


Abstract
We have characterized a novel type of PSI-BLAST error, homologous over-extension (HOE), using embedded PFAM domain queries on searches against a reference library containing Pfam-annotated UniProt sequences and random synthetic sequences. PSI-BLAST makes two types of errors: alignments to non-homologous regions and HOE alignments that begin in a homologous region, but extend beyond the homology into neighboring sequence regions. When the neighboring sequence region contains a non-homologous domain, PSI-BLAST can incorporate the unrelated sequence into its position specific scoring matrix, which then finds non-homologous proteins with significant expectation values. HOE accounts for the largest fraction of the initial false positive (FP) errors, and the largest fraction of FPs at iteration 5. In searches against complete protein sequences, 5–9% of alignments at iteration 5 are non-homologous. HOE frequently begins in a partial protein domain; when partial domains are removed from the library, HOE errors decrease from 16 to 3% of weighted coverage (hard queries; 35–5% for sampled queries) and no-error searches increase from 2 to 58% weighed coverage (hard; 16–78% sampled). When HOE is reduced by not extending previously found sequences, PSI-BLAST specificity improves 4–8-fold, with little loss in sensitivity.
INTRODUCTION
Protein similarity searching is central to genome annotation, characterization of protein families and exploration of distant evolutionary relationships. Similarity searching is effective because proteins that share statistically significant sequence similarity can be inferred to be homologous, and homologous proteins share similar structures and, often, similar functions. Thus, reliable transfer of knowledge between homologous proteins requires accurate identification of homologs.
For pair-wise similarity searches using BLAST (1), FASTA (2) or Smith–Waterman (3,4), the statistical estimates used to infer homology are very accurate; unrelated sequences rarely obtain expectation values lower than expected by chance (5,6). Statistical estimates for iterative methods like PSI-BLAST can be much less accurate (7,8); sometimes sequences that are clearly unrelated (they have different three-dimensional topologies) obtain highly significant expectation values (<10–6) that imply clear homology. These misleading alignments are thought to result from profile or position specific scoring matrix (PSSM) contamination and from the PSI-BLAST sequence weighting strategy; when distant homologs are included in the PSSM, their contribution to the matrix is given a higher weight than when closely related sequences are included. An unfortunate side effect of this weighting occurs when unrelated sequences are included; they also contribute strongly to the new PSSM, which then produces high scores for homologs to the unrelated domain. The corruption of PSSMs leads to the assignment of high scores and statistical significance to biologically-incorrect relationships—a serious problem for any protein characterization effort.
Alternatively, statistical errors in profile-based alignments might reflect inherent limits in distinguishing similarities produced by divergent evolution from those produced by convergence from unrelated proteins (7,9). Currently, the most sensitive structural comparison methods often assign statistically significant similarity to non-homologous structural alignments, and this trend extends to profile–sequence and profile–profile alignments. This inability to distinguish divergent from convergent similarity may reflect the small number of regular structural motifs in proteins. Similarly, PSI-BLAST’s inaccurate statistics may reflect its ability to capture some of this structural information.
Previous refinements to PSI-BLAST have addressed the PSSM corruption issue (8,10–14) by improving the statistical estimates used to evaluate alignment scores. In this article, we describe a novel cause of PSSM contamination: over-extension of a homologous domain into a non-homologous region. Because this problem begins with a homologous alignment, it cannot be addressed with more accurate statistical estimates or more conservative inclusion thresholds; these errors can occur with very significant E()-values (<10−40). It is the alignment, not the score that is inaccurate. However, the problem can be reduced dramatically by limiting alignment extension after a domain is included in the PSSM.
In evaluations of similarity searching programs, alignments are typically scored as true positives (TPs) or false positives (FPs) based on whether the library sequence contains a homologous domain, rather than whether the domain of interest is aligned correctly. For pair-wise sequence comparison, this approach summarizes search performance relatively accurately; incorrect alignment boundaries rarely detract from the identification of the homologous protein, and may reflect difficulties in accurately annotating domain boundaries.
With iterative methods like PSI-BLAST, accurate domain alignments are more important, because inaccurate alignments can cause non-homologous domains to be included in the PSSM used for the next iteration. Therefore, to characterize PSI-BLAST errors, we recorded the beginning and end of the alignments included in the PSSM for the next iteration. Using the domain boundary annotations in our reference library, we could follow the homologous or non-homologous sequence regions that were added to the PSSM. This process allowed us to identify a novel source of PSSM corruption—homologous over-extension (HOE) errors. HOE, particularly of partial domains, is a problem that can affect any iterative strategy for building profiles, PSSMs, or Hidden Markov Models (HMM). We have developed a very simple strategy for reducing HOE errors—once a sequence is included in the PSSM, the boundaries of its alignment do not change. This strategy reduces FP errors by more than a factor of 8, with little loss in search sensitivity.
MATERIALS AND METHODS
Evaluation datasets
We generated two sequence datasets to characterize PSI-BLAST performance: (i) a set of query families and (ii) two target (reference) libraries. The query families are groups of homologous protein domains derived from Pfam (15). The target libraries are collections of full-length sequences used for iterative PSI-BLAST searching.
Query family selection
A subset of domain families from Pfam version 21.0, originally ∼9000 domain families, was selected and filtered down to 320 domain families meeting the following criteria: (i) at least one PDB structure in the family, (ii) HMM models longer than 200 residues, (iii) more than 100 members in the family, (iv) families were taxonomically-broad, with members from organisms in two of the three domains of life; (v) only one family from each clan superfamily was chosen; (vi) the family contained only non-nested (contiguous) domains. Large families were reduced to 1500 members by random removal of members, to avoid an out-of-memory problem when PSI-BLAST stored large numbers of alignments.
Query sequence selection
For each of the 320 Pfam domain families meeting these criteria, two domain members were chosen as queries based on their location on the family phylogenetic tree. One query was selected from a populated area and another from a deserted area of the tree. Each of the 640 queries was compared to the target database using BLAST, and the 50 queries producing the lowest family coverage at E() < 0.001 were chosen for the hard query set. In addition, 50 queries were chosen at random with replacement to be part of the sampled query set. A third set of 50 families (100 queries) was selected to give strong differences between the populated and deserted regions of the tree; one query was taken from each region for the 50 families.
