

Abstract
Human E1 is a key player in protein ubiquitination, however the E1 structure is not available. In this paper, we describe the derivation of a human E1 structure using molecular modelling based on the crystal structure of S. cerevisiae E1 and M. Musculus E1. Key interactions between our E1 model and ubiquitin are also discussed.
Keywords: ubiquitination, human E1, ubiquitin activating enzyme, homology modelling
INTRODUCTION
Ubiquitination is the process of tagging a target protein with ubiquitin (Ub) or a polyubiquitin chain as part of post-translational modifications leading to regulatory roles such as protein degradation in the proteosome [1]. The process of ubiquitination is mediated by a cascade of three enzymes known as the ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2) and ubiquitin ligase (E3). The main function of E1 ubiquitin activating enzyme is to catalyse the adenylation of ubiquitin at the expense of one ATP molecule. The resulting adenylated ubiquitin is then transferred to the active site cysteine residue of E1 through formation of a thioester bond, which is then transferred to the active site cysteine of E2 ligase, the next enzyme in the ubiquitination catalytic cycle, as reported in Fig. (1) [2–4].
Figure 1.
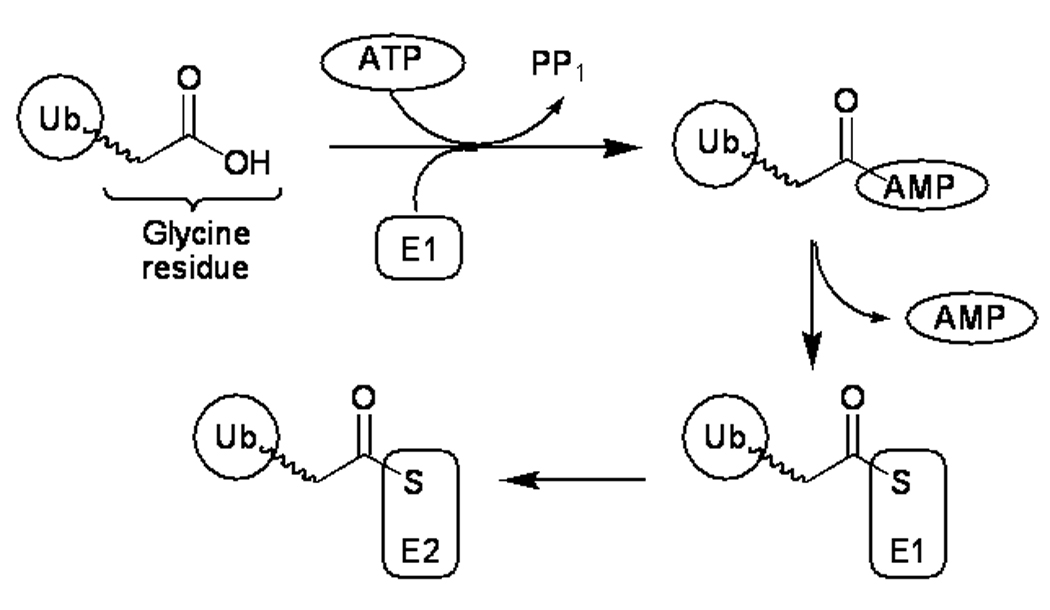
Schematic overview of the role of ubiquitin-activating enzyme, E1. The E1 reaction sequence begins with adenylation of a free ubiquitin molecule with concomitant ATP hydrolysis. The adenylated ubiquitin is then transferred to the active site cysteine residue of E1, followed by transfer to the active site cysteine residue of an E2 ubiquitin ligase, the next enzyme in the catalytic chain.
Until recently, it was generally accepted that there is only a single subtype of human E1 (Ube1; Uba1 in yeast). However in recent work by Harper and co-workers, it was shown that another E1 subtype exists that uses a different pool of E2 ubiquitin conjugating enzymes [5]. Advances in structural biology have led to the X-ray crystallographic/NMR elucidation of multiple structures of E2 and E3 enzymes in their free form as well as in complex with other partners, helping to uncover the mechanisms of ubiquitination enzymes at the molecular level [6,7]. However at the time of writing, the structure of E1 ubiquitin activating enzyme remains unknown, although E1 enzymes for neddylation and sumoylation have been resolved [4, 8–11]. The processes share significant similarities that have allowed extrapolation of the data to better understand the biological function of human E1 ubiquitin activating enzyme.
The roles of deregulated ubiquitin ligases, particularly in relation to cancer development, have meant that members of the ubiquitin ligase family present a multitude of potential therapeutic targets, and some ubiquitin ligases (particularly E3) have been exploited for therapeutic development. For example Mdm2 (protein from murine double minute oncogene) is a protein-protein binding partner [12] of the p53 tumour suppressor that acts as an E3 ubiquitin ligase. In the absence of cellular stress stimuli, Mdm2 keeps p53 levels low by catalysing the ubiquitination of p53, thereby directing p53 for proteosomal degradation. Since elevation of p53 activity is associated with an antitumour effect, attempts have been made to elevate cellular p53 levels by designing molecules that block the protein-protein interaction between p53 and Mdm2 [13]. Amongst the most promising inhibitors of p53-Mdm2 interaction in drug development are the highly potent imidazoline-based Nutlin compounds (e.g. Nutlin 3; 1; Fig. (2)) [14].
