

Abstract
A wavelength and solvent dependent study of a photochromic indolylfulgide is presented. The ring-closure reaction is characterized using stationary and time-resolved spectroscopy with femtosecond time resolution. After excitation into the first excited singlet state (S1) the photoprocesses proceed on ultrafast timescales (0.3–0.45 ps) in both polar and non-polar solvents. Excitation into higher electronic states results in similar reaction kinetics as found for S1 excitation. A simple kinetic scheme can be established for the photoprocesses under all different experimental conditions: as expected from organic textbooks neither the solvent surroundings nor the excitation wavelength strongly alter the reaction scheme. The experimental study demonstrates that the ring-closure reaction of photochromic indolylfulgides can be considered as a very robust photoprocess: this fact may lead to a great variety of different applications where the reaction dynamics of the molecular switch are not disturbed by any surrounding effects.
Introduction
Photochromic molecules, especially fulgides/fulgimides1–7 or diarylethenes,8 have received great attention due to the number of possible applications. The underlying photochemical processes are electrocyclic reactions as found in the cyclohexadiene/hexatriene system, where two different isomers can be interconverted using light. The ring-opening of cyclohexadiene (CHD) and the ring-closure of hexatriene (HT) often served as textbook examples for the validity of the Woodward–Hoffmann rules9 and have thus been studied extensively.10–13 The molecular properties of the CHD/HT chromophore offer unique applications, as a fast and reversible change of chemical states can be selectively initiated by UV/VIS light and, together with an alteration of the absorption properties, the molecules can be used as molecular switches: optical data storage14 or even ultrafast folding experiments as performed with other photoswitchable molecules15 become possible. Beside thermal stability, high reaction quantum yield, low photochemical degradation and well-separated absorption spectra are key properties of molecular switches, which are achieved by chemical modification of the different compounds.1,2
Indolylfulgides exhibit three thermally stable ground state isomers, namely the E-, Z- and C-forms.2 The three isomers, which are shown in Scheme 1, can react according to the photochromism type P:16 thereby the Z-isomer interconverts into the E-form and into the C-form under UV radiation with a very low efficiency for the Z/E isomerization.17 Visible illumination allows the C-isomer to reform the Z-isomer in an electrocyclic ring-opening reaction. Reactions from and into the E-isomer (UV radiation) can be neglected in the case of the studied indolylfulgide as the ratio of E/Z-isomer is not greater than ~2% in any photostationary state.17
Scheme 1.

Structures of the three different isomers (E/Z/C) of the investigated indolylfulgide.
Different studies were able to demonstrate the timescale and reaction kinetics of the electrocyclic reactions of fulgides and fulgimides.3,4,6,7,18–20 Transient absorption experiments in the visible6 and infrared3,21 were used to monitor the structural and electronic changes, which occur after photoexcitation, independently. Pronounced differences for the ring-closure and the ring-opening reaction of an indolylfulgimide were found in a study using time-resolved fluorescence, which contradicted a reaction model with a common pericyclic minimum.4 It was thereby shown that the ring-closure process occurs on fast timescales <1 ps, while the ring-opening is a slower reaction on the timescale of several picoseconds. As the overall reaction times for both processes are of ultrafast nature, funnels or conical intersections (CoIn) seem to control the isomerization speed.4,12,13,22 The ring-opening reaction also proved to be sensitive to solvent effects, the environment temperature and the specific excitation wavelength.5,23
Here, we present an excitation wavelength and solvent dependent study of the ring-closure process of a trifluorinated indolylfulgide. This paper characterizes the photochemical pathway of the ring-closure in real time and confirms or rather experimentally verifies the textbook belief that electrocyclic reactions are only weakly influenced by surrounding effects.24 Therefore dynamics of the ring-closure reaction of the trifluorinated indolylfulgide were recorded in cyclohexane, 1,4-dioxane and acetonitrile using time-resolved spectroscopy with femtosecond time-resolution. The influence of the excitation conditions, e.g. excitation into the first excited state or higher excited states, are also investigated. These data establish a detailed reaction model for the ring-closure reaction and show that pronounced differences exist between the two electrocyclic reactions of indolylfulgides: ring-closure and ring-opening.
Materials and methods
The synthesis of the investigated trifluorinated indolylfulgide has been described elsewhere.17,25 The fulgide was dissolved in the respective solvent (acetonitrile (ACN), 1,4-dioxane (DIO), cyclohexane (CH), Merck, spectroscopic grade >99%) with concentrations ranging from 2–5 mM for all experiments.
