

Abstract
Existing methods for the prediction of immunologically active T-cell epitopes are based on the amino acid sequence or structure of pathogen proteins. Additional information regarding the locations of epitopes may be acquired by considering the evolution of viruses in hosts with different immune backgrounds. In particular, immune-dependent evolutionary patterns at sites within or near T-cell epitopes can be used to enhance epitope identification. We have developed a mutation–selection model of T-cell epitope evolution that allows the human leukocyte antigen (HLA) genotype of the host to influence the evolutionary process. This is one of the first examples of the incorporation of environmental parameters into a phylogenetic model and has many other potential applications where the selection pressures exerted on an organism can be related directly to environmental factors. We combine this novel evolutionary model with a hidden Markov model to identify contiguous amino acid positions that appear to evolve under immune pressure in the presence of specific host immune alleles and that therefore represent potential epitopes. This phylogenetic hidden Markov model provides a rigorous probabilistic framework that can be combined with sequence or structural information to improve epitope prediction. As a demonstration, we apply the model to a data set of HIV-1 protein-coding sequences and host HLA genotypes.
Keywords: epitopes, immune-mediated evolution, phylogenetic hidden Markov model
Introduction
An effective cellular immune response depends on the presentation and recognition of foreign peptides called epitopes. Antigens from endogenous pathogens, such as a virus, are degraded by the proteosome within the cytoplasm and transported to the endoplasmic reticulum (ER) by the transporter associated with processing. In the lumen of the ER, the peptide fragments undergo N-terminal trimming before attaching to human leukocyte antigen (HLA) class I molecules for delivery to the cell surface and presentation to cytotoxic T lymphocytes (CTLs). The subsequent binding of the epitope/HLA class I complex to the T-cell receptor and CD8 coreceptor on a CTL induces clonal expansion and the release of cytotoxins that lyse the virus-infected cell (Vyas et al. 2008). Mutations of amino acid residues in or around epitopes may inhibit any of these stages in the antigen processing pathway, enabling viral escape from the immune response. This is particularly evident in rapidly evolving viruses, such as the HIV that undergoes error-prone reverse transcription before integration as a provirus in the host's DNA.
The genes encoding HLA molecules are highly polymorphic with distinct alleles having different peptide-binding domains. HLA class I molecules are capable of presenting peptide sequences of 8–11 amino acids with different HLA alleles having distinct peptide-binding preferences. The antigenic peptides that are ultimately presented to CTLs therefore differ between individuals with different sets of HLA alleles. Consequently, viral escape from CTL recognition will depend on the HLA genotype of the host and is thus said to be “HLA restricted.” Individuals expressing a common HLA allele will have the potential to present a common set of epitopes and thus to harbor similar escape variants of the virus. Indeed, several studies have reported robust correlations between polymorphisms within or near HIV epitopes and the HLA genotype of the host (Brumme et al. 2008; Carlson and Brumme 2008; Navis et al. 2008; Peters et al. 2008; Kawashima et al. 2009; Rousseau et al. 2008). These studies highlight the important role of the cellular immune response in driving HIV evolution at sites in and around HLA class I epitopes, resulting in adaptation of the virus to HLA alleles both at the level of individual-infected hosts and at the population level.
Accurate prediction of CTL epitopes is a key goal in immunoinformatics (Yu et al. 2002; Flower 2003; Larsen et al. 2005). Common approaches to this problem include motif-based methods (Schuler et al. 2007), structure-based methods (Rognan et al. 1999; Brusicet al. 2004), and machine-learning algorithms (Nielsen et al. 2003; Zhao et al. 2003; Yanover and Hertz 2005; Heckerman et al. 2006). However, none of the tools proposed thus far consider immune-mediated evolution as a distinguishing feature of epitopes. Here, we develop a mutation–selection model of viral escape from host CTL responses, which is applicable when viral-coding sequence data as well as host HLA genotype data are available. For an alignment of HIV-1 sequences of the same subtype, we assume that there is a single amino acid at each site, which is fittest in the absence of a CTL response targeting the site. We refer to this residue as the wild-type amino acid and approximate it with the most common amino acid observed at a site in the alignment. Versions of this assumption are commonly applied to distinguish immune escape and reversion mutations in HIV-1 (see, e.g., Goonetilleke et al. 2009). In the presence of an immune response targeting a specific site, selective pressure for immune escape can cause a reduction in the frequency of the wild-type amino acid. When a site is known to be within a CTL epitope bound by a specific HLA allele, we find that allowing the frequency of the wild-type amino acid to depend on whether or not the allele is expressed in the host often provides a much better description of the data. When it is not known whether a site is within an epitope for a particular HLA allele, comparison of the fit of a model that allows the wild-type amino acid frequency to depend on the presence of the allele (the epitope model) to the fit of a model that ignores HLA genotype (the nonepitope model) can provide evidence that the site is within or close to an epitope. A contiguous set of sites favoring the epitope model provide stronger evidence for an epitope. We show that this evidence can be combined using a phylogenetic hidden Markov model (phylo-HMM) to make probabilistic epitope predictions based on sequence evolution. Evolutionary information provides a means of epitope prediction, which is orthogonal to existing sequence- and structure-based methods. We demonstrate how this information can be integrated with other prediction methods within the phylo-HMM framework, taking a method based on anchor residue motifs as an example.
