control) exposure.
Mitochondrial membrane potential (Δψm)
Δψm was was was monitored using the potentiometric fluorescent dye tetramethylrhodamine methyl ester (TMRM; Molecular Probes, Eugene, OR), under non-quench conditions as modified from previous work in our laboratory [17]. TMRM was prepared with dimethyl sulfoxide (DMSO) to a stock concentration of 10-2 M, stored at a concentration of 5 × 10-7 M, and diluted to a working concentration of 5 × 10-10 M in recording buffer. Rat cardiac myocytes were loaded for at least 60-90 minutes with TMRM (5 × 10-10 M), and then imaged with recording buffer that contained equimolar TMRM. The non-quench loading condition was established by administering a range of TMRM concentrations to cultures and confirming a fluorescence decrease after incubation with carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP), a mitochondrial uncoupler. In addition, FCCP (1 μM) was added at the end of each experimental recording to verify non-quench conditions (i.e. a decrease in TMRM fluorescence). Experiments were initiated by placing rat cardiac myocytes on glass cover slips mounted onto an open-bath imaging chamber. The dye was excited at 545 nm using a rhodamine excitation filter (Chroma Technology, Brattleboro, VT), and fluorescence emission was measured using a rhodamine dichroic mirror and emission filter set (590 nm).
Reactive oxygen species (ROS)
ROS generation was assayed similarly to Δψm, with minor modifications using chloromethyl derivative of dihydro X-rosamine (or CM-H2XRos), a modified version of MitoTracker Red (Molecular Probes, Eugene, OR). CM-H2XRos is a reduced, nonfluorescent dye that selectively stains mitochondria in actively respiring cells and increases in fluorescence upon oxidation with increase cellular ROS levels [18; 19]. We also used the ROS scavenger Mn (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP), a cell-permeable superoxide dismutase (SOD) mimetic and peroxynitrite scavenger (Calbiochem, La Jolla, CA). The fluorescent properties of CM-H2XRos are similar to TMRM fluorescence, and therefore the same microscope settings and filters were used for both dyes. Another indicator of ROS, dihydroethidium (DHE), was used to verify the data obtained with CM-H2XRos. The fluorescent properties of DHE are similar to TMRM and CM-H2XRos.
Statistics
For single comparisons, Student's t-test was performed. For multiple comparisons, either one-way analysis of variance or a repeated measures analysis of variance was carried out. Post-hoc analysis was performed using the Student-Newman-Keuls test. P<0.05 was considered significant.
Results
Cell death
We performed concentration-response experiments to obtain a working concentration of H2O2 that routinely causes approximately 50% cell death (Figure 1A). In experimental buffer lacking bovine serum albumin (BSA), 100 μM H2O2 caused approximately 60% cell death. In the presence of BSA, which is necessary for maintaining viable cultured myocytes in experimental media, cell death was significantly prevented. Therefore, we increased H2O2 to 1 mM to reach the goal of 50% cell mortality. Viable cells exhibited rod-shaped morphology, and viability was verified by the exclusion of the vital stain, trypan blue. Dead cells exhibited a contracted morphology and stained with trypan blue. PQQ pretreatment (10 μM) substantially prevented cell demise (Figure 1B). Representative images of live and dead cardiac myocytes are shown in Figure 1C.
ROS in intact cardiac myocytes
Imaging of adult cardiomyocytes stained with the reduced version of CM-H2XRos revealed low intracellular fluorescence throughout the cells. Figure 2 shows sample traces of CM-H2XRos -loaded myocytes subjected to H2O2, which demonstrated an increase in fluorescence as a direct correlate of an increase in cellular ROS levels. Treatment with PQQ abrogated the increase in fluorescence, indicating that PQQ prevented the increase in cellular levels of ROS, suggesting that PQQ may be a ROS scavenger.
Figure 2.