Stationary absorption spectra were recorded using a spectrophotometer (Perkin-Elmer, Lambda19). The photochemical quantum yields of the ring-closure reaction were determined as described in a former publication:26 a change in the sample composition was induced by actinic light within a certain time period using the following light sources for illumination: light at a wavelength of 414 nm (S1 excitation) was obtained from a violet laser module (VLMA-1, Roithner, 0.3 mW). A Hg/Xe lamp (Hamamatsu, Lightningcure LC4, Japan) combined with suitable filtering (liquid Backström filter for 313 nm) was used as UV source at 313 nm (SN excitation). The induced absorption changes in a wavelength range of predominant absorption of the C-isomer (560 nm) were recorded for different illumination periods. The changes in the concentration of the Z/C-isomer and therefore the number of product molecules were calculated from the recorded absorption changes using the absolute extinction coefficient of the C-isomer (ε(C)571 = 6400 l mol−1 cm−1)17 at the detection wavelength.
The femtosecond measurements were performed under photostationary conditions: therefore the sample solution of the fulgide compound was illuminated with a cold light source (KLC2500, Schott, Mainz filtered by a 3 mm thick OG570). This resulted in the formation of a photostationary state (PSS570) containing approximately >98% Z-isomer and <2% E-isomer. The sample solution was pumped through a fused silica cuvette (Hellma) to ascertain defined starting conditions.
Femtosecond pump–probe spectroscopy in the visible (VIS) and near-ultraviolet (UV) spectral range was used to investigate the dynamics of the photochemical reactions for different solvent surroundings and excitation conditions. The experiments were conducted using two different laser systems: a home-built Ti:sapphire-based regenerative amplifier laser system and a commercial Spitfire Pro XP (Spectra Physics, Darmstadt) generated ultrashort light pulses at 800 nm with a pulse duration of ~90 fs (home-built)/~70 fs (Spitfire) at a repetition rate of 1 kHz. Pump pulses at 400 nm were generated by frequency doubling the laser fundamental in a 0.5 mm thick non-linear β-barium borate crystal (BBO, type I, 29°). Tunable pump pulses in the visible were generated in a non-collinear optical parametric amplifier (NOPA).27 The NOPA was adjusted to produce light pulses at 540 nm, which were subsequently frequency-doubled to 270 nm in a 100 μm thick BBO crystal (type I, 35°). Excitation energies for the pump–probe measurements varied from 100 to 400 nJ. A white light continuum, generated in CaF2 was used as probe light (350–660 nm).28 The transient absorption changes in the white light continuum induced by the pump pulse were recorded at various delay times using a multi-channel detection setup.29 The temporal delay between pump and probe pulses was varied by a mechanical delay stage. The time resolution of the pump–probe experiment for excitation in the UV range was determined to be ~250 fs. A better time resolution (~100 fs) was achieved for pump pulses centered at 400 nm. The spot diameter of the pump light at the sample location was in the range of ~100 μm providing a homogeneous excitation density for the probe pulse (diameter 30–40 μm). In order to avoid effects of rotational diffusion,30 the polarization of pump and probe pulses was at magic angle. A global fit analysis was used to extract decay associated spectra from the transient absorption data set. Further details concerning the transient absorption spectrometer and the laser system can be found in ref. 23b and 26.
Results and spectroscopic interpretation
Stationary measurements
The photochromic properties of the investigated indolylfulgide arise from a photoinduced ring-closure/ring-opening reaction: upon light illumination the open form (Z-isomer) can undergo an electrocyclic ring-closure as shown in Fig. 1.‡ The formed C-isomer exhibits a cyclohexadiene motif, while the Z-form incorporates a hexatriene structure. Both motifs are highlighted in Fig. 1.
Fig. 1.
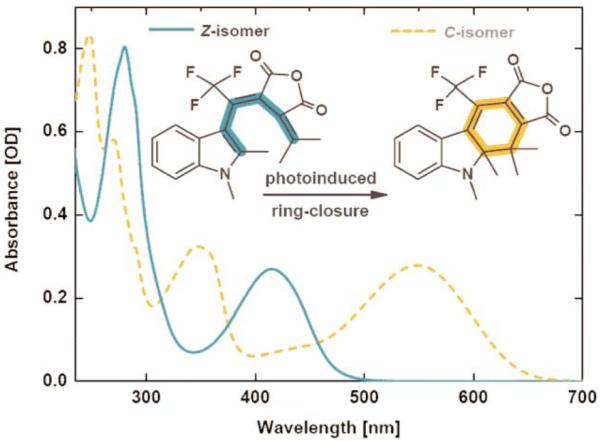
Stationary absorption spectra of two isomeric forms of the investigated fulgide (solvent cyclohexane) and schematic representation of the electrocyclic ring-closure. The dashed curve shows a photostationary state with ~95% C-isomer.