Figure 2.
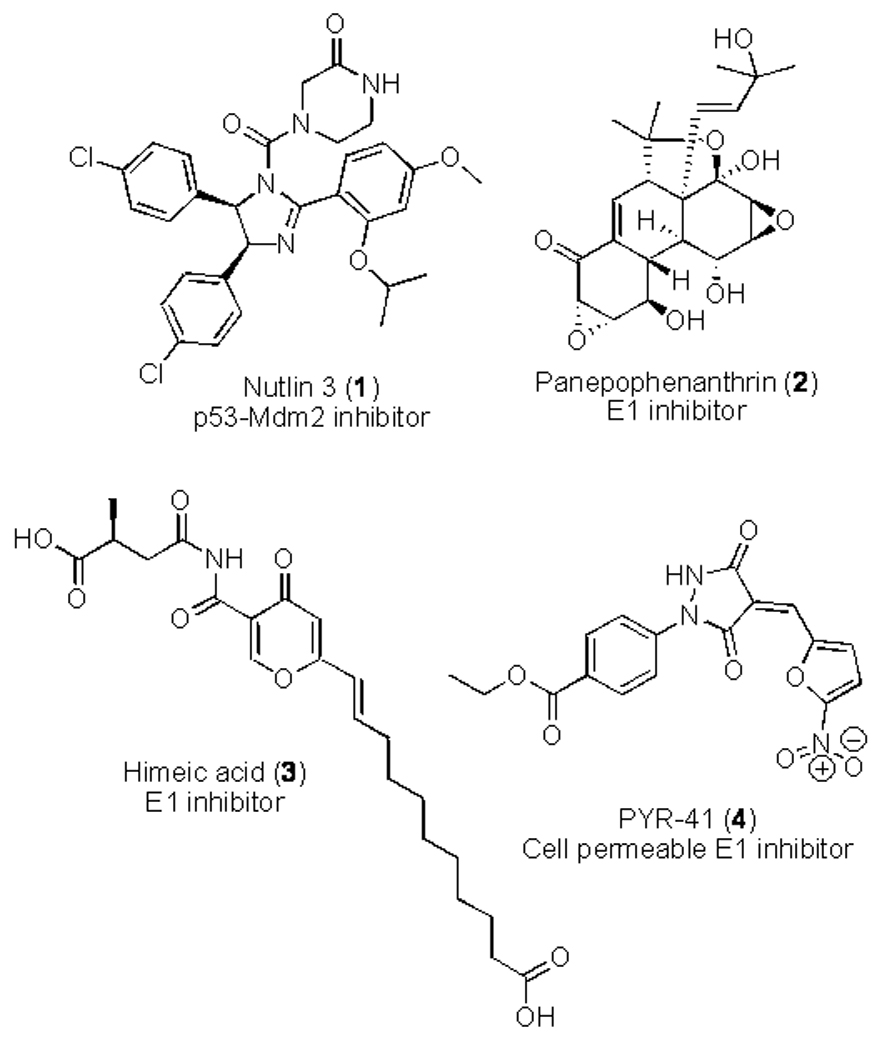
Structures of E1-ubiquitin activating enzyme inhibitors.
The discovery of inhibitors of ubiquitin-activating enzyme E1 has received less attention than inhibitors of the large family of E3 ligases, and the potential therapeutic value of selective small molecule E1 inhibitors is still a topical area of discussion [15–17]. The fundamental role of ubiquitin activating enzyme E1 could theoretically mean that E1 inhibitors would block all ubiquitination and have profound effects in fundamental cellular processes, producing unacceptable E1 inhibitor side effects that preclude therapeutic development. However the identification in recent years of small molecule E1 inhibitors from both natural product and synthetic sources, and study of their antitumour properties suggest that inhibition of E1 may present a viable therapeutic option in cancer. In addition to having potential cancer therapeutic activity, future selective E1 inhibitors will be important further study the roles of the E1 enzyme and related ubiquitin ligase biology.
The first reported inhibitor of E1 (expressed in E. coli) was a microbial secondary metabolite natural product known as panepophenanthrin (2; Fig. (2)) that inhibited the formation of the E1-ubiquitin complex in a dose dependent manner with an IC50 value of 17.0 mg/mL in a cell-free assay [15]. Unfortunately no effects in intact cells were observed up to concentration of 50 mg/mL. A further ubiquitin activating enzyme inhibitor known as himeic acid A (3; Fig. (2)) was isolated from a marine-derived fungus (Aspergillus sp.), but again potent effects in intact cell-based systems have not been reported [16].
