

Abstract
Understanding how cells handle and dispose of misfolded proteins is of paramount importance because protein misfolding and aggregation underlie the pathogenesis of many neurodegenerative disorders, including PD (Parkinson's disease) and Alzheimer's disease. In addition to the ubiquitin–proteasome system, the aggresome–autophagy pathway has emerged as another crucial cellular defence system against toxic build-up of misfolded proteins. In contrast with basal autophagy that mediates non-selective, bulk clearance of misfolded proteins along with normal cellular proteins and organelles, the aggresome–autophagy pathway is increasingly recognized as a specialized type of induced autophagy that mediates selective clearance of misfolded and aggregated proteins under the conditions of proteotoxic stress. Recent evidence implicates PD-linked E3 ligase parkin as a key regulator of the aggresome–autophagy pathway and indicates a signalling role for Lys63-linked polyubiquitination in the regulation of aggresome formation and autophagy. The present review summarizes the current knowledge of the aggresome–autophagy pathway, its regulation by parkin-mediated Lys63-linked polyubiquitination, and its dysfunction in neurodegenerative diseases.
Keywords: aggresome, autophagy, misfolded protein, parkin, Parkinson's disease, ubiquitin-protein ligase
Introduction
PD (Parkinson's disease) and AD (Alzheimer's disease) as well as many other neurodegenerative disorders are often referred to as ‘protein misfolding diseases’ because their pathogenesis involves protein misfolding and aggregation [1,2]. The accumulation of misfolded proteins in these diseases probably occurs due to a chronic imbalance in the generation and clearance of misfolded proteins, and it suggests that the failure of cells to cope with excess misfolded proteins may be a common pathological mechanism linking these clinically distinct diseases. Protein misfolding can occur as a result of genetic mutations, environmental insults or oxidative damage [3]. Misfolded proteins are often prone to aggregation into oligomers and aggregates, and they can impair cell function and viability through a variety of mechanisms, including pore formation, proteasome inhibition and disruption of intracellular transport [1,3].
Growing evidence indicates that, when the production of misfolded proteins exceeds the capacity of the molecular chaperone system and the Ub (ubiquitin)–proteasome pathway, misfolded and aggregated proteins are actively sequestered in a microscopically visible, pericentriolar structure called an aggresome [3,4] and are subsequently degraded by macroautophagy (hereafter referred to as autophagy), a lysosome-dependent process that mediates bulk clearance of cytosolic proteins and organelles [5,6]. Here, we review recent evidence for the involvement of the aggresome–autophagy pathway in protection against misfolded protein accumulation and neurodegeneration and discuss the role of Lys63-linked polyubiquitination by PD-linked E3 ligase parkin in regulation of misfolded protein handling by this pathway.
The aggresome–autophagy pathway
The aggresome–autophagy pathway is increasingly recognized as a key cellular defence system against accumulation of misfolded and aggregated proteins when the proteasome is overwhelmed or impaired [3,4]. In this system, misfolded and aggregated proteins are selectively recognized and delivered via dynein-mediated, microtubule-based retrograde transport towards the MTOC (microtubule-organizing centre) to form aggresomes at the pericentriolar region (Figure 1). Accumulating evidence indicates that aggresome formation not only protects cells by sequestering cytotoxic misfolded and aggregated proteins but also serves as a mechanism for concentrating misfolded and aggregated proteins for subsequent clearance by autophagy [3,7].
Figure 1. The aggresome–autophagy pathway and its regulation by parkin-mediated Lys63-linked polyubiquitination.