Materials and Methods
Data
We analyzed a multiple alignment of 506 HIV-1 subtype C Gag p17 and p24 sequences from a Durban, South Africa cohort, contributed to the Los Alamos HIV Molecular Immunology Database by Rousseau et al. (2008). The sequences and HLA genotypes of the subjects were determined as described in Rousseau et al. (2008). The data utilized in this study correspond to HXB2 Gag positions 1–358, excluding 16 amino acid sites at the end of p17 (positions 112–127) that appeared to be misaligned (most sites contained more than 90% gaps). Two HIV-1 subtype A and subtype B sequences as well as an intersubtype recombinant were removed from the original data set. The alignment was screened for intrasubtype recombination with GARD (Kosakovsky Pond et al. 2006), but we were unable to detect departures from the star phylogeny within estimated recombination breakpoints due to the large number of taxa relative to the number of codon sites between breakpoints. Consequently, we do not account for intrasubtype recombination in our analyses, although different phylogenies across the alignment could be accommodated straightforwardly in principle. HLA-binding motifs and lists of optimally defined epitopes were obtained from the Los Alamos HIV Molecular Immunology Database (http://www.hiv.lanl.gov/content/immunology).
Model of Immune Escape
We take account of immune-mediated selection pressure by developing a codon substitution model with site-specific equilibrium frequencies that reflect the dependence of the wild-type amino acid at a site on the HLA genotype of the host. Our model is based on that of Halpern and Bruno (1998) in which differences in the fitness of codons are modeled in terms of site-specific codon equilibrium frequencies. In this model, the instantaneous rate of substitution from codon I to codon J, where we require that I and J are both sense codons, is defined as follows:
 |
(1) |
where νJ is the equilibrium frequency of codon J at a particular site in the alignment, is the mutation rate from nucleotide i to nucleotide j in the kth codon position, and is proportional to the fixation probability of the mutation (Halpern and Bruno 1998). In our case, we wish to have this fixation probability depend on the HLA genotype of the infected host at a given point on the viral phylogeny whenever the mutation involves the replacement of a wild-type amino acid with a non–wild type residue (escape mutation) or vice versa (reversion mutation). In order to achieve this, we parameterize the site-specific codon equilibrium frequencies in terms of the equilibrium frequency of the wild-type residue, γ, at a site and define
 |
(2) |
where W represents the set of codons that encode the wild-type amino acid and πJ is the equilibrium frequency of codon J estimated empirically from the entire alignment as described in Kosakovsky Pond and Muse (2005). An increase in the equilibrium frequency of the wild-type residue, γ, therefore translates into an increase in the equilibrium frequencies, , of codons that encode the wild-type residue. The factors involving the πJ parameters ensure that the effects of codon usage bias, estimated across the whole alignment, are retained in the model. To allow γ to depend on the HLA genotype of the host, we set with , where p0 (p1) denotes the probability that a specific HLA allele is absent (present) in the host, and γ0 (γ1) represents the equilibrium frequency of the wild-type residue given that the HLA allele is absent (present). We make the simplifying assumption that the presence () or absence () of a specific HLA allele during the entire period represented by a branch on the phylogeny is dictated by the HLA genotype of the host at the tip of that branch. When it is unknown whether or not the HLA allele is expressed in the host at a vertex in the phylogeny, such as at all ancestral nodes, we set p0 and p1 equal to the proportion of HLA-genotyped individuals in the study for which the particular HLA allele is absent and present, respectively. We also constrain because HLA-mediated selection pressure is expected to increase the frequency of escape variants over wild-type amino acids.