The absorption maximum of the Z-form is located in the near UV range. In the solvent cyclohexane (CH) the absorption band assigned to the S0 → S1 transition peaks at 416 nm. Higher energetic excitation at 280 nm is associated with the S0 → SN transition (Fig. 1). Illumination into one of these bands results in strong changes throughout the complete spectrum: the band around 416 nm is reduced while two new absorption features centered at 350 nm and 550 nm grow in. These new bands are related to the formation of the C-isomer. The quantum efficiency for the ring-closure process after S1 excitation in CH is determined to be 17% (Table 1). Excitation into the SN band does not significantly change this value (20%, Table 1). The strong visible absorption of the C-isomer at 550 nm is due to a planar geometry and a strongly enlarged π-electron system.2 The flexible Z-structure allows a dihedral angle of ~45°, separating the π-electron system of the indolyl moiety and the maleic imide, resulting in a strongly blue-shifted absorption compared to the C-isomer.31
Table 1.
Stationary absorption data (Z-isomer) and reaction quantum yields for the ring-closure reaction in different solvents and for different excitation conditions
| Absorption maximum/nm |
Reaction quantum yield (%) |
|||
|---|---|---|---|---|
| Solvent | S1 | SN | S1 | SN |
| Cyclohexane | 416 | 280 | 17 ±1 | 20 ± 5 |
| 1,4-Dioxane | 425 | 280 | 15 ±1 | — |
| Acetonitrile | 430 | 280 | 10 ±1 | — |
The absorption properties of the Z-isomer in a changing solvent environment behave as follows: increasing the solvent polarity from CH → DIO → ACN shifts the peak maximum of the S0 → S1 transition from 416 nm → 425 nm → 430 nm (Table 1). However, this positive solvatochromism is not found for the S0 → SN transition, where the peak remains at a spectral position of 280 nm for all different solvents (Table 1). This behavior clearly shows that the Franck–Condon state (FC*) of the S0 → S1 transition is more polar than the corresponding ground state, while the FC* state of the S0 → SN transition features similar properties compared to its electronic ground state.
The photochemical quantum yield of the ring-closure reaction is 17% in CH (excitation to S1). According to Kasha’s rule32 this value is very similar for SN excitation (20%). However, the changing solvent polarity alters the reaction quantum yields from Z → C in a systematic way: the highest value is found for CH with 17% and decreases to 15% in DIO down to 10% for ACN (Table 1). All results from stationary measurements are summarized in Table 1.
Time-resolved spectroscopy
The ultrafast dynamics of the ring-closure reaction are investigated by means of transient absorption spectroscopy in the visible spectral range. Fig. 2 shows an overview of the reaction dynamics of the indolylfulgide in the solvent CH under different excitation conditions.
Fig. 2.

Transient absorption data of the investigated indolylfulgide in the solvent cyclohexane for different excitation conditions: (a) λexc = 400 nm, the results (b) λexc = 270 nm. The upper panel shows transient spectra at certain delay times between pump and probe pulse. The lower panel presents the results (decay associated spectra and corresponding time constants) of a global fitting routine of the complete data sets.
S1 excitation in CH
The upper panel of Fig. 2a shows an excerpt of the transient spectra for various delay times between pump and probe pulse. We observe strong absorbance changes throughout the investigated spectral range (Fig. 2a, tD = 0.2 ps). Ground state bleaching (GSB) reduces the induced absorption in a wavelength range which can be assigned to the absorption of the ground state Z-isomer (λmax = 416 nm). Pronounced signal contributions due to stimulated emission (SE) are not observable most likely due to overlapping signals originating from excited state absorption (ESA). These different signals result in an ESA band centered around 520 nm. This band partially decays on fast timescales (Fig. 2a, tD = 0.2 ps vs. 0.5 ps vs. 1.0 ps). Subsequently, the complete signal with ESA and GSB decays into a constant offset (Fig. 2a, tD = 200 ps) on the timescale of several ten picoseconds (Fig. 2a, tD = 1.0 ps vs. 5.0 ps vs. 20 ps). This offset nicely resembles the steady-state difference between the C- and Z-isomer absorption (Fig. 1). Additionally, this fact gives no indication for the formation of a significant amount of E isomer molecules, as Z and E isomers differ in their absorption maximum by ~15 nm.17
A multi-exponential fit allows reproducing the complete data set with three decay components together with an additional offset (Fig. 2a, lower panel). The time constants are found to be τ1 = 0.45 ps, τ2 = = 4.1 ps and τ3 = 32 ps, where the fastest process shows the strongest amplitude (Fig. 2a, τ1 = 0.45 ps). The decay associated spectrum (DAS) of this component is dominated by positive signals with a maximum around 550 nm and a minimum at 420 nm. Its large amplitude and the spectral characteristics assign this process to the decay of the excited singlet state, together with a repopulation of the ground state of both product and reactant molecules. This fast internal conversion process (τ1 = 0.45 ps) generates molecules in a vibrationally hot ground state. As a consequence subsequent processes partially show sigmoidal signatures which are typical for vibrational cooling processes in the electronic ground state (Fig. 2a, τ2, τ3 lower panel). The spectral signature of the cooling process is expected to have minor amplitude for formed product molecules (~20%) and a dominant signature of the reformed reactant molecules (~80%).