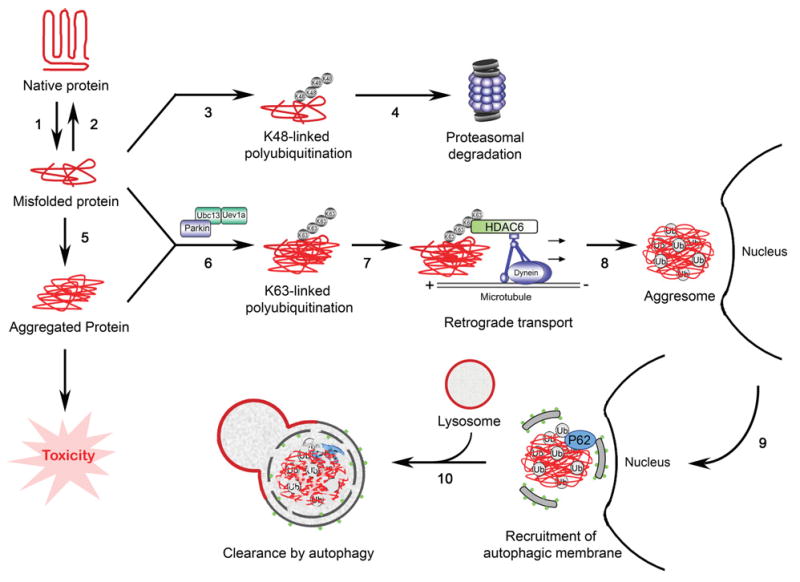
Protein misfolding can occur as a result of genetic mutations or oxidative damage (1). Once formed, misfolded proteins may be refolded by chaperones (2) or tagged with Lys48-linked polyubiquitin chains (3) for degradation by the proteasome (4). When the chaperone and proteasome systems fail or are overwhelmed, misfolded proteins form oligomers and aggregates (5) that can cause cytotoxicity. Recent evidence indicates that, under conditions of proteasomal impairment, PD-linked E3 ligase parkin co-operates with the E2 enzyme Ubc13/Uev1a to mediate Lys63-linked polyubiquitination of misfolded proteins (6). The Lys63-linked polyubiquitin chains promote binding to HDAC6 (7) and thereby link the misfolded proteins to the dynein motor complex for retrograde transport towards the MTOC to form the aggresome (8). Lys63-linked polyubiquitination may also promote binding to p62 and thereby facilitate the recruitment of autophagic membrane to the aggresome for the formation of an autophagosome (9). Subsequent fusion of the autophagosome with the lysosome allows the degradation of misfolded and aggregated proteins by lysosomal hydrolases (10).
Autophagy is a multistep process characterized by the formation of an isolation membrane called a phagophore that expands to sequester a portion of the cytoplasm, leading to the formation of the double-membrane autophagosome, which subsequently fuses with the lysosome for degradation of the sequestered cytoplasmic cargo [5,6]. Unlike the proteasome, autophagy does not require unfolding of the substrates and is able to break down large protein complexes, protein aggregates and entire organelles. Autophagy is a highly regulated process involving the co-ordinated action of a large number of proteins encoded by Atg (autophagy-related) genes [5,6]. Recent studies have shown that autophagy is induced in response to oxidative stress or proteasome impairment and participates directly in the clearance of aggresomes [6–10].
Although autophagy is generally thought to be a non-selective, bulk degradation process, increasing evidence indicates the presence of selective autophagy that mediates clearance of specific cargos, such as mitophagy, reticulophagy, pexophagy and xenophagy [6]. Emerging evidence suggests that selective autophagy is involved in the clearance of misfolded and aggregated proteins, as inhibition of autophagy preferentially affects the degradation of several neurodegenerative disease-associated mutant proteins but not their wild-type counterparts [8,11]. Although the mechanism underlying the selective autophagic clearance of misfolded proteins is not understood, the selective sequestration of misfolded proteins in aggresomes offers one method of preferential clearance of the abnormal proteins by autophagy. Aggresomes have also been shown to participate in the induction of autophagy by sequestering the endogenous autophagy suppressor mTOR (mammalian target of rapamycin) kinase [3,9]. Together, current data suggest that, unlike basal autophagy that mediates non-selective clearance of misfolded proteins along with normal cellular proteins, the aggresome–autophagy pathway is a specialized type of induced autophagy that mediates selective clearance of misfolded and aggregated proteins under the conditions of proteotoxic stress.