The most significant inhibitor of ubiquitin-activating enzyme E1 reported to date is 4[4-(5-nitro-furan-2-ylmethylene)-3,5-dioxo-pyrazolidin-1-yl]benzoic acid ethyl ester (4; PYR-41; Fig. (2)) [17]. PYR-41 was found to have a range of useful antitumour cellular functions, including attenuation of cytokine-mediated nuclear factor-κB (NF-κB) activation through prevention of proteosomal degradation of IκBa, the inhibitory subunit of NF-κB. PYR-41 (4) was also found to inhibit p53 degradation, leading to differential activity in p53-expressing cancer cells. PYR-41 and related E1 inhibitors therefore have potential as lead compounds for further development, or as molecular tools to inform E1 cell biology research.
Since a complete crystal or NMR structure for human ubiquitin-activating enzyme E1 is not available at the time of writing, we here describe the use of computer modelling as an alternative to access the structure. A model for a protein structure can be built if the 3D structure of a homologous protein that shares high sequence similarity is available through NMR or crystallography. Higher sequence similarity between the two structures usually leads to better quality homology models. In this paper, we report a homology model for human E1 ubiquitin activating enzyme based on the crystal structure of E1 from S. cerevisiae (PDB entry: 3CMM) [18] and an E1 fragment from mouse (PDB entry: 1Z7L) [19].
METHODS
All molecular modelling studies were performed on a MacPro dual 2.66GHz Xeon running Ubuntu 8. Homology modelling was performed using MOE (Molecular Operating Environment) 2008/10 [20]. Minimisations were performed using the AMBER99 forcefield until a RMSD (Root Mean Square Deviation) gradient of 0.1 kcal mol−1 Å−1 was reached. Molecular dynamics simulations were performed using GROMACS 4.0.4 [21] using the NPT environment (300K; 1 atm; 0.002 ps timestep). The protein complex was initially soaked in a triclinic water box and neutralised by the addition of 29 sodium ions. The system was then minimized with GROMACS using a steepest descent algorithm. The initial 200 ps of the dynamics (equilibration phase) were carried out applying a position constraint on all bonds. A further 4 ns of simulation (production phase) were then performed with constraints on bonds that involved H-atoms.
HOMOLOGY MODELLING
The primary sequence of human E1 was downloaded from GenBank under the accession numbers AAA61246 [22]. The 1010 amino acid sequence was compared to the templates and the sequence alignment confirmed that the mouse protein 1Z7L shares the highest percentage residue identity with the query sequence (96%). Unfortunately, this small fragment consists only of 276 amino acids from the protein SCCH domain and it could not be used to build the full E1 human protein.
A more significant crystal structure of a complex structure of E1 enzyme and UBL from S. cerevisiae was solved by Imsang et al. [18]. The authors claimed that the S. cerevisiae E1 shares 50 % identity with human E1, and this was confirmed by the sequence alignment we performed with MOE (53%). It should be also noted that the sequence identity between the yeast E1 and the human E1 increases significantly in the ubiquitin binding site region, rising to 88% when the residues in direct contact with ubquitin are considered, as shown in Fig. (3). We decided to build the human E1 model using both the yeast protein and the mouse fragment as templates: the yeast E1 was used as the main template and the mouse fragment used to model only the corresponding SCCH domain. Ten models were calculated by MOE and the one with the best score was energy minimised.
Figure 3.

Sequence alignment between human E1, yeast E1 (PDB: 3CMM) and mouse E1 (PDB: 1Z7L). Represented in red are the residues that define the ubiquitin binding site.
The minimised model was then analysed further and validated using Ramachandran plots obtained from the RAMPAGE server [23]. The amino acid environment was evaluated using ERRAT plots [24], which assess the distribution of different types of atoms with respect to one another in the protein model. The main yeast E1 template was also evaluated and compared with the model (Table 1).
Table 1.
Comparison of our minimised E1 model with the yeast E1 template.
| Protein | Ramachandran plot | ERRAT |
|---|---|---|
| Our human E1 model | 95.0% | 94% |
| Yeast E1 (template) | 94.8% | 95% |
From the results obtained it is possible to observe that the stereochemical qualities of the model backbone and the amino acid side chain environment are virtually identical to the template, indicating a relatively accurate model. Furthermore, superimposition of the model with the two templates showed a very low RMSD (with yeast E1: 1.009; with mouse E1: 0.832), suggesting a high similarity between them (Fig. (4) and Fig. (5)).
Figure 4.