Query embedding
While our goal is to simulate searches performed with full-length proteins against a full-length protein database (the typical use of PSI-BLAST), our query sequences are not complete proteins; they contain a single Pfam domain (complete proteins can contain multiple domains, with complex homology relationships). To more accurately simulate searches with full-length proteins, we embedded bare Pfam domains in random synthetic sequences. The embeddings were created by randomly shuffling the domain, splitting it in half, and placing each shuffled flank before and after the real domain. We confirmed that the random embedding sequences did not produce significant alignments by comparing them to the Swiss-Prot database (16). Embedding the domains also allowed HOE to occur in the query sequence, since alignments could extend beyond the Pfam domain boundaries.
Target library construction
The standard search library was constructed by bringing together full-length UniProt (17) proteins containing domains from the query families (excluding viral sequences), supplemented with an equal number of randomly generated sequences. The random sequences were generated by shuffling each full-length protein sequence. A second long-domain-only library was constructed from UniProt sequences containing family domains that were at least 75% of their respective Pfam model length.
Annotating the datasets
The target library sequence homology annotations specified by Pfam (v.21) were supplemented to reduce the number of unannotated homologs and to extend domain boundaries in some cases. Because Pfam identifies homologs by alignment with the model HMM, some library sequences contain cryptic domains that share strong similarity to a domain in the family that is distant from the model HMM, so that the cryptic domains do not meet the Pfam threshold. Potentially cryptic homologs were identified by reverse-searching apparent ‘non-homologs’ with E()-values <10−4 against the target library using SSEARCH v36 (3,4). Additional unannotated homologs were identified by examining Pfam v.23, SCOP (18) and CATH (19) classifications. Domain boundaries were adjusted using GLSEARCH, which produces an alignment that is global in the query domain and local in the library full-length sequence. The detailed curation of the database will be described elsewhere (Gonzalez and Pearson, manuscript in preparation). The database may be accessed at http://faculty.virginia.edu/wrpearson/fasta/PUBS/gonzalez09a.
As a result of Pfam family consolidation based on updates in later versions of Pfam, examination of SCOP and CATH classifications, and additional searches, a number of our initial Pfam families were merged. In the standard library, the distribution of query hard and randomly sampled domain family sizes (the number of homologous domains, or TPs) ranged from 84 to 11 435 with a median family size of 847 and first and third quartiles at 473 and 1514 members. For the long domain library, family sizes (TPs) ranged from 29 to 6048, with a median of 425 and quartiles of 249 and 870 members.
PSI-BLAST searches
PSI-BLAST (blastpgp 2.2.19) searches were performed with test queries against the standard and long-domain libraries at four different inclusion thresholds [E() < 0.01; <0.005, the blastpgp default; <0.001; and <0.0001]. Each query family was evaluated by searching with the bare domain and with 10 different embedding replicates (the same domain embedded in different shuffles). Searches ran to convergence or were stopped at 20 iterations. All other parameters used the default values.
By default, blastpgp 2.2.19 uses a composition-based score adjustment (−t 2) described in ref. (14). However, for our modification of PSI-BLAST, we interrupted the program after each iteration, extracted the aligned sequences, and built a new PSSM for the sequences using the appropriate alignment boundaries. Because the program was stopped and restarted using its checkpoint facility, a different composition-based score adjustment [−t 1, ref. (8)] is required by blastpgp.
PSI-BLAST noExt
We implemented a simple modification to PSI-BLAST to prevent the propagation of HOE errors. For the first iteration, (i) we search with PSI-BLAST against the standard search library for two rounds; (ii) the first-iteration PSSM and all significant [E() < 0.005] alignments are stored. For the second iteration, (iii) we search with PSI-BLAST against the standard search library for one round, using the first-iteration PSSM. (iv) The significant [E() < 0.005] alignments that overlap with previously found high-scoring segment pairs (HSP) are replaced by the original alignments, while all new HSP alignments are stored. (v) We build a significant-hits library: a formatdb PSI-BLAST compatible library of all significant (new plus HOE-corrected) HSP sequences plus 10 000 randomized sequences—the latter set provides a large sequence sample for more accurate E-value calculation. (vi) We run PSI-BLAST for two iterations against the significant-hits library with the first-iteration PSSM. (vii) We use all alignments with E() < 10−6 to build the HOE-corrected second-iteration PSSM. (viii) We continue to repeat steps (iii)–(vii) using the HOE-corrected PSSM from the previous iteration (e.g. for iteration 3, we use the HOE-corrected second-iteration PSSM) for a user-defined number of times or through convergence. All PSI-BLAST searches use the −t 1 composition-based score adjustment (8), which is the only permitted option when re-starting from a checkpoint PSSM file.
Classification of PSI-BLAST errors
Error types were classified based on differences between the known query embeddings, the annotated domain boundaries, and the query and library sequence boundaries in the PSI-BLAST alignment. TPs: in a TP alignment, at least 50% of the residues in the alignment overlapped the query domain and the homologous region in the library sequence, as defined by the annotated Pfam boundaries (Figure 1A). Because of the 50% overlap requirement for TPs, two types of FP errors can occur: (i) non-homologous FP alignments (NH-FP), which align the query domain to either a random sequence (Fig. 1B, left), a non-domain region (Fig. 1B middle), or a non-homologous domain (Fig. 1B right); (2) HOE FPs (Figure 1C). HOEs begin with a TP alignment but extend it so that more than 50% of the alignment is outside the homologous domains. Extensions can occur on the library domain (Figure 1C, left and middle) and on the query domain (Figure 1C, right). Each different error type for a sequence was recorded; multiple errors of the same type were counted once for the sequence. For coverage at iteration 5 and the sensitivity/specificity analysis, HOE errors were counted both as TPs and FPs. Likewise, non-homologous (NH) alignments due to a library sequence domain that had appeared in an earlier HOE alignment were scored as HOE alignments, but only as FPs.
Figure 1.

True positive and two types of PSI-BLAST errors. PSI-BLAST alignments are classified after comparing the alignment boundaries to the embedding boundaries in the query sequence, and to the annotated domain boundaries in the library sequence. (A) TP—an alignment is classified as a TP if at least 50% of the aligned residues overlap the Pfam annotation. Two types of FP errors can occur: (B) non-homologous FP alignments (NH-FP) and (C) homologous over-extension (HOE) FPs. Non-homologous FPs map entirely to random (and thus unrelated) sequences (B, left), to non-domain regions of the protein (B, middle), or to regions covered by unrelated Pfam domains (B, right). Homologous over-extension FPs occur when two homologous domains align, but the alignments overextend, so that more than 50% of the alignment is outside the homologous region. Both the library domain (C, left and middle) and the query domain (C, right) can overextend.