§ The fraction of molecules which has not undergone the ring-closure reaction shows up as a positive signal at 475 nm and a negative signal at 420 nm (Fig. 2a, τ2 4.1 ps). The minor fraction of product molecules (~20%) is found=in the long wavelength part (Fig. 2a, τ2 = 4.1 ps, λmax: 650 nm, λmin: 560 nm) and is partially overlapped by spectral signatures of hot reactant molecules—the cooling signature of the DAS behaves exactly as expected from stationary measurements. The non-exponential behavior of the cooling processes is thereby approximated using a biexponential fit with the time constants 4.1 ps and 32 ps. The large amplitudes of the DAS, related to these time constants, may also be interpreted in a way that some molecules remain trapped in the excited state and return into the ground state on longer timescales. Further on, the spectral shape of the 32 ps component allows no distinct assignment of this process to cooling. We disregard this kind of interpretation in the present paper as infrared experiments of a related compound show that the major fraction of product molecules is already formed on a sub-picosecond timescale and displays cooling dynamics on the timescale of several ten picoseconds.3,21
The transient behavior presented above can be summarized in a kinetic reaction scheme, which is already known from the literature of related compounds: after light excitation at 400 nm, the molecules reach the Franck–Condon (FC*) region. It has been shown using time-resolved fluorescence measurements on fulgimide compounds that the originally populated region (FC*) is left with a short time constant <0.1 ps most likely together with a change of electronic states. This transition strongly alters the electronic properties of the molecules and results in the formation of a fluorescently dark state.4 These initial processes do not show up in our experiments, which may be explained by the time resolution of ~100 fs or coherent artifacts in the pump–probe signal. The subsequent photoreaction can occur from this dark state with a time constant τ1 = 0.45 ps (Fig. 2a)—thereby the hot ground states of reactant and product molecules are populated. This interpretation is strongly supported by fluorescence and IR absorption measurements on related fulgimide compounds.3,4,21 On later timescales vibrational cooling to the solvent takes place with time constants of τ2/3 = 4.1 ps/32 ps (Fig. 2a). The actual product formation is seen as a constant offset. We can conclude that both the experimental data and a close inspection of literature data3,4,21 point to an ultrafast ring-closure process on a sub-picosecond timescale with a quantum yield of ~17%.
SN excitation in CH
Fig. 2b shows the transient absorption data induced by excitation in the UV range. We find an overall similar behavior for the ring-closure at 400 nm (S1)/270 nm (SN), therefore compare Fig. 2a and 2b: again ESA is overlapped by GSB (~415 nm) at early delay times (Fig. 2b, tD = 0.2 ps). The signal decays with a fast component on the sub-picosecond timescale (Fig. 2b, tD = 0.2 ps vs. 0.5 ps vs. 1.0 ps), shows slight modifications and decay of the induced absorption in the time range of several picoseconds (Fig. 2b, tD = 1.0 ps vs. 5 ps vs. 20 ps) and finally leaves a constant offset (Fig. 2b, tD = 200 ps). The data set is well reproduced by an exponential model with three time constants of 0.4 ps, 7.6 ps and 46 ps (Fig. 2b, lower panel). In order to relate this data set to the known behavior of S1 excitation in Fig. 2a the additional time constant of 46 ps with a very small amplitude is used. Ignoring this time constant reduces the quality of the fit only moderately. Nevertheless, we chose to use the third time constant in order to get smaller errors and to obtain a better comparison with the data for S1 excitation.
Slight differences between the two data sets (400 nm/270 nm excitation) can be observed at early delay times (Fig. 2a and 2b, tD = 0.2 ps vs. 0.5 ps). These are most likely due to the lower time resolution of ~250 fs for 270 nm excitation, which may distort the transient spectra in this time range.
The interpretation of the data set leads to an overall similar picture for both excitation conditions: we do not observe any spectral signature of the SN state after absorption of a 270 nm photon. Therefore we conclude that the molecule converts back into the first excited singlet state within our time resolution (Fig. 2b, tD = 0.2 ps vs. 0.5 ps). Differences in the two data sets at early delay times can hence be referred to a lower time resolution for higher energetic excitation. The subsequent decay of the excited state proceeds with the fast time constant of 0.4 ps, where the DAS shows the strongest amplitude showing typical spectral features of the S1 state (Fig. 2b, τ1 = 0.4 ps). The spectral signature of the DAS and the value of the time constant are nearly equivalent compared to S1 excitation. Again a pronounced cooling signature is found (Fig. 2b, τ2 = 7.6 ps, τ3 = 46 ps). The main component (Fig. 2b, τ2 = 7.6 ps) is nearly identical to the cooling signature found after 400 nm excitation. The longer value (τ2 ps 4.1 for 400 nm excitation, τ2 = 7.6 ps for 270 nm excitation) and the tiny amplitude of the τ3 = 46 ps component is attributed to a higher amount of excess energy. The kinetic reaction scheme for higher electronic excitation is hence very similar to the S1 excitation case. Within our time resolution an ultrafast decay of the SN state into the first excited state according to Kasha’s rule32 takes place. The photoreaction after 270 nm excitation proceeds with the fast time constant τ1 = 0.4 ps via the reaction scheme discussed above for 400 nm excitation. Subsequent cooling processes on the timescale of several picoseconds mark the completion of the ring-closure reaction after SN excitation.