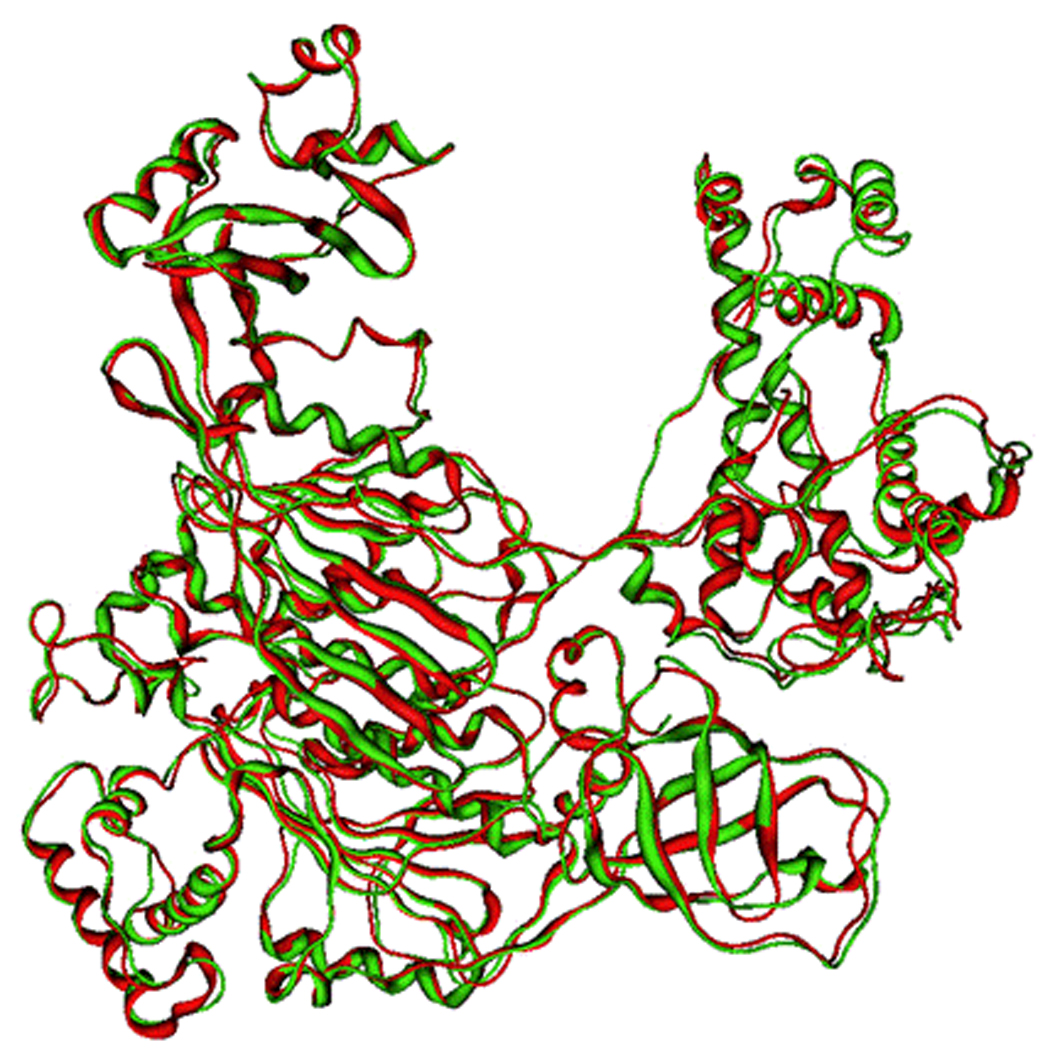
Superposition between the human E1 model and yeast E1 template (human E1 in green; yeast E1 in red).
Figure 5.
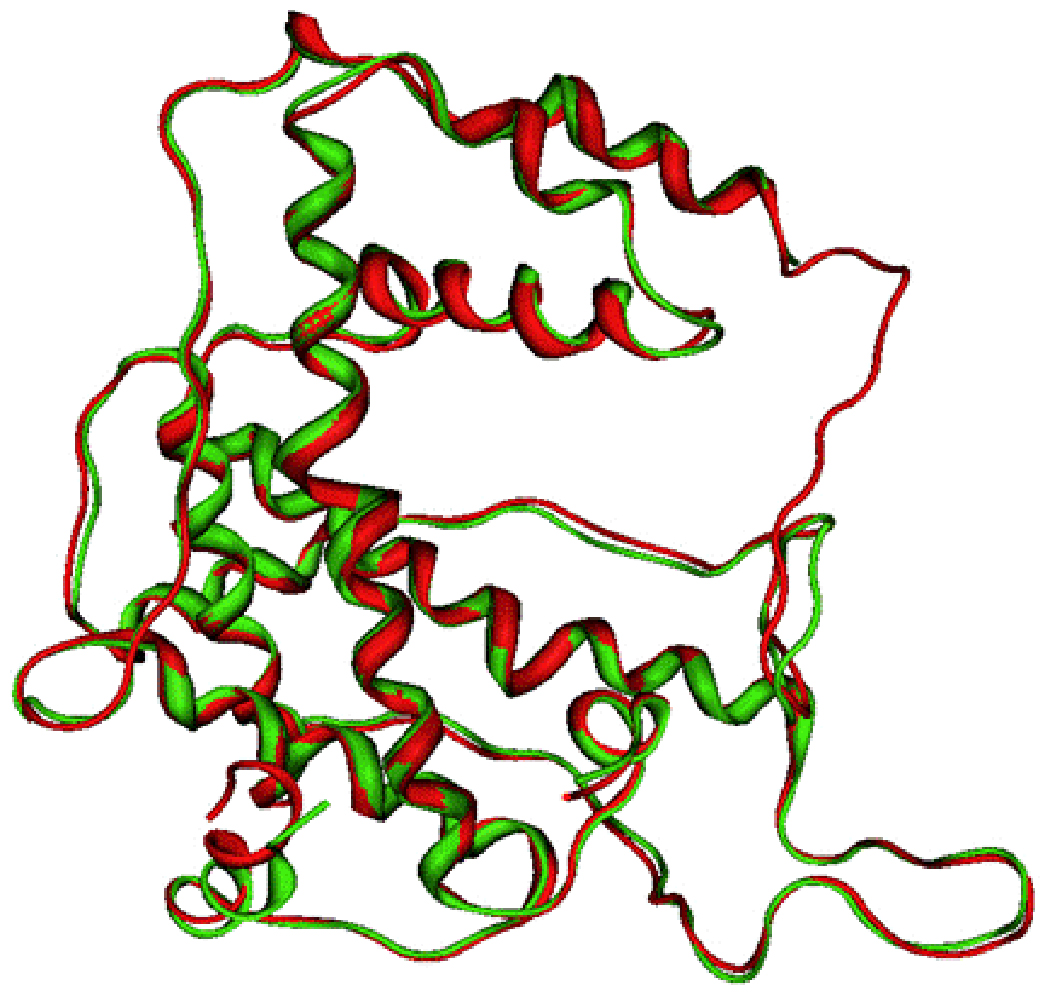
Superposition of the SCCH domains from the model of human and mouse E1 (human E1 in red; mouse E1 in green).
THE HUMAN E1-UBIQUITIN (Ub) COMPLEX
The adenylation site on E1 is located on a major groove. As might be expected, ubiquitin activation requires the formation of an E1-Ub complex, which is maintained by non-covalent interactions. We were interested in analysing these interactions, thus we built the E1-Ub complex starting from our human E1 model. The structure of human ubiquitin is available [25], but in the unbound conformation. For this reason, it would have been difficult to use this ubiquitin structure in building the complex. Given that human and yeast Ub differ only in two residues [Pro19/Ser19], and [Ala28/Ser28], we decided to build the model with the yeast Ub, which is present in the bound conformation in the yeast E1 structure used previously as primary template (PDB: 3CMM; chain B). Furthermore, we