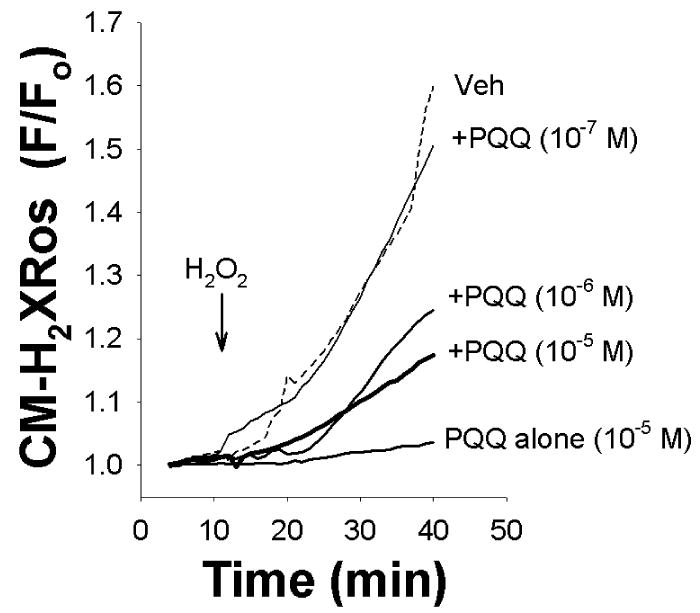
Sample traces of ROS-mediated oxidation of CM-H2XRos plotted as an increase in fluorescence over time. H2O2 caused an increase in CM-H2XRos fluorescence (dotted line), which was dose-dependently reduced by increasing PQQ concentrations. PQQ alone (10 μM) did not cause any significant change in CM-H2XRos fluorescence (solid line, bottom trace).
Average fluorescence values of CM-H2XRos for each experimental group were plotted over time (Figure 3A), and these data showed that PQQ significantly reduced the increase in CM-H2XRos fluorescence 40 minutes after H2O2 exposure (Figure 3B). To verify this finding, we tested the effect of the cell permeable superoxide dismutase mimetic Mn (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP), which is a potent free radical scavenger [20]. As shown, MnTBAP (10 μM) significantly prevented the increase in CM-H2XRos fluorescence (Figure 3A, B).
Figure 3.
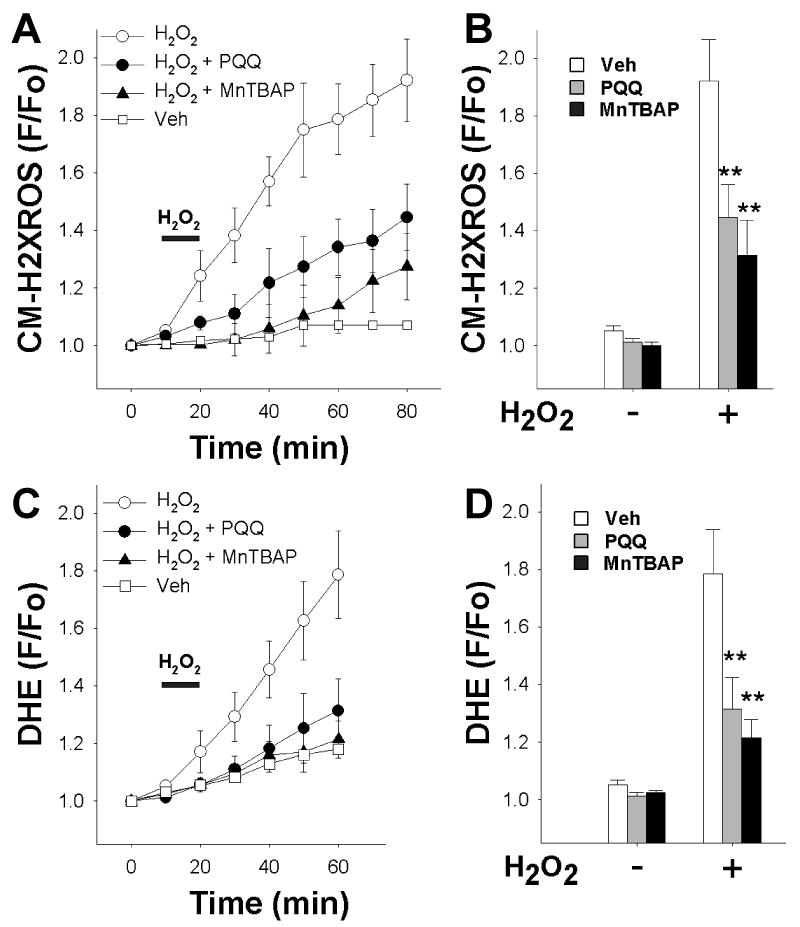
PQQ protects against an increase in cellular reactive oxygen species (ROS) in acutely isolated adult rat cardiac myocytes.