Family and tree coverage
Because homologous target library domains are rarely uniformly distributed across their phylogeny, simply counting the fraction of the homologous sequences identified in a search can obscure the evaluation of a method’s success in identifying distant homologs. Thus, we measured both family coverage, the fraction of the homologous domains identified, and tree coverage, the weighted fraction of tree partitions covered in the search. A phylogenetic tree for each query domain family was generated using Quicktree [v. 1.1, ref. (20)]. The multiple sequence alignments were built with HMMER [v. 2.3.2, ref. (21)] and the Pfam HMM model. Tree coverage is the weighted sum of the branch lengths of the sub-tree covered by the homologs found by the search, as illustrated in Supplementary Figure S1.
RESULTS
PSI-BLAST makes two distinct types of errors
Characterizing the sources of PSI-BLAST errors, in particular the process by which a PSI-BLAST PSSM becomes contaminated, is challenging. PSI-BLAST searches proceed iteratively, with the addition of new sequences that can either improve the sensitivity of the PSSM, or, with the inclusion of unrelated sequences, that can turn the PSSM toward an unrelated family. Moreover, sequence inclusions that take place early may not have a major effect until several iterations later. To try to follow the process of sequence inclusion and PSSM contamination, we searched a mixture of real, full-length, UniProt proteins and random synthetic sequences using two sets of query sequences that contained a single Pfam domain. The boundaries of each significant alignment, at each iteration, were recorded and classified by alignment type (Figure 1). TPs align the domain in the query sequence to homologous domains in the reference library, while FPs align the query domain to a non-homologous region. Examination of FP alignments revealed two distinct error morphologies: alignments on non-homologous regions (NH-FP, Figure 1B) and alignments that begin in a homologous domain, but that extend into a non-homologous region (HOE, Figure 1C).
Non-homologous FP errors are well recognized. These errors involve alignments either to random sequences (Figure 1B, left), to regions of the protein that do not contain Pfam domain annotation (Figure1B, middle), or to regions that contain unrelated domains (Figure 1B, right). Non-homologous errors have been the target of the improved PSI-BLAST statistical estimates. Non-homologous regions align by chance, so methods that reduce chance alignments, either with more accurate statistics or stricter inclusion thresholds, will reduce non-homologous errors. This is an effective approach because the E()-values for the first NH-FP errors are often near the threshold for inclusion of a sequence for the next iteration. In our hard and randomly sampled data sets, the first NH-FP errors had expectation values ranging E() < 10−7–10−3 (Figure 2A and C).
Figure 2.

Distribution of initial alignment errors. The expectation values for the first FP errors are shown for each of 50 hard (A and B) or randomly-sampled (C and D) searches, classified by error type (i.e. HOE-L, HOE-Q and NH). FP E()-values are plotted from lowest (most-significant) to highest in each of the four panels; thus, query families are ordered differently in each panel. The iteration number for the first independent FP type (when two FP types occur for a query in the same iteration, the less significant FP is not considered independent) with the lowest E()-value (expectation) for each error type is also shown. (A) FP E()-values and error types with embedded hard queries against the standard domain library. (B) Searches with non-embedded hard queries against the long-domain library. (C) Searches with the embedded randomly sampled queries against the standard domain library [the E()-value for the lowest HOE-L first FP in this panel is E() = 5 × 10−70, but it is graphed at E() = 10−50]. (D) Searches with non-embedded randomly sampled queries against the long-domain library.
We were initially surprised that in our comparisons, some FPs had extremely significant similarity scores, with E()-values (expectations) < 10−70–10−40 (Figure 2A and C). Examination of these alignments provided a simple explanation; they began in a homologous region, which sometimes produced a high similarity score, but extended into a non-homologous region. In contrast to non-homologous alignments, HOE cannot be reduced with better statistics or reasonable inclusion thresholds; a threshold that excluded alignments with E()-values less than E() < 10−40 would exclude most homologs. HOE is not caused by inaccurate statistical estimates; it is caused by inaccurate alignments.
In our searches, HOE errors can occur on the library sequence (HOE-L: Figure 1C, left and middle) and on the query sequence (HOE-Q: Figure 1C, right). But, library sequence over-extensions tend to cause more problems, because, in the library sequence, the (HOE-L) extension can continue into a non-homologous domain, which, in turn, can recruit additional homologs to that unrelated domain. HOE-Q errors are found at all inclusion thresholds, but not as frequently as HOE-L errors: HOE-L errors occur 10-times more often than HOE-Q errors (Figure 2A and C). HOE-Q errors cannot occur when searching with non-embedded domains (Figure 2B and D).
PSI-BLAST searches have a history that affects the number of errors
The pathological consequences of HOE are illustrated in Figure 3 and Table 1, which follow an over-extension that brings a new domain into the PSSM, so that the unrelated domain eventually crowds out the signal from the original homology. Here, the query domain is a 298-residue region from 2 to 300 from Q8KR72_PHOLU, a condensation domain in the photobactin biosynthesis protein PhG. For this PF00668 query, the first error occurs at iteration 3 when an initially homologous alignment extends onto the unrelated PF00550 domain. As a result of the initial over-extension, by iteration 5, most of the alignments only contain the PF00550 domain (Figure 3, Table 1). In fact, while these two families are similar in size (PF00668: 2528 members; PF00550: 2855 members) and the actual query was a member of the PF00668 family, the search at iteration 5 covered ∼50% of the non-homologous PF00550 family and only 13% of the PF00668 family. PF00668 and PF00550 have clearly distinct structures; the PF00668 (Chloramphenicol Acetyl Transferase fold) domain is characterized by a 2-layer αβ sandwich fold, while the PF00550 acyl carrier protein domain is an all α structure.
Figure 3.

Iterative growth of a homologous over-extension. (A) The raw PSI-BLAST output of a search querying a PF00668 embedded domain against the standard curated Pfam library at iteration 5. The portion of the alignment that contains the PF00668 homologous domain is shown in blue, while the over-extension onto the structurally unrelated PF00550 domain is shown in red. (B) A diagram that tracks the progression of the alignments shown in (A) from iterations 2 through 5. The alignment on the partial PF00668 domain begins as the first FP in the search at iteration 3, and continues to overextend further onto the unrelated PF00550 domain (in red) in subsequent iterations. By iteration 5, the entire unrelated PF00550 domain is covered by the overextended alignment.
Table 1.