mutated the two residues above to the corresponding residues seen in the human UB with the rotamer explorer function of MOE. It is worth noting that the side chain conformation of the two mutated residue is comparable to the conformation of the corresponding residues on the human ubiquitin.
At this point, to further improve the model of the human E1-Ub complex, we performed a molecular dynamics simulation in water. It is possible to observe that during the 4ns production phase the system remains very stable. As shown in Fig. (7), the energy quickly reaches a plateau and the backbone RMSD shows little variation. The final structure from the simulation, shown in Fig. (8), was minimized with MOE and the interactions between the two proteins were examined.
Figure 7.
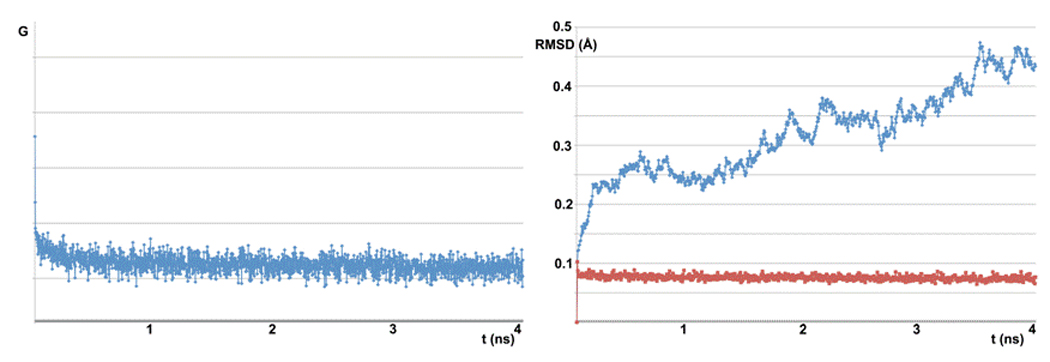
Molecular dynamics simulation plots. On the left is the energy variation during the simulation. On the right is the RMSD variation during the simulation (in red the RMSD value of each step compared to the previous one; in cyan the RMSD value of each step compared to the initial step).
Figure 8.