We parameterize the mutation rate according to the MG94 × HKY85 Dual GDD 3 × 3 model of Kosakovsky Pond and Muse (2005) and use an F3 × 4 estimator for πJ. Substituting these parameters into equation (1) yields the following:
 |
(3) |
where πjk denotes the equilibrium frequency of target nucleotide j in codon position k, κ is the transition/transversion rate ratio, α is the synonymous substitution rate, β is the nonsynonymous substitution rate, and . Notice that the factors involving γ only appear in the instantaneous rate matrix for substitutions that result in the replacement of a wild-type residue with a non–wild type residue or vice versa. Consequently, the selective influence of the host's HLA genotype is limited to these substitutions only as desired.
Equation (3) defines a time-reversible substitution process for each branch in the phylogeny. However, the equilibrium frequencies, and hence the instantaneous rate matrix describing this model, will differ from branch to branch depending on whether a specified HLA allele is present, absent, or unknown along that branch. Consequently, the substitution process is not time reversible when considered over the entire tree and the location of the root node and the initial codon frequencies must therefore be specified. The problem of identifying the root node is greatly simplified in our case if we assume that the initial codon frequencies are given by equation (2) with an indeterminate HLA genotype. Because the HLA genotypes of donors are always unknown, the subprocess along all interior branches is time reversible and hence any interior node other than the most recent common ancestors of the leaf nodes may be selected to root the tree. Indeed, the root of our HIV-1 subtype C phylogeny did fall within this region of the tree when the two subtype A and subtype B sequences in the original alignment were used as outgroups for tree construction.
Let ψ ′ denote all the parameters except γ and γ0 in the codon substitution process defined by equation (3). Because γ1 is defined in terms of γ and γ0, it is ignored here. The site likelihood at column ℓ in an alignment X may then be computed for any parameter values in and nonnegative real-valued branch lengths along a phylogeny using Felsenstein's pruning algorithm (Felsenstein 1981). We adopt a random effects approach with respect to the parameters γ and γ0 and integrate them out of the site likelihood to obtain
 |
(4) |
where and denote prior distributions on γ0 and γ with hyperparameters ξ0 and ξ, respectively. The subscript E in the likelihood refers to the epitope state and is introduced here to highlight the fact that this site likelihood was computed by allowing the equilibrium frequency of the wild-type amino acid to depend on the HLA genotype of the host, as would be expected in epitope regions of the viral genome. In nonepitope regions, the absence of HLA-mediated selective pressure implies that and site likelihoods may therefore be computed as
| (5) |
This is equivalent to equation (4) with if and otherwise. A comparison of and over a region of the viral genome will reveal localized sets of sites that evolve under immune pressure and are thus indicative of CTL epitopes. We train a hidden Markov model with the state emission probabilities given by equations (4) and (5) and hence are able to infer the epitope and nonepitope states taking account of all the information available across the alignment.
Phylogenetic Hidden Markov Model
A phylo-HMM is defined as the four-tuple consisting of a set of M hidden states with associated phylogenetic models , a state transition probability matrix for and a vector of initial state probabilities (Siepel and Haussler 2005). Here, all hidden states share a common phylogenetic model and differ only in the constraints imposed on the parameters γ and γ0 in the computation of the state emission probabilities. This continuous-time Markov stochastic process is defined by an instantaneous codon substitution rate matrix Q with entries given by equation (3), codon equilibrium frequencies given by equation (2) and a binary tree topology with a corresponding set of branch lengths. The constraints on γ and γ0 determine whether evolution in a particular state is immune mediated or not.
Let denote a path through the phylo-HMM with for in an alignment X with L codon sites. The joint probability of the alignment and the path is
 |
The emission probability is given by equation (4) if ϕℓ corresponds to the epitope state and equation (5) if it corresponds to the nonepitope state. The likelihood of the alignment may be computed with the forward algorithm, whereas the maximum likelihood path may be obtained via Viterbi decoding. The forward–backward algorithm may be used to obtain posterior state probabilities for all ℓ and y (Rabiner 1989).