HOE from PF00668 domain onto the unrelated PF00550
| Iteration | TPs | Family coverage | Tree coverage | FPs | Unrelated Pfam coveragea |
|---|---|---|---|---|---|
| 1 | 271 | 0.11 | 0.14 | 0 | |
| 2 | 998 | 0.49 | 0.93 | 0 | |
| 3 | 478 | 0.19 | 0.93 | 113 | PF00550 (10), CL61(1) |
| 4 | 330 | 0.13 | 0.72 | 1784 | PF00550 (1111) PF08415 (15) CL61 (1) CL28 (52) CL63 (14) PF00501 (4) |
| 5 | 318 | 0.13 | 0.67 | 2174 | PF00550 (1347/2855) PF08415 (19/140) PF02911 (1/8) CL28 (38/2153) CL63 (16/9791) CL46 (6/3586) PF00501 (4/1905) |
aThe number of alignments to the unrelated family in the given iteration is shown in parenthesis. The size of the family is shown for the last iteration. The number of FPs is often less than the sum on the alignments that map to unrelated domains because sometimes over-extension in one sequence covered multiple unrelated domains.
HOE into unrelated domains can have a dramatic effect on alignments in subsequent iterations. As illustrated in Figure 3 and Table 1, when an alignment extends into an adjacent non-homologous domain, many of the unrelated-domain homologs can be found. An extension into a non-domain region or an error that maps entirely to a random sequence can also contaminate the PSSM, but this contamination is less likely to produce additional FPs, since it is much less likely that the non-domain or random sequence has additional homologs in the database. Non-homologous alignments with unrelated domains can cause error propagation in subsequent iterations, but these errors are relatively rare. The level of unrelated error propagation is proportional to the size of the unrelated family: i.e. errors to larger families lead to stronger corruption.
HOE causes the largest number of errors
Because PSI-BLAST errors can propagate in later iterations, we consider the relative importance of non-homologous and HOE alignments from three different perspectives. To focus on the initial cause of the errors, we show that HOE errors are the most common initial error (Figure 2), and that HOE errors account for the greatest loss of error-free family coverage (Figure 4). However, since there is no practical way to recognize the initial error in real searches, we also show that HOE errors produce the largest fraction of FPs at iteration 5, a commonly used PSI-BLAST stopping point (Figure 5).
Figure 4.

Error-free family coverage before the first FP. (A–D) show the weighted fraction of family coverage by FP type—HOE on the library sequence (HOE-L, filled down triangles), HOE on the query sequence (HOE-Q, filled up triangles), non-hom (non-homologous error, filled diamonds)—on two datasets: hard families (A and B) and sampled families (C and D). Performance against the standard library (all domains, panels A and C) and the long-domain library (B and D) is shown. Coverage for searches that converged without any error (no error) is plotted with an ‘X’. Results are weighted so that each of the 50 searches contributes 2% of the coverage. The weighted coverage of each search was calculated as 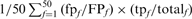 where
where 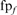 is the number of errors of each type (NH, HOE-Q, HOE-L) in family f;
is the number of errors of each type (NH, HOE-Q, HOE-L) in family f; 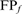 is the total number of FP errors for the family in the error iteration;
is the total number of FP errors for the family in the error iteration; 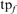 is the number of TPs found before the first error iteration, and
is the number of TPs found before the first error iteration, and 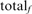 is the total family size in the complete or long-domain database. Thus, a search that achieves 50% family coverage before the first FP and had an equal number of HOE-L and NH errors would contribute 0.005 on both the HOE-L and the NH curves. The total family coverage in the iteration before the first FP for each search is also shown (open squares).
is the total family size in the complete or long-domain database. Thus, a search that achieves 50% family coverage before the first FP and had an equal number of HOE-L and NH errors would contribute 0.005 on both the HOE-L and the NH curves. The total family coverage in the iteration before the first FP for each search is also shown (open squares).
Figure 5.

Family coverage at iteration 5. The frequency of TPs and FPs found at iteration 5, weighted by number of alignments from each search is shown. Both hard (A, B) and randomly sampled (C, D) families were tested against the standard library (all domains, A, C) and the long-domain library (B, D). The weighted FP frequency at iteration 5 is 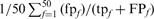 , where
, where 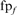 is the number of FPs of the specified type (HOE-Q, HOE-L, NH, or total errors) in family f at iteration 5,
is the number of FPs of the specified type (HOE-Q, HOE-L, NH, or total errors) in family f at iteration 5, 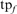 is the total number of FPs at iteration 5, and
is the total number of FPs at iteration 5, and 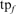 is the number of TPs found for the family at iteration 5. Filled squares plot the total weighted coverage of all three error types: HOE-Q (up-triangle), HOE-L (down-triangle) and NH (diamond). Total family coverage (open squares) is defined as
is the number of TPs found for the family at iteration 5. Filled squares plot the total weighted coverage of all three error types: HOE-Q (up-triangle), HOE-L (down-triangle) and NH (diamond). Total family coverage (open squares) is defined as 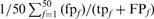 , where
, where 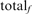 is the total number of homologs in the family. With this weighting, a family that finds all of its homologs without any errors will contribute 0.02 to the coverage; a family that finds half of its homologs and an equal number of non-homologs will get a weighted frequency of (0.02 × 0.333) for the HOE-L, HOE-Q or non-hom. error type. For this figure and Figure 7, an HOE-L or HOE-Q alignment is counted both as a TP, reflecting the homologous alignment, and as a FP, because more than half of the alignment is outside the homologous domain; NH alignments are counted as only as FPs.
is the total number of homologs in the family. With this weighting, a family that finds all of its homologs without any errors will contribute 0.02 to the coverage; a family that finds half of its homologs and an equal number of non-homologs will get a weighted frequency of (0.02 × 0.333) for the HOE-L, HOE-Q or non-hom. error type. For this figure and Figure 7, an HOE-L or HOE-Q alignment is counted both as a TP, reflecting the homologous alignment, and as a FP, because more than half of the alignment is outside the homologous domain; NH alignments are counted as only as FPs.
Figure 2A and C display the E()-value and error type for each of 50 hard and randomly-sampled queries in our test set for a search with the default PSI-BLAST inclusion threshold of E() < 0.005. In this figure, we report the lowest E()-value for each error type in the first iteration that produced an error. Thus, we seek to catch the initial error event, before the error alignment has been incorporated into the PSSM. For the searches with hard families, HOE-L errors occurred first for 43/50 query domains, HOE-Q errors occurred first in 12 searches, and NH errors occurred first in four searches. In addition, three of the searches converged without an error. (These values add up to more than 50 because, in some searches, several first errors occurred in the same iteration.) For the randomly-sampled query searches, the prevalence of HOE-L first errors is similar; 34 queries had first HOE-L errors, 10 queries had HOE-Q first errors, 10 had NH first errors and eight searches had no errors at convergence. Figure 2 also reports the iteration in which the first error of each type occurred. Figure 2 shows clearly the challenge posed by HOE alignments; many have extremely significant E()-values (produced because the alignment starts in a homologous region), which prevent a more conservative statistical approach from excluding these errors.