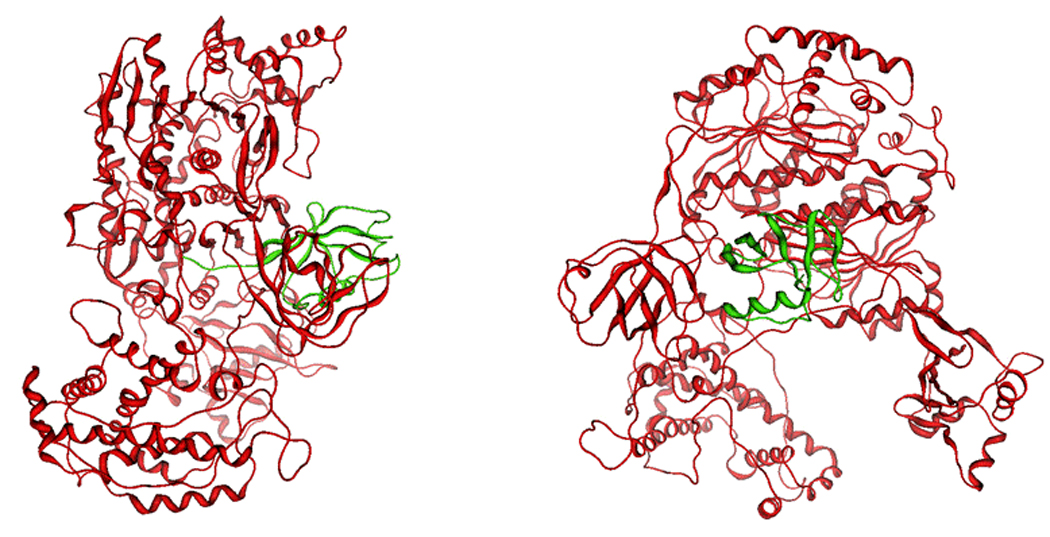
Model of human E1 in complex with ubiquitin viewed from different angles (90° rotation). The E1 model is represented in red; ubiquitin is represented in green.
The surface of ubiquitin that lies within 4.5A proximity of E1 was selected and the residues were identified. As mentioned above, the most remarkable feature is the extension of the ubiquitin C terminus to deliver the terminal glycine to the adenylation site within E1. This segment is made up of seven residues Val70 to Gly76, and makes extensive interactions with E1, as shown in Fig. (9). The most important ubiquitin residue is Arg72 whose guanidine side chain makes multiple hydrogen bonds with nearby E1 residues (Tyr 571, Asp 576). In addition, Arg 72 forms π–π stacking with the Tyr 571 aromatic side chain. This residue is thought to be responsible for the selectivity in recruiting ubiquitin and avoiding other Ubl’s such as SUMO and Nedd8 as shown by mutation studies [9,10]. The glycine at the C-terminus of ubiquitin is directed toward a glycine rich area of E1. This glycine rich sequence is a hallmark of adenylation sites.
Figure 9.
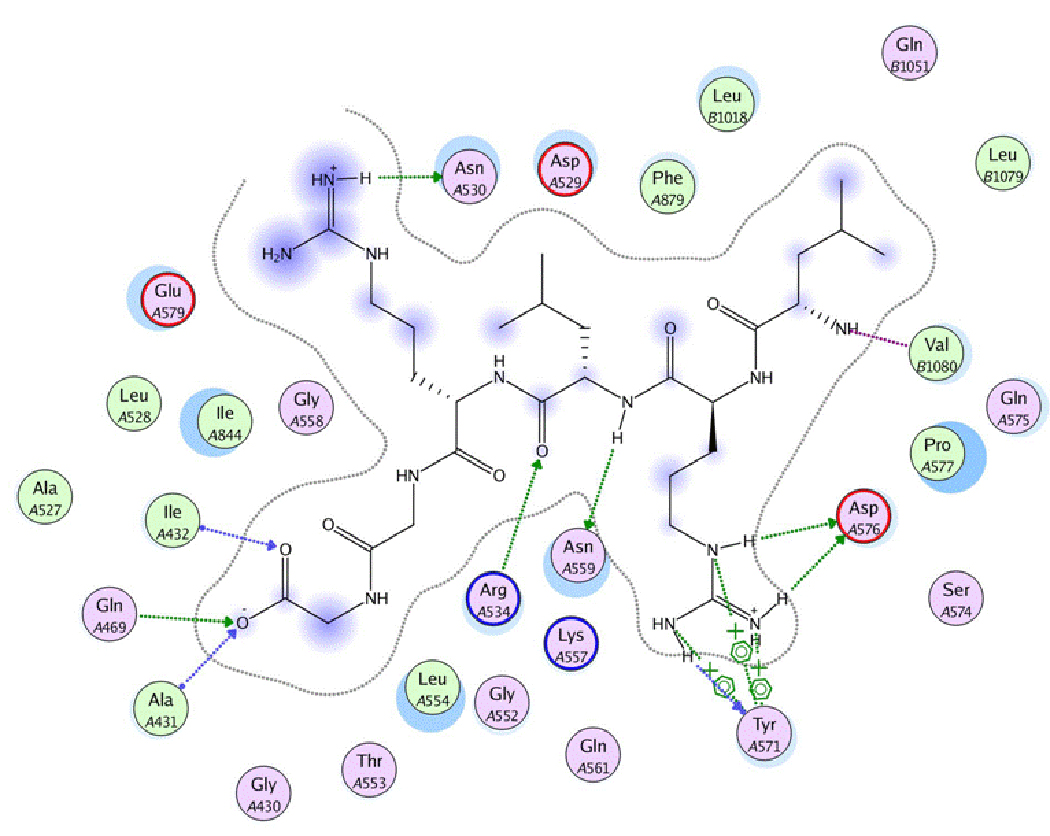
Interactions between ubiquitin c-terminus and E1. Hydrogen bonds are represented as arrows (green: side chain hydrogen bond; blue: backbone hydrogen bond). Blue areas indicate the exposition of the residue to the external solvent. Note the cation-aromatic interaction between Arg72 of ubiquitin and Tyr 571 of E1.