(A) Rat cardiomyocytes were loaded with CM-H2XRos to detect the increase in intracellular levels of ROS. CM-H2XRos fluorescence was collected, and fluorescence (F) at the indicated timepoint was normalized to baseline fluorescence (Fo) and plotted over time. Bar indicates addition of H2O2. Each point represents the mean ± SEM of 3-5 coverslips, each prepared from a separate rat heart. For each coverslip, 1-3 cells were analyzed. Veh – vehicle treatment (control); H2O2 (1 mM); PQQ (10 μM); MnTBAP (10 μM); PQQ and MnTBAP were administered for at least 30 minutes prior to H2O2.
(B) Peak cellular levels of ROS detected as a function of CM-H2XRos normalized to baseline value after 60 min H2O2 exposure. PQQ (gray bars) and MnTBAP (black bars) significantly reduced fluorescence compared to control (white bars).
(C) Cardiomyocytes were loaded with DHE to detect the increase in intracellular levels of ROS. DHE fluorescence was collected, and fluorescence (F) at the indicated timepoint was normalized to baseline fluorescence (Fo) and plotted over time. Arrow indicates addition of H2O2. Each point represents the mean ± SEM of 3-5 coverslips, each derived from a separate rat heart. For each coverslip, 1-3 cells were analyzed. Veh – vehicle treatment (control); H2O2 (1 mM); MnTBAP (10 μM); PQQ and MnTBAP were administered for at least 30 minutes prior to H2O2.
(D) Peak cellular levels of ROS detected as a function of DHE normalized to baseline value after 40 min H2O2 exposure. PQQ (gray bars) and MnTBAP (black bars) significantly reduced fluorescence compared to control (white bars). ** p<0.01 compared to control (H2O2 alone).
In addition, we used dihydroethidium (DHE) as another indicator of ROS in intact cells. Treatment with H2O2 caused an increase in DHE fluorescence over time, indicating an increase in ROS. This increase was reduced to near-control levels by PQQ treatment (Figure 3C). Values at the 40 minute time point after H2O2 treatment are shown in Figure 3D. These results indicate that PQQ protects against oxidative stress by decreasing cellular exposure to ROS.
Depolarization of Δψm by H2O2
Fluorescence imaging of adult cardiomycytes stained with the potentiometric fluorescent dye tetramethylrhodamine methyl ester (TMRM) revealed intracellular fluorescence throughout the cells that was similar to the staining pattern of CM-H2XRos, indicating that TMRM fluorescence was from mitochondria. Cardiac myocytes loaded with TMRM were subjected to H2O2 and demonstrated a decrease in fluorescence as a function of Δψm depolarization 45 minutes after exposure to H2O2. Exposing cardiomyocytes to H2O2 led to a decrease in TMRM fluorescence relative to control, indicating Δψm depolarization (Figure 4A). Treatment with PQQ (10 μM) prevented the decrease in TMRM fluorescence, indicating that PQQ protects against H2O2-induced Δψm depolarization.
Figure 4.
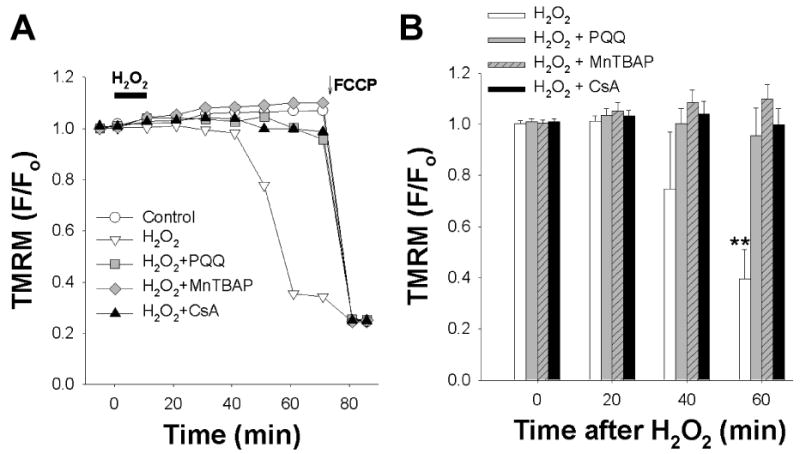
PQQ protects against H2O2-mediated Δψm depolarization in acutely isolated adult rat cardiac myocytes.