Parkin-mediated Ub signalling in regulation of the aggresome–autophagy pathway
Parkin and PD
PD is the most common neurodegenerative movement disorder, characterized by the loss of nigral dopaminergic neurons and the presence of intraneuronal cytoplasmic inclusions called Lewy bodies [12–14]. Homozygous mutations in the gene encoding the E3 Ub–protein ligase parkin cause an autosomal recessive, early onset form of PD that is devoid of Lewy bodies [12,13]. In addition, heterozygous mutations in parkin have been implicated as a significant risk factor in the development of late-onset sporadic PD [15]. Studies in a number of cell and animal model systems have shown that parkin exerts cytoprotective action against a wide variety of cellular stresses, including oxidative stress, proteasome inhibition and proteotoxic stress induced by overexpression of aggregation-prone proteins [14]. However, the molecular mechanisms underlying the cytoprotective action of parkin remain poorly understood.
A mechanistic understanding of the cytoprotective action of parkin requires molecular characterization of parkin E3 ligase function, its substrates and the types and functional consequences of parkin-mediated ubiquitination. Parkin has been reported to regulate Lys48-linked polyubiquitination and proteasomal degradation of several putative substrates [13,14], although the validity of these proteins as physiological parkin substrates remains controversial [16]. Parkin has also been shown to be capable of catalysing monoubiquitination and Lys63-linked polyubiquitination, thereby regulating proteasome-independent cellular processes, such as endocytosis and NF-κB (nuclear factor κB) signalling [17,18]. In addition, parkin has been implicated in regulation of mitochondria dynamics [19,20], although the parkin substrate(s) on the mitochondria remains to be identified.
Parkin-mediated Lys63-linked polyubiquitination and aggresome formation
In a recent study [21], we investigated the specificity of parkin-mediated ubiquitination and its role in cellular management of misfolded proteins using wild-type DJ-1 and L166P mutant DJ-1 as the substrates. DJ-1 is a ubiquitously expressed protein that is mutated in an autosomal recessive, early-onset form of PD [22]. We have previously shown that wild-type DJ-1 is a compactly folded protein with a helix–strand–helix sandwich structure, whereas the PD-linked L166P mutant DJ-1 is a misfolded protein that is efficiently degraded by the Ub–proteasome system under normal conditions [23,24]. The results from our recent study [21] showed that, under conditions of proteasomal impairment, parkin co-operated with the heterodimeric E2 enzyme UbcH13–Uev1a to selectively catalyse Lys63-linked polyubiquitination of misfolded DJ-1, but not wild-type DJ-1 (Figure 1, step 6). Our results [21] further revealed that parkin-mediated Lys63-linked polyubiquitination coupled misfolded DJ-1 with the dynein motor complex via the adaptor protein HDAC6 (histone deacetylase 6) and thereby facilitated its transport to the MTOC for sequestration into the aggresome (Figure 1, steps 7 and 8).
HDAC6 is a Ub-binding protein that can simultaneously bind ubiquitinated proteins via its ZnF-UBP (zinc finger Ub-processing protease) domain and the dynein motor via another domain [25]. Depletion of HDAC6 by siRNAs (small interfering RNA) blocks aggresome formation, and this phenotype can be rescued only with a Ub-binding-competent form of HDAC6 [25]. Our previous study revealed that HDAC6 preferentially bound Lys63-linked polyubiquitinated proteins and that inhibition of Lys63-linked polyubiquitination or targeted disruption of parkin in mice impaired recruitment of misfolded DJ-1 to aggresomes [21]. Our findings suggest that Lys63-linked polyubiquitination by parkin serves as a signal for targeting misfolded proteins to the aggresome [21,26]. Consistent with our results, expression of mutant Ub that can only form Lys63-linked chains was recently shown to promote aggresome formation of several misfolded proteins, including tau protein and SOD-1 (superoxide dismutase 1) mutants [27], providing additional evidence supporting a role for Lys63-linked polyubiquitination in facilitating aggresome formation.
Parkin-mediated Lys63-linked polyubiquitination and autophagy
Our recent work suggests that parkin-mediated Lys63-linked polyubiquitination not only promotes sequestration of misfolded proteins into aggresomes but also facilitates their subsequent clearance by autophagy [21,26]. We found that misfolded DJ-1-containing aggresomes stained with autophagic markers and were tightly encircled by lysosomes, indicating that aggresomes promoted by parkin-mediated Lys63-linked polyubiquitination are active sites of autophagy (Figure 1). Corroborating our findings, Tan et al. [27] demonstrated that the aggresomes formed in cells overexpressing the mutant Ub that can only form Lys63-linked polyubiquitin chains were preferentially cleared when autophagy is induced, whereas those formed in cells expressing Ub mutant that was unable to form Lys63-linked polyubiquitin chains were resistant to autophagic clearance. Thus emerging evidence has begun to suggest that Lys63-linked polyubiquitination has a signalling role in autophagic clearance of aggresomes.