CONCLUSIONS
Until recently, it was believed that there is only one single type of E1 in the cell. However, it was reported recently that there is another E1 subtype [5]. Given the importance of ubiquitination in normal biological function, inhibiting E1 might be expected to significantly derange these functions. Although this might indicate that E1 is not an attractive target for cancer therapy, a paper published recently reported a selective inhibitor of E1 (PYR 41) with antitumor activity [17]. This molecule was reported, amongst other effects, to stabilize p53 and differentially kill transferred cells. The authors suggested that PYR 41 inhibits E1 by covalently interacting with active site cysteine. Other molecules reported include panepophenanthrin and himeic acid [16]. Similarly, the authors suggested that these molecules inhibit the activity of E1 by covalently interacting with active site cysteine, albeit without experimental evidence.
These results suggests that further studies on E1 as anticancer target are required and the model we are reporting here represents a step forward in understanding the structural features of the E1 and the E1-Ub complex, which could be exploited in the design of novel anticancer agents.
Figure 6.
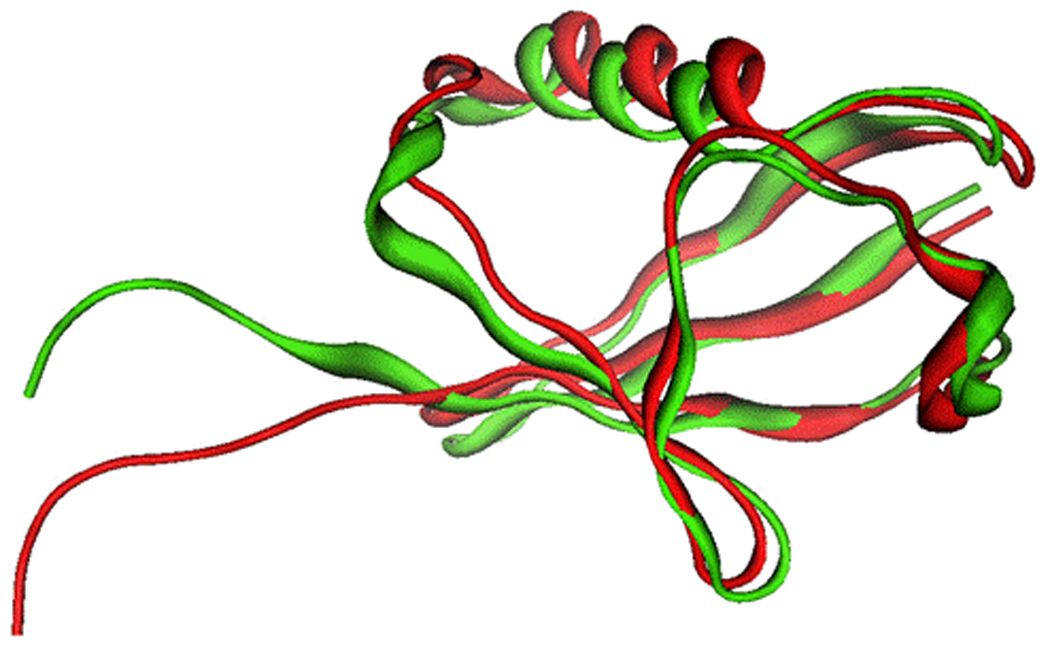
Superposition of free and E1-bound ubiquitin (bound mouse ubiquitin in red; free human ubiquitin in green). The binding of ubiquitin to E1 introduces a significant shift at the ubiquitin C-terminus.
ACKNOWLEDGEMENTS
The authors thank the Algerian Government for funding, in the form of a PhD studentship to G.B. Work in the AMB laboratory is supported by Award Number R01CA127258 from the National Cancer Institute, Developmental Therapeutics Program.
