
Figure 1.
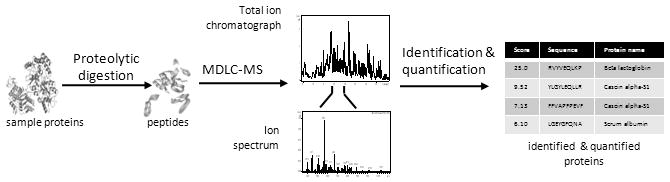
Typical bottom-up proteomics experimental workflow. Proteins are isolated from biological samples and enzymatically digested into peptides. Each protein generates many peptides (30-50 or more), which significantly increases the sample complexity. Peptides are separated using multidimensional liquid chromatography (MDLC) prior to mass spectrometry (MS) analyses. Various bioinformatics tools, such as database search algorithms, are employed for protein identification.