Solvent dependency for S1 excitation
Fig. 3 gives an overview of the transient absorption data of the fulgide in the solvents 1,4-dioxane (DIO) and acetonitrile (ACN) recorded after excitation at 400 nm.
Fig. 3.

Transient absorption data of the investigated indolylfulgide in the solvent (a) 1,4-dioxane and (b) acetonitrile for an excitation wavelength of λexc = 400 nm. The upper panel shows transient spectra at certain delay times between pump and probe pulse. The lower panel presents the results (decay associated spectra and corresponding time constants) of a global fitting routine of the respective data sets.
The transient spectra of the indolylfulgide in DIO and ACN (Fig. 3a and b, upper panel) show a strong similarity to the data found for CH (Fig. 2a). A broad ESA in the complete investigated spectral range is superimposed by GSB around 430 nm depending on the specific ground state absorption of the sample (Fig. 3a and b, tD = 0.2 ps). The complete signal decays multi-exponentially (Fig. 3a and b) into a constant offset spectrum, which is reached after several ten picoseconds (Fig. 3a and b, tD = 200 ps).
A closer inspection of the data sets by a global fitting analysis reveals the best description of the data using three time constants together with an offset. Again, the offset nicely resembles the difference between the absorption spectra of Z- and C-isomer. We find the time constants of 0.35 ps, 4.0 ps and 25 ps for the solvent DIO (Fig. 3a). ACN is best described by 0.3 ps, 1.2 ps and 15 ps (Fig. 3b). The fast time constants τ1 show the largest amplitudes and are related to internal conversion to the electronic ground state (Fig. 3a, τ1 = 0.35 ps; Fig. 3b, τ1 = 0.3 ps): within this time period the ring-closure reaction occurs. The slower decay components τ2/3 can be assigned to cooling processes of vibrationally excited ground state molecules (Fig. 3a, τ2 = 4.0 ps and τ3 = 25 ps; Fig. 3b, τ2 = 1.2 ps and τ3 = 15 ps). The comparison of the presented experimental findings shows that the complete reaction mechanism seems to be nearly identical for all three solvent conditions.
Discussion
The presented steady-state and transient data can be explained in the framework of the schematic reaction model shown in Fig. 4.
Fig. 4.
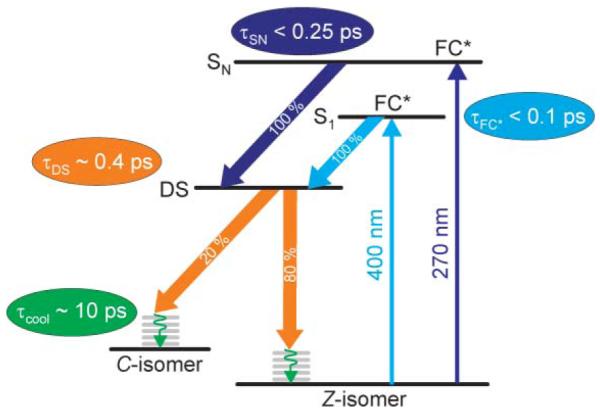
Kinetic reaction scheme of the photoinduced ring-closure of the investigated fluorinated indolylfulgide for all solvents and excitation conditions. The tentative lifetimes of the respective states and approximate yields of internal conversion processes are indicated in the figure.
Light excitation of the open Z-isomer with either 400 nm or 270 nm light populates the respective Franck–Condon region of the addressed electronic state (Fig. 4, FC*). The steady-state measurements show that the absorption maximum of the S0 → S1 transition shifts bathochromic in the different solvents from CH → DIO → ACN (Table 1). So, this FC* state is slightly more polar than the corresponding ground state. In contrast to these findings, the S0 → SN transition is not influenced by the solvent environment (Table 1), which suggests a similar polarity compared to the electronic ground state.