REFERENCES
- 1.Burger AM, Seth AK. The ubiquitin-mediated protein degradation pathway in cancer: therapeutic implications. Eur. J. Cancer. 2004;40:2217–2229. doi: 10.1016/j.ejca.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 2.Lake MW, Wuebbens MM, Rajagopalan KV, Schindelin H. Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB-MoaD complex. Nature. 2001;414:325–329. doi: 10.1038/35104586. [DOI] [PubMed] [Google Scholar]
- 3.Lois LM, Lima CD. Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. EMBO J. 2005;24:439–451. doi: 10.1038/sj.emboj.7600552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang DT, Paydar A, Zhuang M, Waddell MB, Holton JM, Schulman BA. Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8's E1. Mol. Cell. 2005;17:341–350. doi: 10.1016/j.molcel.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 5.Jin J, Li X, Gygi SP, Harper W. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature. 2007;447:1135–1138. doi: 10.1038/nature05902. [DOI] [PubMed] [Google Scholar]
- 6.Reverter D, Lima CD. Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature. 2005;435:687–692. doi: 10.1038/nature03588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108:345–356. doi: 10.1016/s0092-8674(02)00630-x. [DOI] [PubMed] [Google Scholar]
- 8.Walden H, Podgorski MS, Schulman BA. Insights into the ubiquitin transfer cascade from the refined structure of the activating enzyme for NEDD8. Nature. 2003;422:330–334. doi: 10.1038/nature01456. [DOI] [PubMed] [Google Scholar]
- 9.Huang DT, Hunt HW, Zhuang M, Ohi MD, Holton JM, Schulman BA. Basis for a ubiquitin-like protein thioester switch toggling E1-E2 affinity. Nature. 2007;445:394–398. doi: 10.1038/nature05490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang DT, Miller DW, Mathew R, Cassell R, Holton JM, Roussel MF, Schulman BA. A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat. Struct. Mol. Biol. 2004;11:927–935. doi: 10.1038/nsmb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walden H, Podgorski MS, Huang DT, Miller DW, Howard RJ, Minor DL, Holton JM, Schulman BA. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol. Cell. 2003;12:1427–1437. doi: 10.1016/s1097-2765(03)00452-0. [DOI] [PubMed] [Google Scholar]
- 12.White AW, Westwell AD, Brahemi G. Protein-protein interactions as targets for small-molecule therapeutics in cancer. Exp. Rev. Mol. Med. 2008;10:e8, 1–14. doi: 10.1017/S1462399408000641. [DOI] [PubMed] [Google Scholar]
- 13.Vassilev LT. p53 Activation by small molecules: application in oncology. J. Med. Chem. 2005;48:4491–4499. doi: 10.1021/jm058174k. [DOI] [PubMed] [Google Scholar]
- 14.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 15.Sekizawa R, Ikeno S, Nakamura H, Naganawa H, Matsui S, Iinuma H, Takeuchi T. Panepophenanthrin, from a mushroom strain, a novel inhibitor of the ubiquitin-activating enzyme. J. Nat. Prod. 2002;65:1491–1493. doi: 10.1021/np020098q. [DOI] [PubMed] [Google Scholar]
- 16.Tsukamoto S, Hirota H, Imachi M, Fujimuro M, Onuki H, Ohta T, Yokosawa H. Himeic acid A: a new ubiquitin-activating enzyme inhibitor isolated from a marine-derived fungus, Aspergillus sp. Bioorg. Med. Chem. Lett. 2005;15:191–194. doi: 10.1016/j.bmcl.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y, Kitagaki J, Dai R-M, Tsai YC, Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P, Li C-CH, Kenten JH, Beutler JA, Vousden KH, Weissman AM. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007;67:9472–9481. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- 18.Lee I, Schindelin H. Structural insights into E1-catalysed ubiquitin activation and transfer to conjugating enzymes. Cell. 2008;134:268–278. doi: 10.1016/j.cell.2008.05.046. [DOI] [PubMed] [Google Scholar]
- 19.Szczepanowski RH, Filipek R, Bochtler M. Crystal structure of a fragment of mouse ubiquitin activating enzyme. J. Biol. Chem. 2005;280:22006–22011. doi: 10.1074/jbc.M502583200. [DOI] [PubMed] [Google Scholar]
- 20.Molecular Operating Environment (MOE 2008.10) Montreal, Quebec, Canada: Chemical Computing Group, Inc.; http://www.chemcomp.com. [Google Scholar]
- 21.Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comp. Phys. Comm. 1995;91:43–56. [Google Scholar]
- 22.Handley PM, Mueckler M, Siegel NR, Ciechanover A, Schwartz AL. Molecular cloning, sequence, and tissue distribution of the human ubiquitin-activating enzyme E1. Proc. Natl. Acad. Sci. U.S.A. 1991;88:258–262. doi: 10.1073/pnas.88.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. [accessed June 23, 2009];RAMPAGE Server. http://ravenbioccam.ac.uk/rampage.php.
- 24.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vijay-Kumar S, Bugg CE, Cook WJ. Structure of ubiquitin refined at 1.8 Å resolution. J. Mol. Biol. 1987;194:531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]