A potential mechanism by which parkin-mediated Lys63-linked polyubiquitination may facilitate autophagic clearance of misfolded proteins is to promote the recruitment of autophagic membranes and autophagy machinery via binding to the adaptor protein p62 (Figure 1, steps 9 and 10). p62 is a Ub-binding protein that interacts with ubiquitinated proteins via its UBA (Ub-associated) domain and the autophagy machinery component LC3 via a 22-amino-acid LIR (LC3-interacting region) [28,29]. p62 shows preference for binding Lys63-linked polyubiquitin chains [30,31], and deletion of its UBA domain or LIR impairs the packing of ubiquitinated aggregates into autophagosomes [28,29]. Recent evidence indicates that, in addition to facilitating autophagic clearance, p62 also has a role in promoting protein aggregate formation [32]. Further studies are needed to determine whether p62 is indeed a Ub receptor for regulating the processing of Lys63-linked polyubiquitinated misfolded proteins by the aggresome–autophagy pathway.
Recently, parkin was shown to be selectively recruited to damaged mitochondria and promote their clearance by autophagy [20]. Autophagic clearance of mitochondria is often referred to as mitophagy [6], and there is evidence suggesting the involvement of ubiquitination in this process [33,34]. It remains to be determined whether the E3 ligase activity of parkin is required for its action in promoting mitophagy, what the parkin substrates on the mitochondria are and what type of ubiquitination is involved. A tantalizing possibility is that, similar to its action in promoting clearance of misfolded proteins by the aggresome–autophagy pathway (Figure 1), parkin may catalyse Lys63-linked polyubiquitination of misfolded proteins on the damaged mitochondria and this ubiquitination could be the signal for targeting damaged mitochondria for mitophagy.
Dysfunction of the aggresome–autophagy pathway and neurodegeneration
A link between dysfunction of the aggresome–autophagy pathway and neurodegeneration was first suggested by postmortem findings of the accumulation of Ub-positive protein aggregates and autophagosome-like structures in brains of patients with diverse neurodegenerative diseases, including PD and AD [3,35,36]. This link was further strengthened by recent identification of mutations in the aggresome–autophagy pathway components as the genetic defects responsible for several hereditary forms of neurodegenerative disorders (Table 1).
Table 1. The aggresome–autophagy pathway components and neurodegenerative diseases.
Loss-of-function mutations in parkin are a major cause of recessively transmitted early-onset PD [12,13]. Our finding that parkin-mediated Lys63-linked polyubiquitination of misfolded proteins promotes their sequestration into aggresomes and subsequent clearance by autophagy [21,26] provides evidence linking deregulation of the aggresome–autophagy pathway to PD pathogenesis. The impaired aggresome formation observed in cells from parkin-knockout mice [21] is reminiscent of the lack of Lewy bodies in parkin-associated human PD cases [13,14], suggesting that Lys63-linked polyubiquitination by parkin may be directly involved in the formation of Lewy bodies and that the inability to form these protective inclusion bodies may underlie the rapid disease onset and progression observed in patients with mutations in parkin. Accumulation of Lys63-linked polyubiquitinated proteins was recently detected in brains of human HD (Huntington's disease) patients [37], further supporting a connection between Lys63-linked polyubiquitination and the formation of pathological inclusion bodies.