While Figure 2 shows that HOE errors occur early, often, and with very significant E()-values, Figure 2 does not show the effect of HOE errors on search sensitivity (family coverage). HOE errors are not only important because they happen early, but also because early errors dramatically reduce the sensitivity of the search. Figure 4 provides an alternative perspective on the searches shown in Figure 2; for exactly the same searches, it reports the fractional error-free family coverage before the first FP. We measure error-free coverage as the family coverage achieved at the iteration before the first FP, and distribute the coverage among the three different types of errors. Thus, each of the 50 queries contributes 2% to the graph if all of its homologs are found. Here again (Figure 4A and C), we see that HOE-L and HOE-Q errors are responsible for the largest reduction in error-free coverage. NH errors have a small effect on the level of error-free coverage. With hard queries, at a PSI-BLAST inclusion threshold of 0.01, 3.7% of fractional error-free coverage ends with an NH error; this drops to 2.2% at E() <10−4. For the randomly sampled query families, the reduction in NH errors is more dramatic as the PSSM inclusion threshold becomes more stringent; NH errors terminate weighted coverage at 7.2% for E()<0.01 dropping to 0.9% at E()<10−4. As expected, NH first-errors drop with more stringent statistical thresholds.
Implementing more stringent inclusion thresholds has little effect on HOE errors (Figure 4). For hard queries, HOE-L fractional error-free coverage remains steady between 12 and 14% across the threshold range, and HOE-Q error-free coverage ranges between 2 and 3%. For the randomly-sampled queries, 29–32% of error-free coverage ends with an HOE-L while 6–7% of error-free coverage ends with an HOE-Q error. Thus, not only do HOE-L and HOE-Q errors happen early, HOE errors dramatically reduce the number of homologs that can be found before the first error.
The plots in Figures 2 and 4 describe what happens when no errors are made; Figure 4 reports sensitivity (family coverage) at very high specificity, i.e. before the first error occurs. In general, researchers are comfortable with a modest number of errors if that reduction in specificity allows more homologs to be detected (increased sensitivity). Indeed, for the hard families, average family coverage increases from ∼20 (Figure 4A) to 76% at iteration 5 (Figure 5A). However, that increased sensitivity comes with a cost; selectivity drops from zero FPs (Figure 4) to from 3% [E() <10−4] to 5% [E() <0.01] FPs. For the randomly sampled queries, coverage increases from 59% (Figure 4C) to 86% coverage (Figure 5C), with ∼9% FPs.
HOE is also a major source of error at iteration 5, although its predominance among the other error types is not as dramatic as it is at first FP; HOE errors were found 1.5–3 times as frequently as the NH errors (Figure 5A and C). Our HOE measurements are underestimates. We only record HOE errors once they cover 50% of the alignment; a shorter over-extension might include a non-homologous domain, but the errors caused by that inclusion would be counted as NH errors. If HOE alignments are classified after 25% over-extension, HOEs are identified sooner, their frequency increases, and error-free coverage is reduced even more. However, we used a 50% alignment threshold to focus on clear over-extensions.
HOE errors largely occur because of partial domains in proteins
HOE has been recognized for more than 10 years in genomic DNA sequence alignments; a strong similarity in an exon alignment can cause the alignment to extend into a non-homologous intron or flanking sequence (22). Because local alignment algorithms, like Smith–Waterman, FASTA, BLAST and PSI-BLAST, determine their alignment boundaries to maximize the similarity score, local alignment over-extension can occur adjacent to any high-scoring region. However, the problem may be exaggerated with profile-based methods like PSI-BLAST, which can produce higher positive scoring matrix values in conserved regions, and may weight non-homologous aligned residues to encourage alignment across the entire PSSM. (Indeed, the drop-off threshold was introduced in BLAST (1), which is implemented as the x-drop parameter in BLAST2 and PSI-BLAST (8), to reduce this tendency.) Thus, we expect that many HOE alignments occur between a full-length domain (such as our query sequences), and partial homologous domains in other proteins.
We examined the importance of partial domains in nucleating HOE by comparing our query domains to a reference library with a reduced number of partial domains. In this long-domain library, Uniprot sequences containing query homologs were excluded if their homologous domains were shorter than 75% of the Pfam model length. Our standard domain library contains 234 505 UniProt sequences (and an equal number of randomly shuffled sequences). Our long-domain library contains 139 165 sequences (and an equal number of random sequences).
Searches against the long-domain library dramatically illustrate the importance of HOE from partial domains. Out of 50 hard searches, 45 generate HOE errors against the standard library. Against the long-domain library, the number of searches with initial HOE errors decreases to 17, and to only one when the embedding is removed (Figure 2B). In the randomly sampled families, HOE errors decrease from 36 (standard library) to 7 (long domain library) to 1 (long-domain library + non-embedded domains, Figure 2D). However, the long-domain library also has better performance because long domains are easier to find than short domains. Error free coverage improves by ∼15% (from 21 to 25% for hard queries, and from 56 to 67% for sampled queries) when coverage of only long domains is calculated (data not shown).
A similar trend is seen with weighted coverage before the first FP (Figure 4B and D) and with coverage and fraction of FPs at iteration 5 (Figure 5B and D). In both cases, when the long-domain library is searched, the number of HOE errors drops dramatically (from 16 to 3% of weighted error free coverage for hard queries, 35–5% for randomly sampled queries), the no-error at convergence coverage increases (from 2 to 58%), and total coverage increases as well (from 20 to 65% for hard queries before the first FP).
HOE happens frequently because partial domains occur in many proteins. Domains <75% of Pfam model length are found in 95 340 (41%) of our protein sequences, and removing them from the library removes 47% of all domains. Twenty percent of the proteins in our standard library contain a domain that is half the Pfam model length or less. The distribution of query domain homolog lengths in our reference libraries is shown in Supplementary Figure S2.