Two-State Epitope Model
We first consider a model with where E denotes an epitope state and N denotes a nonepitope state. The emission probabilities for the epitope and nonepitope states are given by equations (4) and (5), respectively. To compute these probabilities, we define as a beta distribution on and as a beta distribution with support , where is an empirical estimate of the proportion of individuals who do not express the HLA allele under consideration. The support of ensures that . In order to alleviate the computational burden of numerical integration, we utilize discretized versions of these distributions that allows the double integration to be approximated by summation. For each , we compute the likelihood at seven equally spaced values of γ0 in the interval for the epitope state. A better approximation to the integration may be attained with a finer discretization over the space, particularly near the support boundaries when the beta densities are convex; however, our experience suggests that the incremental gain in predictive accuracy is not worth the increase in computational time needed to calculate the likelihood function for the additional pairs at every site. Given estimates of the hyperparameters ξ and ξ0, the density of each pair is computed and normalized to obtain its joint probability. The probability of the epitope state emitting alignment column Xℓ is then calculated as the sum of the conditional probabilities weighted by the joint probabilities of γ and γ0. The emission probability for the nonepitope state is computed analogously with respect to the discretized marginal distribution on γ. This model is illustrated in figure 1A.
FIG. 1.

(A) Two-state epitope model. (B) Phylo-HMMwD+M for HLA-B*57. Modifications to the phylo-HMMwD are indicated with dashed lines.
Epitope Model with Duration
The two-state model ignores several known sequence and structural features of epitopes. The requirement that an epitope be between 8 and 11 amino acids in length in order to bind to an HLA class I molecule is perhaps the simplest constraint that is not considered in the two-state model. We incorporate this restriction by defining a phylo-HMM with duration (phylo-HMMwD) on the state space , where denotes the ith amino acid in the epitope state for . The architecture of this model is illustrated with solid nodes and edges in figure 1B. The phylo-HMMwD provides the foundation for incorporating other spatial features relevant to epitope prediction. In what follows, we show how to integrate motif-based prediction with sequence evolution within this framework.
Phylo-HMMwD Integrated with Motif-Based Prediction
Motif-based methods of epitope prediction exploit the fact that the binding affinity of an antigenic peptide and HLA molecule is critically dependent on a few anchor residues in the peptide sequence. For example, ligands of HLA-B*57 molecules typically have the form X[ATS]XXXXXX[FW], where the amino acids in square brackets indicate dominant anchor residues and X denotes an indeterminate amino acid (Marsh et al. 2000). The region between the two anchors is of variable length and affects the conformational structure of the bound peptide-HLA complex (Stewart-Jones et al. 2005). We demonstrate how to extend our phylo-HMMwD to incorporate motif-based epitope prediction using HLA-B*57 as an example. We include the anchor motif in the model by replacing states and with anchor states and in figure 1B. Because anchor motifs are HLA specific, the architecture of the graphical model describing the sequence of hidden states will differ between HLA alleles. Epitope prediction may also be informed by processing escape mutations that have been reported in the N-terminal flanking regions of some epitopes (Draenert et al. 2004). We therefore introduce three optional flanking states , , and prior to entry into the first epitope state in figure 1B. We will refer to this model as a phylo-HMMwD+M, where the suffix “+M” indicates the combining of a phylo-HMMwD with motif-based epitope prediction.
To take account of the anchor motifs, we define an indicator variable , or for short, which equals one if the proportion r of observed amino acids corresponding to anchor residue i at site ℓ is greater than or equal to a predefined threshold λ and zero otherwise. In the case of HLA-B*57, for example, indicates whether the proportion of sequences with Ala, Thr, or Ser at site ℓ exceeds λ. The indicator variable is similarly defined with respect to Phe and Trp. The probability of emitting column Xℓ under the ith anchor state is then
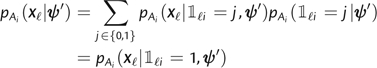 |
because and . Setting , we obtain
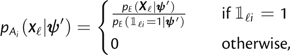 |
because when and thus . The probability is given by equation (4) and we approximate with the proportion of alignment sites at which . Note that if , at all sites and the anchor residues will not inform epitope prediction. Larger values of λ will result in anchor state emission probabilities of zero at some sites and thereby restrict epitope predictions to those regions where the anchor motifs are observed in at least some of the aligned sequences. In our analyses, we set λ = 0.3 and will therefore not detect epitopes when the anchor residues are only present in less than 30% of sequences. It is worth noting that at almost 90% of the sites in our alignment, the most common amino acid occurred at a frequency greater than 80% and our results are consequently robust to the choice of λ.
Implementation
Given an inferred topology (see below) and assuming F3 × 4 codon equilibrium frequencies, all other mutational parameters in equation (3) and the branch lengths were estimated by maximum likelihood in HyPhy (Kosakovsky Pond et al. 2005) under a standard MG94 × HKY85 Dual GDD 3 × 3 model of codon substitution (i.e., without the factor involving ζ in equation (3)). Site likelihoods were then computed under the Halpern–Bruno model defined by equation (3) for discrete values of γ and γ0 given the previously inferred mutational parameters and branch lengths. The hyperparameters of the prior distributions on γ and γ0 and the transition probabilities between hidden states were optimized with the Baum–Welch algorithm implemented in the R Language and Environment for Statistical Computing (R Development Core Team 2009). The initial state probabilities were constrained such that the Viterbi path always begins in the nonepitope state. The computer code is freely available from the corresponding author.