Mutations in the Ub-binding domain of p62, an adaptor that binds Lys63-linked polyubiquitin chains and promotes autophagic clearance of protein aggregates [28–30], cause Paget disease [38]. Although Paget disease is primarily a bone disorder, knockout studies in mice revealed age-dependent accumulation of Lys63-polyubiquitinated protein aggregates and neurodegeneration in p62−/− brains [39]. Additional support for the involvement of the aggresome–autophagy pathway dysfunction in neurodegeneration comes from the following: (i) the identification of mutations in dynactin subunit p150Glued, a component of the dynein/dynactin motor that plays a critical role in aggresome formation as well as autophagy [40,41], as the cause for human motor neuron disease [42]; and (ii) animal model studies showing that disruption of the dynein/dynactin motor function leads to motor neuron degeneration [43,44] and enhanced toxicity of aggregation-prone proteins [41]. Dysfunction of the aggresome–autophagy pathway has also been implicated in the pathogenesis of two human neurodegenerative diseases, frontotemporal dementia [45] and ALS (amyotrophic lateral sclerosis) [46], by the findings that the disease-causing mutations in the ESCRT (endosomal sorting complexes required for transport)-III subunit CHMP2B (charged multivesicular body protein 2B) cause impairments in autolysosome formation and autophagic clearance, leading to accumulation of Ub-positive protein aggregates and neuronal cell death [47,48].
Previously, it was reported that overexpression of HDAC6, a key regulator of the aggresome–autophagy pathway [10,25], is able to suppress neurodegeneration in Drosophila induced by proteasome impairment or by expression of the spinobulbar muscular atrophy-associated mutant protein [49]. Furthermore, pharmacological activation of autophagy with mTOR inhibitors has been shown to reduce neurotoxicity of misfolded and aggregated proteins in cell and animal models of neurodegenerative diseases [8,9,50]. Together, these findings point to a critical role for the aggresome–autophagy pathway in the protection against misfolded protein accumulation and neurodegeneration, and they suggest that targeting this pathway may have therapeutic benefits for treating neurodegenerative disorders.
Conclusions and perspectives
While the role of the Ub–proteasome system in misfolded protein degradation has long been appreciated, the involvement of the aggresome–autophagy pathway in cellular defence against cytotoxic accumulation of misfolded proteins has only recently been recognized. Lys63-linked polyubiquitination by parkin has emerged as a signal for targeting misfolded proteins into aggresomes and facilitating their selective clearance by autophagy [21,26]. Increasing evidence suggests that the Ub-binding proteins HDAC6 and p62 may function as cargo receptors for recognizing Lys63-linked polyubiquitinated misfolded proteins and facilitating their processing through the aggresome–autophagy pathway (Figure 1). Given the recently reported role of parkin in mitophagy [20], it is tempting to speculate that parkin-mediated Lys63-linked polyubiquitination of mitochondria-localized misfolded proteins may act as a signal for targeting damaged mitochondria for mitophagy. Further studies of the molecular mechanisms by which parkin promotes clearance of misfolded proteins and damaged mitochondria should facilitate the development of novel therapies for treating PD as well as other neurodegenerative diseases.
Acknowledgments
Funding: This work was supported by grants from the National Institutes of Health to L.-S.C. [grant numbers NS050650 and AG034126] and to L.L. [grant numbers ES015813 and GM082828].
Abbreviations used
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- CHMP2B
charged multivesicular body protein 2B
- ESCRT
endosomal sorting complexes required for transport
- HDAC6
histone deacetylase 6
- LIR
LC3-interacting region
- MTOC
microtubule-organizing centre
- mTOR
mammalian target of rapamycin
- PD
Parkinson's disease
- Ub
ubiquitin
References
- 1.Gregersen N. Protein misfolding disorders: pathogenesis and intervention. J Inherit Metab Dis. 2006;29:456–470. doi: 10.1007/s10545-006-0301-4. [DOI] [PubMed] [Google Scholar]
- 2.Whatley BR, Li L, Chin LS. The ubiquitin–proteasome system in spongiform degenerative disorders. Biochim Biophys Acta. 2008;1782:700–712. doi: 10.1016/j.bbadis.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olzmann JA, Li L, Chin LS. Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr Med Chem. 2008;15:47–60. doi: 10.2174/092986708783330692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 5.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 6.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, Kopito RR. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci USA. 2005;102:13135–13140. doi: 10.1073/pnas.0505801102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 9.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 10.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated Huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 11.Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem. 2006;281:14474–14485. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- 12.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 13.Hattori N, Mizuno Y. Pathogenetic mechanisms of parkin in Parkinson's disease. Lancet. 2004;364:722–724. doi: 10.1016/S0140-6736(04)16901-8. [DOI] [PubMed] [Google Scholar]