After excitation with 400 nm the FC* state decays ultrafast into a dark state (Fig. 4, DS) on the excited state potential energy surface (Fig. 4, τFC* < 0.1 ps). Thereby possible interactions with the first solvent shell and relaxational processes may occur on the timescale of τFC* < 0.1 ps. The transfer of the molecules into a fluorescently dark state within several femtoseconds has been observed using time-resolved fluorescence experiments on related fulgimide compounds.4 Higher energetic excitation at 270 nm populates the FC* region of the state SN (Fig. 4). Transient absorption experiments do not allow observing this state. Hence, we consider it as extremely short lived (Fig. 4, τSN < 0.25 ps). After relaxation into S1 it transforms into the DS state on the same route as found for S1 excitation. All these findings are in good agreement with Kasha’s rule.32
Independent of the excitation wavelength and solvent surroundings, the electrocyclic ring-closure and hence the internal conversion back to the C-form start from the relaxed S1 state: the excited state is left with a time constant of τDS ~ 0.4 ps. Within this time period vibrationally hot product and reactant molecules are formed in the electronic ground state (Fig. 4, τDS ~ 0.4 ps). The time constants for these processes show only a very weak dependence on the solvent polarity: thereby τDS (describing the photoreaction and internal conversion) increases from the polar solvent ACN from 0.3 ps to 0.35 ps in DIO up to 0.45 ps in the non-polar CH. The reaction yield shows a corresponding behavior where the lowest value is found for ACN (Table 1). We interpret this in terms of slight changes in the shape of the potential energy surface. This may alter the reaction quantum yield of the processes and the excited state lifetime as observed in the transient absorption experiments in the different solvents.
The ring-closure itself is followed by vibrational cooling on the timescale of τcool ~ 10 ps. The results from a global fitting routine (Fig. 2, lower panel; Fig. 3, lower panel) suggest that the cooling processes are fastest in the polar solvent ACN while the non-polar CH shows the slowest cooling kinetics. Again, these findings are in good agreement with data from the literature.33 There it was shown that the cooling rate is strongly correlated to the solvent polarity: non-polar solvents (hexane) show significantly slower cooling times than polar solvents as methanol.33
The presented data and their interpretation show that the reaction pathway of the ring-closure strongly differs from the ring-opening reaction: it was recently demonstrated that the ring-opening reaction is altered by both solvent effects3 and excitation wavelength.23 Hence, the reaction dynamics of the ring-opening process are heavily dependent on the specific experimental conditions.
Therefore the final paragraphs of this article discuss the strong differences between the ring-opening and the ring-closure process: quantum-chemical calculations12,13,22 and experiments4,21,23 suggest that the strong differences between the two processes (ring-opening/ring-closure) arise from a differing potential energy landscape of the Z/C-isomers in the excited state:
The photoreaction starting from the Z-isomer: the ring-closure reaction
(i) The Z-isomer exhibits one accessible conical intersection (CoIn) connecting the lowest excited state and the ground state. This CoIn allows both the transfer of molecules into the reactant and product state, it can hence be considered as a branching point. Furthermore, this photoreaction is not thermally activated as very fast reaction times on the sub-picosecond timescale were found—the CoIn is easily accessible on very fast timescales. So, changes of the potential energy surface, induced by solvents, alter the reaction dynamics and the quantum yield of the processes only slightly. (ii) Kasha’s rule is fulfilled in the case of the ring-closure reaction as fast population transfer between SN and the lowest excited singlet state is possible. The ring-closure reaction of indolylfulgides can hence be considered as a textbook example of an electrocyclic reaction:24 the reaction is not strongly influenced by its solvent surroundings and excitation conditions.
The photoreaction starting from the C-isomer: the ring-opening reaction
In contrast, the ring-opening reaction is influenced by different experimental conditions: (i) The photoprocesses after C-isomer excitation (S1) can proceed via two different CoIns in the first excited state as demonstrated in ref. 23. Here, one CoIn is tilted allowing only back transfer to the C-form. The other CoIn branches into Z/C-form and is hence responsible for the photoreaction. The access to both CoIns is activated and the photochemical quantum yield of the process thus strongly depends on the solvent surroundings and excitation wavelength.23 (ii) Higher energetic excitation (SN state) results in a strongly increased reaction quantum yield and a faster photoreaction as the molecule can reach the branching point into product/reactant more easily.23
Conclusions
The wavelength and solvent dependence of the electrocyclic ring-closure reaction of a trifluorinated indolylfulgide was investigated using stationary and transient absorption spectroscopy in the visible spectral range. The ultrafast photoreaction on the sub-picosecond timescale proved to be nearly unaffected by its solvent environment and excitation wavelength as expected from organic textbooks. A simple reaction scheme could be established according to former studies on related compounds. The presented results clearly demonstrate that indolylfulgides are widely applicable as photoswitches, where the ring-closure reaction is not disturbed by any surrounding effects.
Acknowledgements
The work was supported by the Deutsche Forschungsgemeinschaft through the DFG-Cluster of Excellence Munich-Centre for Advanced Photonics and SFB 749. Financial support to WJL from the NIH/NIGMS programs (S06GM008205 and SC3GM084752) is gratefully acknowledged. The authors thank W. Zinth and B. Heinz for stimulating discussions and careful reading of the manuscript.