(A) Rat cardiomyocytes were loaded with TMRM to quantify Δψm depolarization. TMRM fluorescence was collected, and fluorescence (F) at indicated timepoint was normalized to baseline fluorescence (Fo) and plotted over time. Bar indicates addition of H2O2. Representative traces from 5 separate experiments are shown. Control – vehicle treatment; H2O2 (1 mM); PQQ (10 μM); MnTBAP (10 μM); CsA (200 nM). PQQ, MnTBAP, and CsA were administered for at least 30 minutes prior to H2O2. Cells were treated with the mitochondrial uncoupler FCCP (1 μM) at the end of each experiment.
(B) Bar graph of average TMRM fluorescence (F) normalized to baseline (Fo) at indicated timepoints after H2O2 exposure and treatment with PQQ (gray bars) or MnTBAP (black bars). ** p<0.01 compared to time=0, n=3-5.
We then tested the effect of the SOD mimetic and free-radical scavenger MnTBAP. Treatment with MnTBAP (10 μM) similarly prevented Δψm depolarization, thereby verifying the antioxidant effect of PQQ against H2O2-induced injury. To confirm that the Δψm depolarization was via mitochondrial permeability transition (MPT) induction, cells were treated with the MPT inhibitor cyclosporine-A (CsA). CsA (200 nM) similarly prevented Δψm depolarization. Cells were treated with the mitochondrial uncoupler FCCP (1 μM) at the end of the experiment to depolarize mitochondria.
Figure 4B shows a summary data for PQQ (10 μM), MnTBAP (10 μM), and CsA (200 nM) each of which prevented the decrease in TMRM fluorescence. These results indicate that PQQ protected against oxidative stress by preventing enhanced MPT via Δψm depolarization.
Discussion
The first major finding of this study is that PQQ substantially reduced cell death in cultured adult rat cardiac myocytes subjected to oxidative damage with H2O2. PQQ protection against oxidant-induced cell death is consistent with our previous reports demonstrating that PQQ is a highly effective in reducing infarct size and in improving hemodynamics in intact rats subjected to I/R injury [14; 15]. As a dietary supplement, PQQ has also been found to be neuroprotective in a rodent stroke model [11; 13].
The concentration of H2O2 used in these studies is within the range used by others [21; 22]. In addition, the experimental buffer requires the presence of serum albumin, which scavenges free radicals [23]. Because BSA effectively protected against H2O2 toxicity at 100 μM, we had to use 1 mM to match the efficacy of H2O2 in serum albumin-free buffer (Fig. 1A).
Previously we also reported that PQQ reduced lipid peroxidation measured by malondialdehyde levels [14]. As lipid peroxidation is the result of excessive free radical generation, the initial cell survival data provided a basis to test the hypothesis that PQQ would impair excessive mitochondrial ROS generation.
The second major finding of this study is that PQQ indeed decreased mitochondrial ROS generation. Under physiological conditions and normal mitochondrial respiration, detrimental oxidative systems coexist with anti-oxidative machinery in a dynamic equilibrium. However, ROS generation is increased in cardiac myocytes during post-ischemic reperfusion [24; 25]. These ROS species include (but are not limited to) hydrogen peroxide, superoxide, singlet oxygen, and hydroxyl anion radical. The pernicious feature of these molecules is their high reactivity as oxidative agents that target redox-sensitive cellular constituents such as lipids, proteins, and nucleic acids. The finding that PQQ reduced intracellular ROS levels is consistent with the hypothesis that it acts directly or indirectly as a potent free radical scavenger. Whether its site of action is intramitochondrial or in the cytoplasm, or both, has yet to be determined.