- 14.Moore DJ. Parkin: a multifaceted ubiquitin ligase. Biochem Soc Trans. 2006;34:749–753. doi: 10.1042/BST0340749. [DOI] [PubMed] [Google Scholar]
- 15.Tan EK, Skipper LM. Pathogenic mutations in Parkinson disease. Hum Mutat. 2007;28:641–653. doi: 10.1002/humu.20507. [DOI] [PubMed] [Google Scholar]
- 16.Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- 17.Fallon L, Belanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, Voortman J, Haber M, Rouleau G, Thorarinsdottir T, et al. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K–Akt signalling. Nat Cell Biol. 2006;8:834–842. doi: 10.1038/ncb1441. [DOI] [PubMed] [Google Scholar]
- 18.Henn IH, Bouman L, Schlehe JS, Schlierf A, Schramm JE, Wegener E, Nakaso K, Culmsee C, Berninger B, Krappmann D, et al. Parkin mediates neuroprotection through activation of IκB kinase/nuclear factor-κB signaling. J Neurosci. 2007;27:1868–1878. doi: 10.1523/JNEUROSCI.5537-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 20.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD, Chin LS. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J Cell Biol. 2007;178:1025–1038. doi: 10.1083/jcb.200611128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 23.Olzmann JA, Brown K, Wilkinson KD, Rees HD, Huai Q, Ke H, Levey AI, Li L, Chin LS. Familial Parkinson's disease-associated L166P mutation disrupts DJ-1 protein folding and function. J Biol Chem. 2004;279:8506–8515. doi: 10.1074/jbc.M311017200. [DOI] [PubMed] [Google Scholar]
- 24.Huai Q, Sun Y, Wang H, Chin LS, Li L, Robinson H, Ke H. Crystal structure of DJ-1/RS and implication on familial Parkinson's disease. FEBS Lett. 2003;549:171–175. doi: 10.1016/s0014-5793(03)00764-6. [DOI] [PubMed] [Google Scholar]
- 25.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 26.Olzmann JA, Chin LS. Parkin-mediated K63-linked polyubiquitination: a signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy. 2008;4:85–87. doi: 10.4161/auto.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17:431–439. doi: 10.1093/hmg/ddm320. [DOI] [PubMed] [Google Scholar]
- 28.Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on Huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 30.Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wooten MW, Geetha T, Babu JR, Seibenhener ML, Peng J, Cox N, Diaz-Meco MT, Moscat J. Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J Biol Chem. 2008;283:6783–6789. doi: 10.1074/jbc.M709496200. [DOI] [PubMed] [Google Scholar]
- 32.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 33.Rapoport S, Dubiel W, Muller M. Proteolysis of mitochondria in reticulocytes during maturation is ubiquitin-dependent and is accompanied by a high rate of ATP hydrolysis. FEBS Lett. 1985;180:249–252. doi: 10.1016/0014-5793(85)81080-2. [DOI] [PubMed] [Google Scholar]
- 34.Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitin tag for sperm mitochondria. Nature. 1999;402:371–372. doi: 10.1038/46466. [DOI] [PubMed] [Google Scholar]
- 35.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 36.Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci. 2006;29:528–535. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 37.Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in Huntington's disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- 38.Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70:1582–1588. doi: 10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom AL, Kemppainen R, Cox N, Zhu H, Wooten MC, Diaz-Meco MT, Moscat J, et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008;106:107–120. doi: 10.1111/j.1471-4159.2008.05340.x. [DOI] [PubMed] [Google Scholar]
- 40.Johnston JA, Illing ME, Kopito RR. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil Cytoskeleton. 2002;53:26–38. doi: 10.1002/cm.10057. [DOI] [PubMed] [Google Scholar]
- 41.Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ, Brown SD, Rubinsztein DC. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. 2005;37:771–776. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- 42.Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 43.Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS, Hummerich H, Nicholson S, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- 44.LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 45.Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–808. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- 46.Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM, Morrison KE, Pall HS, Hardiman O, Collinge J, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B) Neurology. 2006;67:1074–1077. doi: 10.1212/01.wnl.0000231510.89311.8b. [DOI] [PubMed] [Google Scholar]
- 47.Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179:485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol. 2007;17:1561–1567. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 49.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 50.Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]