Footnotes
At this point it is important to mention that we assume that only a very small percentage of the Z-isomer undergoes Z → E isomerization when the sample is illuminated with near UV-light. This is supported by quantum yield measurements that estimate the efficiency for E-isomer formation to be about 1%.17
The ratio is rougly 20/80 because the photochemical quantum yield for the formation of the C-isomer was found to be 17% in stationary experiments.
References
- 1.Tamai N, Miyasaka H. Ultrafast dynamics of photochromic systems. Chem. Rev. 2000;100:1875–1890. doi: 10.1021/cr9800816. [DOI] [PubMed] [Google Scholar]
- 2.Yokoyama Y. Fulgides for memories and switches. Chem. Rev. 2000;100:1717–1739. doi: 10.1021/cr980070c. [DOI] [PubMed] [Google Scholar]
- 3.Koller FO, Schreier WJ, Schrader TE, Malkmus S, Schulz C, Dietrich S, Rück-Braun K, Braun M. Ultrafast ring-closure reaction of photochromic indolylfulgimides studied with UV-pump - IR-probe spectroscopy. J. Phys. Chem. A. 2008;112:210–214. doi: 10.1021/jp073545p. [DOI] [PubMed] [Google Scholar]
- 4.Heinz B, Malkmus S, Laimgruber S, Dietrich S, Schulz C, Rück-Braun K, Braun M, Zinth W, Gilch P. Comparing a Photo-induced Pericyclic Ring Opening and Closure: Differences in the Excited State Pathways. J. Am. Chem. Soc. 2007;129:8577–8584. doi: 10.1021/ja071396i. [DOI] [PubMed] [Google Scholar]
- 5.Brust T, Draxler S, Malkmus S, Schulz C, Zastrow M, Rück-Braun K, Zinth W, Braun M. Ultrafast dynamics and temperature effects on the quantum efficiency of the ring-opening reaction of a photochromic indolylfulgide. J. Mol. Liq. 2008;141:137–139. [Google Scholar]
- 6.Malkmus S, Koller FO, Heinz B, Schreier WJ, Schrader TE, Zinth W, Schulz C, Dietrich S, Rück-Braun K, Braun M. Ultrafast ring opening reaction of a photochromic indolyl-fulgimide. Chem. Phys. Lett. 2006;417:266–271. [Google Scholar]
- 7.Malkmus S, Koller FO, Draxler S, Schrader TE, Schreier WJ, Brust T, DiGirolamo JA, Lees WJ, Zinth W, Braun M. All-optical operation cycle on molecular bits with 250-GHz clock-rate based on photochromic fulgides. Adv. Funct. Mater. 2007;17:3657–3662. [Google Scholar]
- 8.Irie M. Diarylethenes for memories and switches. Chem. Rev. 2000;100:1685–1716. doi: 10.1021/cr980069d. [DOI] [PubMed] [Google Scholar]
- 9.Woodward RB, Hoffmann R. Conservation of Orbital Symmetry. Angew. Chem. Int. Ed. 1969;8:781–853. [Google Scholar]
- 10.Fuss W, Schmid WE, Trushin SA. Time-resolved dissociative intense-laser field ionization for probing dynamics: Femtosecond photochemical ring opening of 1,3-cyclohexadiene. J. Chem. Phys. 2000;112:8347–8362. [Google Scholar]
- 11.Pullen S, Walker LA, Donovan B, Sension RJ. Femtosecond Transient Absorption Study of the Ring-Opening Reaction of 1,3-Cyclohexadiene. Chem. Phys. Lett. 1995;242:415–420. [Google Scholar]
- 12.Hofmann A, de Vivie-Riedle R. Quantum dynamics of photoexcited cyclohexadiene introducing reactive coordinates. J. Chem. Phys. 2000;112:5054–5059. [Google Scholar]
- 13.Geppert D, de Vivie-Riedle R. Control strategies for reactive processes involving vibrationally hot product states. J. Photochem. Photobiol. A-Chem. 2006;180:282–288. [Google Scholar]
- 14.Seibold M, Port H. Mid-infrared recognition of the reversible photoswitching of fulgides. Chem. Phys. Lett. 1996;252:135–140. [Google Scholar]
- 15.Schrader TE, Schreier WJ, Cordes T, Koller FO, Babitzki G, Denschlag R, Renner C, Dong SL, Löweneck M, Moroder L, Tavan P, Zinth W. Light triggered β-hairpin folding and unfolding. Proc. Natl. Acad. Sci. U. S. A. 2007;104:15729–15734. doi: 10.1073/pnas.0707322104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouas-Laurent H, Dürr H. Organic photochromism. Pure Appl. Chem. 2001;73:639–665. [Google Scholar]
- 17.Wolak MA, Thomas CJ, Gillespie NB, Birge RR, Lees WJ. Tuning the optical properties of fluorinated indolylfulgimides. J. Org. Chem. 2003;68:319–326. doi: 10.1021/jo026374n. [DOI] [PubMed] [Google Scholar]