The third major finding of this study is that PQQ delayed mitochondrial permeability transition, which was determined by assaying Δψm. Injury after I/R is considered to be mediated by MPT induction, a phenomenon characterized by increased permeability across the mitochondrial membrane. Evidence for the role of MPT in I/R injury is exemplified by the cytoprotection conferred by the immunosuppressive compound and classical inhibitor of MPT, cyclosporine A (CsA) [26; 27].
There is contradictory evidence regarding the role of ROS in MPT induction. In isolated mitochondria, calcium ions can directly lead to MPT induction [28; 29], independent of the presence of ROS. However, others have shown that ROS can directly enhance MPT [30]. Our data are consistent with (but do not prove) the possibility that ROS generation and augmented MPT are linked, as PQQ was able both to reduce ROS generation and prevent or significantly delay MPT.
In summary, we have for the first time explored the effects of the naturally occurring quinone PQQ on cellular and subcellular levels. Our data indicate that the cardioprotection conferred by PQQ can at least in part be explained by a reduction in free radical generation and suppression of MTP. Taken together with the cardioprotection shown in an in vivo model [14; 15], these observations suggest that PQQ could serve as an effective cardioprotective agent before or during acute ischemia, such as occurs during cardiopulmonary bypass, thrombolysis and/or stent deployment in acute coronary syndromes, or as prophylaxis in high risk patients undergoing noncardiac surgery.
Acknowledgments
Grant support: NIH PO1 HL68738 (JSK), VA Merit Awards (CCA), Charitable Leadership Foundation (JSK), and American Heart Association (CCA, JZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jolly SR, Kane WJ, Bailie MB, Abrams GD, Lucchesi BR. Canine myocardial reperfusion injury. Its reduction by the combined administration of superoxide dismutase and catalase. Circ Res. 1984;54:85. doi: 10.1161/01.res.54.3.277. [DOI] [PubMed] [Google Scholar]
- 2.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61:70. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 3.Levraut J, Iwase H, Shao ZH, Vanden Hoek TL, Schumacker PT. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol. 2003;284:H549–58. doi: 10.1152/ajpheart.00708.2002. [DOI] [PubMed] [Google Scholar]
- 4.Morin D, Hauet T, Spedding M, Tillement J. Mitochondria as target for antiischemic drugs. Adv Drug Deliv Rev. 2001;49:74. doi: 10.1016/s0169-409x(01)00132-6. [DOI] [PubMed] [Google Scholar]
- 5.Marczin N, El-Habashi N, Hoare GS, Bundy RE, Yacoub M. Antioxidants in myocardial ischemia-reperfusion injury: therapeutic potential and basic mechanisms. Arch Biochem Biophys. 2003;420:36. doi: 10.1016/j.abb.2003.08.037. [DOI] [PubMed] [Google Scholar]
- 6.McIntire WS. Newly discovered redox cofactors: possible nutritional, medical, and pharmacological relevance to higher animals. Annu Rev Nutr. 1998;18:77. doi: 10.1146/annurev.nutr.18.1.145. [DOI] [PubMed] [Google Scholar]
- 7.Misra HS, Khairnar NP, Barik A, Indira Priyadarsini K, Mohan H, Apte SK. Pyrroloquinoline-quinone: a reactive oxygen species scavenger in bacteria. FEBS Lett. 2004;578:30. doi: 10.1016/j.febslet.2004.10.061. [DOI] [PubMed] [Google Scholar]
- 8.He K, Nukada H, Urakami T, Murphy MP. Antioxidant and pro-oxidant properties of pyrroloquinoline quinone (PQQ): implications for its function in biological systems. Biochem Pharmacol. 2003;65:74. doi: 10.1016/s0006-2952(02)01453-3. [DOI] [PubMed] [Google Scholar]
- 9.Smidt CR, Steinberg FM, Rucker RB. Physiologic importance of pyrroloquinoline quinone. Proc Soc Exp Biol Med. 1991;197:26. doi: 10.3181/00379727-197-43218. [DOI] [PubMed] [Google Scholar]
- 10.Bishop A, Gallop PM, Karnovsky ML. Pyrroloquinoline quinone: a novel vitamin? Nutr Rev. 1998;56:93. doi: 10.1111/j.1753-4887.1998.tb01661.x. [DOI] [PubMed] [Google Scholar]