While many HOEs of library sequences (HOE-Ls) start in partial domains, HOE of the query sequence (HOE-Qs) cannot involve partial sequences; all our query sequences are full-length domains. For example, in Figure 3, while the library sequence contains a partial domain, the homology over-extends into the random embedding sequence around the query domain. The role of HOE-Q extension can be estimated by preventing over-extension of the query sequence by removing the embedding. For the hard query sequences, searching with non-embedded queries improves sensitivity (weighted coverage) from 20 to 29% in the standard library and from 63 to 75% in the long-domain library. Reducing HOE-L extension with the long-domain library and HOE-Q extension with non-embedded query domains increases no-error at convergence from 2% (hard) and 16% (sampled) weighted coverage at E() < 0.005 to 67% (hard) and 90% (sampled) no error weighted coverage on the long-domain library without embedding.
While embedding the query domains usually reduces search coverage (Figure 4), we were surprised to find that sometimes query embedding can improve search performance (Table 2). The data in Figures 2, 4 and 5 reflect the result from a single random embedding for each of the query domains, but searches were done with 10 different embeddings. In 20% of the hard queries and 14% of the sampled ones, family coverage at first FP differs by more than 50% across the 10 embeddings. And for 6 and 20% of the hard and sampled queries, respectively, the number of FPs at iteration 5 also varies by more than 50% across the embedding replicates. For example, one embedding of the PF01018 domain finds 98% of the homologs in the library and makes very few errors; while the another embedding of the same domain, only finds 22% of the family and 78% of the results that PSI-BLAST reports are FPs. The random embeddings alone do not produce any statistically significant scores, but different embeddings can produce slightly different over-extension alignments.
Table 2.
Performance of different embeddings of PF01018 at iteration 5
| Embedding replicate performance | TPs | TP coveragea | FPs | FP frequencya |
|---|---|---|---|---|
| Best | 507 | 0.98 | 67 | 0.07 |
| Worse | 116 | 0.22 | 419 | 0.78 |
aTP coverage is calculated by dividing the frequency of TPs at iteration 5 by family size. FP frequency is defined as the number of FPs found divided by the number of significant alignments found by the search.
Indeed, for one query set, HOE of the query improved PSI-BLAST performance because the random over-extension allowed a marginally insignificant homolog to score within the statistical threshold. Thus, for searches with a PF00589 query (Q1YWW7_PHOPR, a phage integrase domain), in nine out of 10 different embedded replicate searches the query could only find itself, causing search convergence after just two iterations. For one of the 10 embedding replicates, the alignment of the only recovered sequence at iteration 1 extends 11 residues into the unrelated embedding. This extended alignment allowed homologs to be found, so that a PSSM could be built. As a result, that one search in 10 achieved 95% family coverage at iteration 5.
PSI-BLAST searches follow an opportunistic phylogenetic path
The traditional measure of search sensitivity, the fraction of related homologs found in a search, can obscure the performance of searches in different protein families. Many protein families in Pfam and UniProt are sampled non-uniformly. Many vertebrate homologs might be available, but few bacteria or archea are present in the databases. Thus, a query sequence from a vertebrate might achieve high coverage, or apparent sensitivity, while an archeal sequence might be much less successful in identifying homologs.
Tree coverage scoring complements the traditional family coverage evaluation of similarity searching methods. Here, we count how completely the results from a search cover a phylogenetic tree of the family members. For the example above, a vertebrate query might find 80% of the family members, but miss a large fraction of the tree, while the archeal query sequence might find homologs in prokaryotes and eukaryotes, and in animals, plants and fungi, and thus sample the phylogenetic tree much more completely. Tree coverage is measured by counting the branch-length-weighted partitions traversed by the searches at each iteration (Supplementary Figure S1).
To control for the different phylogenetic distributions of different domain families, we tested queries that were chosen based on their location on the family tree; one from a relatively populated region of the tree, a second from a deserted region. In the early iterations, searches from deserted parts of the family phylogeny traverse the tree in a ‘depth-first’ order, while searches that begin in populated regions use an initial ‘breadth-first’ approach.
By traveling deep on the tree initially, queries from deserted areas achieve better tree coverage, but worse family coverage, than queries from populated areas in early iterations. At iteration 1, 78% of the deserted (and 48% of the populated) queries have better tree coverage than family coverage (Figure 6). Tree location was not used as a criterion to choose the hard and sampled queries. Yet, 84% of the hard queries were found to come from deserted areas of the tree while the sampled queries were split almost evenly across both locations (48% populated and 52% deserted). As one might expect, the limitations in family coverage observed for the hard families (Figures 4 and 5), are likely explained by their patterns of tree coverage.
Figure 6.

Difference between tree coverage and family coverage by iteration. A comparison between family and tree coverage for the non-embedded searches of two queries from each of 50 families against the long-domain library. Two queries per family were chosen based on tree location: one domain query from a populated and another from a deserted area of the tree. The number of searches where tree coverage was larger than family coverage is plotted by iteration.
Despite the difference in tree coverage paths between ‘deserted’ and ‘populated’ queries, their family coverage in the late iterations is similar. At first FP, tree and family coverage did not differ significantly in searches with queries from deserted and populated areas of the tree. Likewise, by iteration 5, 86 and 89% of the homologs have been found by the deserted and populated queries, respectively (Figure 6).
Preventing alignment extension improves PSI-BLAST specificity
We implemented a small modification to PSI-BLAST to reduce the effect of HOE that prevents alignments from being extended once they have been included in the PSSM (PSI-BLAST noExt; see ‘Materials and Methods’ section). With this modification, the alignment boundaries calculated when a library sequence is first included in the PSSM are not extended when that library sequence is aligned in subsequent iterations. In some cases, the non-extension may decrease the sensitivity of the search because the initial alignment was too short, but the no-extension modification provides a simple strategy for reducing over-extension. With the PSI-BLAST default E() <0.005 threshold, the no extension modification dramatically improves PSI-BLAST specificity with a very small cost in sensitivity (Figure 7). At 50% family coverage, the PSI-BLAST noExt reduces the weighted fraction of errors from 0.02% of TPs (hard families) to 0.0036%, a 5.6-fold reduction in FPs. At the end of iteration 5 with hard families, PSI-BLAST noExt achieves 93% of the family coverage of unmodified PSI-BLAST, with one-quarter the FPs. For the randomly sampled families, the improvement is even greater. For the randomly sampled queries, PSI-BLAST noExt finds 98% of the homologs found by conventional PSI-BLAST, but reduces the FP errors 8.3-fold at maximum coverage and 14.7-fold at 50% family coverage (Figure 7).
Figure 7.