- 18.Port H, Gärtner P, Hennrich M, Ramsteiner I, Schöck T. Ultrafast photochromic reactions of fulgide photoswitches. Mol. Cryst. Liq. Cryst. 2005;430:15–21. [Google Scholar]
- 19.Renth F, Foca M, Petter A, Temps F. Ultrafast transient absorption spectroscopy of the photo-induced Z-E isomerization of a photochromic furylfulgide. Chem. Phys. Lett. 2006;428:62–67. [Google Scholar]
- 20.Koller FO, Schreier WJ, Schrader TE, Sieg A, Malkmus S, Schulz C, Dietrich S, Rück-Braun K, Zinth W, Braun M. Ultrafast structural dynamics of photochromic indolylfulgimides studied by vibrational spectroscopy and DFT calculations. J. Phys. Chem. A. 2006;110:12769–12776. doi: 10.1021/jp0657787. [DOI] [PubMed] [Google Scholar]
- 21.Draxler S, Brust T, Malkmus S, Koller FO, Heinz B, Laimgruber S, Schulz C, Dietrich S, Rück-Braun K, Zinth W, Braun M. Ultrafast reaction dynamics of the complete photo cycle of an indolylfulgimide studied by absorption, fluorescence and vibrational spectroscopy. J. Mol. Liq. 2008;141:130–136. [Google Scholar]
- 22.Geppert D, Seyfarth L, de Vivie-Riedle R. Laser control schemes for molecular switches. Appl. Phys. B-Lasers Opt. 2004;79:987–992. [Google Scholar]
- 23(a).Ishibashi Y, Murakami M, Miyasaka H, Kobatake S, Irie M, Yokoyama Y. Laser Multiphoton-Gated Photochromic Reaction of a Fulgide Derivative. J. Phys. Chem. C. 2007;111:2730–2737. [Google Scholar]; (b) Cordes T, Malkmus S, DiGirolamo JA, Lees WJ, Nenov A, de Vivie-Riedle R, Braun M, Zinth W. Accelerated and Efficient Photochemistry from Higher Excited Electronic States in Fulgide Molecules. J. Phys. Chem. A. 2008;112:13364–13371. doi: 10.1021/jp807097w. [DOI] [PubMed] [Google Scholar]
- 24.Smith MB, March J. March’s Advanced Organic Chemistry. 5 edn Wiley-Interscience; New York: 2001. [Google Scholar]
- 25.Yokoyama Y, Takahashi K. Trifluoromethyl-substituted photochromic indolylfulgide. A remarkably durable fulgide towards photochemical and thermal treatments. Chem. Lett. 1996:1037–1038. [Google Scholar]
- 26.Cordes T, Weinrich D, Kempa S, Riesselmann K, Herre S, Hoppmann C, Rück-Braun K, Zinth W. Hemithioindigo-based photoswitches as ultrafast light trigger in chromopeptides. Chem. Phys. Lett. 2006;428:167–173. [Google Scholar]
- 27.Riedle E, Beutter M, Lochbrunner S, Piel J, Schenkl S, Spörlein S, Zinth W. Generation of 10 to 50 fs pulses tunable through all of the visible and the NIR. Appl. Phys. B-Lasers Opt. 2000;71:457–465. [Google Scholar]
- 28.Huber R, Satzger H, Zinth W, Wachtveitl J. Noncollinear optical parametric amplifiers with output parameters improved by the application of a white light continuum generated in CaF2. Opt. Commun. 2001;194:443–448. [Google Scholar]
- 29.Seel M, Wildermuth E, Zinth W. A multichannel detection system for application in ultra-fast spectroscopy. Meas. Sci. Technol. 1997;8:449–452. [Google Scholar]
- 30.Fleming GR. Chemical Applications of Ultrafast Spectroscopy. Oxford University Press; New York: 1986. [Google Scholar]
- 31.Wolak MA, Finn RC, Rarig RS, Thomas CJ, Hammond RP, Birge RR, Zubieta J, Lees WJ. Structural properties of a series of photochromic fluorinated indolyl-fulgides. Acta Cryst. C. 2002;58:o389–o393. doi: 10.1107/s0108270102008041. [DOI] [PubMed] [Google Scholar]
- 32.Kasha M. Characterization of Electronic Transitions in Complex Molecules. Farad. Disc. 1950:14–19. [Google Scholar]
- 33(a).Horng ML, Gardecki JA, Papazyan A, Maroncelli M. Sub-picosecond Measurements of Polar Solvation Dynamics - Coumarin-153 Revisited. J. Phys. Chem. 1995;99:17311–17337. [Google Scholar]; (b) Iwata K, Hamaguchi H. Microscopic Mechanism of Solute – Solvent Energy Dissipation Probed by Picosecond Time-Resolved Raman Spectroscopy. J. Phys. Chem. A. 1997;101:632–637. [Google Scholar]