- 11.Jensen FE, Gardner GJ, Williams AP, Gallop PM, Aizenman E, Rosenberg PA. The putative essential nutrient pyrroloquinoline quinone is neuroprotective in a rodent model of hypoxic/ischemic brain injury. Neuroscience. 1994;62:406. doi: 10.1016/0306-4522(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Feustel PJ, Kimelberg HK. Neuroprotection by pyrroloquinoline quinone (PQQ) in reversible middle cerebral artery occlusion in the adult rat. Brain Res. 2006;1094:6. doi: 10.1016/j.brainres.2006.03.111. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Rosenberg PA. The essential nutrient pyrroloquinoline quinone may act as a neuroprotectant by suppressing peroxynitrite formation. Eur J Neurosci. 2002;16:24. doi: 10.1046/j.1460-9568.2002.02169.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhu BQ, Zhou HZ, Teerlink JR, Karliner JS. Pyrroloquinoline quinone (PQQ) decreases myocardial infarct size and improves cardiac function in rat models of ischemia and ischemia/reperfusion. Cardiovasc Drugs Ther. 2004;18:31. doi: 10.1007/s10557-004-6219-x. [DOI] [PubMed] [Google Scholar]
- 15.Zhu BQ, Simonis U, Cecchini G, Zhou HZ, Li L, Teerlink JR, Karliner JS. Comparison of pyrroloquinoline quinone and/or metoprolol on myocardial infarct size and mitochondrial damage in a rat model of ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther. 2006;11:28. doi: 10.1177/1074248406288757. [DOI] [PubMed] [Google Scholar]
- 16.Tao R, Zhang J, Vessey DA, Honbo N, Karliner JS. Deletion of the sphingosine kinase-1 gene influences cell fate during hypoxia and glucose deprivation in adult mouse cardiomyocytes. Cardiovasc Res. 2007;74:63. doi: 10.1016/j.cardiores.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 17.Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:902. doi: 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- 18.Shanker G, Aschner JL, Syversen T, Aschner M. Free radical formation in cerebral cortical astrocytes in culture induced by methylmercury. Brain Res Mol Brain Res. 2004;128:57. doi: 10.1016/j.molbrainres.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 19.Itoh S, Lemay S, Osawa M, Che W, Duan Y, Tompkins A, Brookes PS, Sheu SS, Abe J. Mitochondrial Dok-4 recruits Src kinase and regulates NF-kappaB activation in endothelial cells. J Biol Chem. 2005;280:96. doi: 10.1074/jbc.M410262200. [DOI] [PubMed] [Google Scholar]
- 20.Konorev EA, Kennedy MC, Kalyanaraman B. Cell-permeable superoxide dismutase and glutathione peroxidase mimetics afford superior protection against doxorubicin-induced cardiotoxicity: the role of reactive oxygen and nitrogen intermediates. Arch Biochem Biophys. 1999;368:8. doi: 10.1006/abbi.1999.1337. [DOI] [PubMed] [Google Scholar]
- 21.Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, Shiojima I, Hiroi Y, Yazaki Y. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100:21. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H(2)O(2) regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol. 2003;35:21. doi: 10.1016/s0022-2828(03)00084-1. [DOI] [PubMed] [Google Scholar]
- 23.Gum ET, Swanson RA, Alano C, Liu J, Hong S, Weinstein PR, Panter SS. Human serum albumin and its N-terminal tetrapeptide (DAHK) block oxidant-induced neuronal death. Stroke. 2004;35:5. doi: 10.1161/01.STR.0000110790.05859.DA. [DOI] [PubMed] [Google Scholar]
- 24.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 25.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:54. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 26.Di Lisa F, Canton M, Menabo R, Dodoni G, Bernardi P. Mitochondria and reperfusion injury. The role of permeability transition. Basic Res Cardiol. 2003;98:41. doi: 10.1007/s00395-003-0415-x. [DOI] [PubMed] [Google Scholar]
- 27.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60:25. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 28.Kristian T, Siesjo BK. Calcium in ischemic cell death. Stroke. 1998;29:18. doi: 10.1161/01.str.29.3.705. [DOI] [PubMed] [Google Scholar]
- 29.Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, Brenner DA. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4:81. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- 30.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:14. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]