Sensitivity and Specificity of PSI-BLAST and PSI-BLAST noExt. Weighted fractional family coverage for TPs is plotted as a function of weighted fractional FPs at iteration five using a threshold of E() <0.005. (A) Performance of unmodified PSI-BLAST with the −t 1 composition adjustment (long dashed line), unmodified PSI-BLAST with the default −t 2 composition adjustment (short dashed line), and PSI-BLAST noExt (solid line) on the hard queries. (B) Performance of PSI-BLAST −t 1, PSI-BLAST −t 2 and PSI-BLAST noExt on the randomly sampled queries. As in Figures 4 and 5, each family contributes 2% to the fraction of TPs (weighted). On the y-axis, fraction TPs (weighted) is calculated as 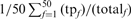 where
where 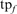 is the number of TPs at iteration 5, and
is the number of TPs at iteration 5, and 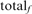 is the total number of homologs in family f. Likewise, fraction FPs (weighted) is calculated as
is the total number of homologs in family f. Likewise, fraction FPs (weighted) is calculated as 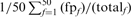 , where
, where 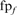 is the number of FPs at iteration 5. For this figure and Figure 5, an HOE-L and HOE-Q alignment is counted both as a TP and a FP.
is the number of FPs at iteration 5. For this figure and Figure 5, an HOE-L and HOE-Q alignment is counted both as a TP and a FP.
PSI-BLAST noExt also performs well by the performance measures displayed in Figures 2, 4 and 5. Since PSI-BLAST noExt does not modify the initial alignment boundaries, its performance on the first FP measures should be similar to unmodified PSI-BLAST, and this is largely true. On the hard queries, PSI-BLAST noExt (threshold E() <0.005) has a first FP at E() <10-73, similar to the unmodified PSI-BLAST. But with the sampled queries the worst first FP is E() <10−28, a dramatic improvement over the unmodified PSI-BLAST E() <10−70 (Figure 2). PSI-BLAST noExt coverage [threshold E() <0.005] at first FP is 21.4% (hard) and 60.9% (sampled), which is slightly better than unmodified PSI-BLAST (20.8 and 56.5%, Figure 4). For the sampled families, the coverage by searches with no-error at convergence increases from 15.9 to 28.9% and it is about the same (2%) for both methods on the hard families. For the hard families, PSI-BLAST noExt coverage at iteration 5 is 74.6% (hard) and 89.0% (sampled) with 1.4 and 2.0% FPs, compared with 77.3 and 85.3% coverage, and 5 and 9% FPs (Figure 5). The no-extension strategy does not reduce over-extension when it first occurs, but it dramatically improves specificity, at a small cost in sensitivity, by preventing additional extension in subsequent iterations.
DISCUSSION
For iterative searches that build a PSSM, HOE, is an important source of error caused by alignment errors. On our data set, HOE errors account for the largest fraction of PSI-BLAST errors (Figure 2), the greatest reduction of error-free coverage (Figure 4), and the largest fractions of FPs at iteration 5 (Figure 5). Moreover, we exclude nested or discontinuous domains from our queries; such domains are likely to cause even more HOE errors.
We used several strategies to reduce the possibility that HOE errors are an artifact of domain boundary annotation in the query and library sequences. For the queries, we embedded the domain queries in random flanking sequences to clearly specify the boundary between the homologous and non-homologous (random) regions. Yet, long over-extensions from the query sequence account for about a quarter as much error-free coverage loss as is caused by over-extension of homologous library domains. While we are less certain of the accuracy of the library sequence domain boundaries, we extended the annotations on the library sequences in an attempt to reduce error misclassification. We carefully examined the HOE-L errors with very low E()-values shown in Figure 2, and are confident that they involve alignments between sequences that are structurally unrelated at the fold (SCOP) or topology (CATH) level. While over-extensions onto non-domain regions may be attributed to conservative homology annotations, we are confident that over-extensions onto unrelated domains are true errors.
HOE provides an explanation for the observation that PSSMs built from manually curated Pfam HMMs perform significantly better than that same PSSMs supplemented with iterative searches against ‘nr’ [Figure 2 in ref. (12)]. The ‘nr’ proteins contain partial domains; HOE from these domains may be responsible for the reduction in search specificity.
HOE is an alignment error, rather than a statistical error. The high similarity scores that nucleate a HOE reflect genuine homologies. Thus, HOE errors occur early and with extremely low E-values; they cannot be fixed with better statistics or by lowering the inclusion threshold. Since its release in 1997, adjustments to the construction of PSSMs (8,11,14) and improved estimates of the statistical parameters used in the scoring function (12,23) have been described to address PSI-BLAST’s susceptibility to PSSM corruption. Such refinements have improved the sensitivity and specificity on the evaluation test sets, which are often sets of proteins from yeast or Astral that contain few multiple-domain proteins or partial domains. Likewise, the approach suggested by Lee et al. to resolve PSSM corruption does not address the HOE problem (10,13). Lee et al. suggest that the results from the first two iterations can be used to discriminate TPs from FPs at later iterations, but our results show that dramatic HOE errors are often seen at iteration 2, with expectation values as low as E() <10−70– <10−40 (Figure 2).
HOE is a natural consequence of an alignment strategy that is very effective in conventional pair-wise similarity searching. Because a longer homologous alignment produces a higher score, modern alignment programs use gap penalties that are both as low as possible and still allow local sequence alignments against unrelated sequences, yet tend to produce near-global alignments in homologous sequences or domains. While this strategy improves sensitivity and has little effect on specificity for pair-wise alignments (which are often bounded by the ends of the sequences), the ‘longer alignment’ strategy can recruit unrelated domains in iterative searches. Alignment boundaries are less important for pair-wise comparison, where errors do not propagate, and less common in datasets that contain relatively few shuffled domains (e.g. the yeast genome) or domain-sized sequences [PDB (24), Astral (25) and CATH (19) sequences]. In contrast, more comprehensive databases of complete proteins contain many partial domains and domains in different combinations.
While we have focused on PSI-BLAST because it is the most popular iterative program for building PSSMs, HOE will affect any automatic iterative profile searching method (PSSM, HMM, etc.). The HMMs constructed for Pfam (26) are curated and aligned manually, and thus less sensitive to the over-extension problem, and the HMMER package provides two scoring strategies, −ls (global) and –fs (local), which may more accurately delimit partial domain boundaries. But even the hmm-fs (local) scoring system may have a tendency to over-extend (domain) alignment boundaries, particularly if some parts of the domain are highly conserved while others are more neutral. Domain over-extension is a problem for every sequence alignment, but when erroneous alignments are iteratively incorporated, over-extension is more pathological.
We can imagine three strategies for reducing the effect of iterative HOE and tested a crude implementation of one of these strategies. A simple strategy is to search with a domain against a library of domains, rather than against a library of complete protein sequences. Pfam domain assignments are available for a large fraction of UniProt sequences (15); iterative searches against the proteins broken at domain boundaries, so that both domain and inter-domain sequences are aligned, but separately, might dramatically reduce PSSM corruption.
We have implemented a second strategy—limiting the extension of an alignment once it has been included in the PSSM. Currently, PSI-BLAST searches a database, identifies all the statistically significant sequences in the database, aligns them, builds a PSSM, and then repeats the process, re-adjusting the boundaries of every sequence found in the database at every iteration. HOE takes place over multiple iterations. Thus, to reduce it, we set the alignment boundaries when a sequence is first brought into the PSSM alignment, and preserve the alignment boundaries for that sequence in subsequent iterations. Our initial implementation of this strategy is effective; for hard and sampled queries, the strategy reduces the number of FPs 4–8-fold, with little reduction in sensitivity (Figure 7). However, while this strategy reduces iterative over-extension, there is room for improvement: initial alignments often extend beyond the domain boundaries.
A third, more sophisticated strategy might use localized PSSM/HMM scoring parameters to terminate alignments sooner, which should, in turn, reduce over-extension. Scoring matrices have preferred alignment lengths. Evolutionarily-deep matrices (e.g. BLOSUM45/50) generate longer alignments against random (unrelated) sequences than shallower matrices (e.g. BLOSUM62/80) (27). In diverse families, a single PSSM or HMM model will always be distant from the leaves of the family’s phylogenetic tree. Thus, as the model incorporates more diverse homologs, the evolutionary distances become larger and, as a result, these profiles produce longer alignments against unrelated sequences. Having multiple shallower scoring models near the leaves of a tree, much like those SATCHMO (28) generates, might make it possible to design PSMMs/HMMs that are more likely to generate shorter alignments against unrelated sequences. Alternatively, initial alignment boundaries might be set by pair-wise alignment to the closest homolog in the family using the appropriate position-independent scoring matrix.
In addition to suggesting strategies for dramatically improving PSI-BLAST specificity, our results suggest that sensitive profile-based comparison methods can produce accurate statistical estimates. One could argue that, as profile comparison methods approach the sensitivity of structure comparison, the reduced variety of structural motifs might make it more difficult to distinguish divergent from convergent similarities. While this may be true, the current inaccuracies in PSI-BLAST are more easily explained by HOE.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Library of Medicine, LM04969. Funding for open access charge: National Library of Medicine, R01 grant (LM04969).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 2.Pearson WR. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol. Biol. 2000;132:185–219. doi: 10.1385/1-59259-192-2:185. [DOI] [PubMed] [Google Scholar]
- 3.Pearson WR. Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms. Genomics. 1991;11:635–650. doi: 10.1016/0888-7543(91)90071-l. [DOI] [PubMed] [Google Scholar]
- 4.Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 5.Brenner SE, Chothia C, Hubbard TJ. Assessing sequence comparison methods with reliable structurally identified distant evolutionary relationships. Proc. Natl Acad. Sci. USA. 1998;95:6073–6078. doi: 10.1073/pnas.95.11.6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearson WR. Comparison of methods for searching protein sequence databases. Protein Sci. 1995;4:1145–1160. doi: 10.1002/pro.5560040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearson WR, Sierk ML. The limits of protein sequence comparison? Curr. Opin. Struct. Biol. 2005;15:254–260. doi: 10.1016/j.sbi.2005.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001;29:2994–3005. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sierk ML, Pearson WR. Sensitivity and selectivity in protein structure comparison. Protein Sci. 2004;13:773–785. doi: 10.1110/ps.03328504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee MM, Chan MK, Bundschuh R. SIB-BLAST: a web server for improved delineation of true and false positives in PSI-BLAST searches. Nucleic Acids Res. 2009;37:W53–W56. doi: 10.1093/nar/gkp301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altschul SF, Gertz EM, Agarwala R, Schaffer AA, Yu YK. PSI-BLAST pseudocounts and the minimum description length principle. Nucleic Acids Res. 2009;37:815–824. doi: 10.1093/nar/gkn981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stojmirovic A, Gertz EM, Altschul SF, Yu YK. The effectiveness of position- and composition-specific gap costs for protein similarity searches. Bioinformatics. 2008;24:i15–i23. doi: 10.1093/bioinformatics/btn171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee MM, Chan MK, Bundschuh R. Simple is beautiful: a straightforward approach to improve the delineation of true and false positives in PSI-BLAST searches. Bioinformatics. 2008;24:1339–1343. doi: 10.1093/bioinformatics/btn130. [DOI] [PubMed] [Google Scholar]
- 14.Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schaffer AA, Yu YK. Protein database searches using compositionally adjusted substitution matrices. Febs J. 2005;272:5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bairoch A, Apweiler R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL. Nucleic Acids Res. 1997;25:31–36. doi: 10.1093/nar/25.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.UniProt Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2009;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 19.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH—a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 20.Howe K, Bateman A, Durbin R. QuickTree: building huge neighbour-joining trees of protein sequences. Bioinformatics. 2002;18:1546–1547. doi: 10.1093/bioinformatics/18.11.1546. [DOI] [PubMed] [Google Scholar]
- 21.Durbin R, Eddy S, Krogh A, Mitchison G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, UK: Cambridge University Press; 1998. [Google Scholar]
- 22.Zhang Z, Berman P, Wiehe T, Miller W. Post-processing long pairwise alignments. Bioinformatics. 1999;15:1012–1019. doi: 10.1093/bioinformatics/15.12.1012. [DOI] [PubMed] [Google Scholar]
- 23.Altschul SF, Bundschuh R, Olsen R, Hwa T. The estimation of statistical parameters for local alignment score distributions. Nucleic Acids Res. 2001;29:351–361. doi: 10.1093/nar/29.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chandonia JM, Hon G, Walker NS, Lo Conte L, Koehl P, Levitt M, Brenner SE. The ASTRAL compendium in 2004. Nucleic Acids Res. 2004;32:D189–D192. doi: 10.1093/nar/gkh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altschul SF. Amino acid substitution matrices from an information theoretic perspective. J. Mol. Biol. 1991;219:555–565. doi: 10.1016/0022-2836(91)90193-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edgar RC, Sjolander K. SATCHMO: sequence alignment and tree construction using hidden Markov models. Bioinformatics. 2003;19:1404–1411. doi: 10.1093/bioinformatics/btg158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.