In order to determine the extent to which our results depend on the inferred phylogeny, we estimated several maximum likelihood topologies using PhyML (Guindon and Gascuel 2003) and GARLI (Zwickl 2006). Fifteen trees were estimated with different starting topologies in PhyML by specifying a general time reversible model of nucleotide substitution with variable substitution rates modeled by a four-category discrete gamma distribution with unit mean. Branch swaps were achieved by subtree pruning and regrafting and nearest neighbor interchange. A further 20 trees were inferred with GARLI under a GY94 × HKY85 codon substitution model with nonsynonymous rates drawn from a three-category general discrete distribution. We trained our phylo-HMMs using several of the highest scoring topologies and did not find any substantive differences in our results. The phylogeny ultimately used here was resolved in PhyML and had the highest likelihood score computed in HyPhy under the MG94 × HKY85 Dual GDD 3 × 3 model of codon substitution.
Results
We applied our models to a multiple alignment of 506 HIV-1 subtype C Gag p17 and p24 sequences described in Materials and Methods. All amino acid positions discussed below correspond to those of the HXB2 reference strain.
Predicted HLA-B*57-restricted epitopes
We begin by considering the specific case of HIV-1 evolution in the context of the HLA-B*57 allele for which immune-mediated escape and compensatory mutations have been extensively documented. Twenty-six of the 502 HLA-typed individuals in this study were HLA-B*57 positive (HLA-B*57+). The majority (15/26) of these individuals expressed the HLA-B*5703 molecule as expected in African populations (Cao et al. 2004), whereas 7 expressed HLA-B*5702, and the remainder were typed at two-digit resolution only. Molecular differences between HLA-B*57 alleles do not, however, significantly affect peptide-binding affinity and we therefore expect a considerable overlap in their bound peptide repertoires (Stewart-Jones et al. 2005), although differential T-cell receptor recruitment may result in subtype-specific escape (Yu et al. 2007).
A comparison of the integrated site likelihoods of the HLA-B*57-restricted epitope model and nonepitope model is presented in figure 2. For each site in Gag p17 and p24, we computed the Bayes factor
assuming flat priors on γ and γ0 given γ and plotted , which is on the same scale as the likelihood ratio test statistic (Kass and Raftery 1995). The results of the two-state phylo-HMM and phylo-HMMwD+M are illustrated in figure 3 for a region of the p24 protein known to contain immunodominant HLA-B*57-restricted epitopes. The results for the entire Gag p17 and p24 region are given in Supplementary figures S1 and S2, Supplementary Material online.
FIG. 2.

Bayes factors comparing the HLA-B*57-restricted epitope model to the nonepitope model at each site in HIV-1 Gag p17 and p24. Shaded regions indicate the degree of evidence in favor of the epitope model: white indicates negligible evidence, light gray indicates positive evidence, intermediate gray indicates strong evidence, and dark gray indicates definitive evidence (Kass and Raftery 1995). Prominent escape mutations known to be associated with the HLA-B*57 allele are annotated. Black dots indicate sites with a significant (q < 0.2) association between host HLA and HIV mutations as determined by the phylogenetically corrected method described in Bhattacharya et al. (2007).
FIG. 3.

Posterior probabilities of the epitope state for HLA-B*57 in HIV-1 Gag p24 based on the two-state epitope model. Gray-shaded regions indicate epitopes predicted by Viterbi decoding of the phylo-HMMwD+M with the posterior probability of each epitope given on the top right-hand corner. Predicted anchors are shaded in dark grey and flanking regions are shaded in light gray. Black dots indicate epitopes predicted by Viterbi decoding of the two-state model. The best defined or “A-list” epitopes described in Yusim et al. (2009) are indicated with white text against a black background.
Strong HLA-B*57-mediated selection pressure has been described in and around three peptides in the Gag p24 capsid region (Chopera et al. 2008; Crawford et al. 2009), namely ISW9 (147ISPRTLNAW155), TW10 (240TSTLQEQIAW249), and KF11 (162KAFSPEVIPMF172) (anchor residues are underscored). Escape from ISW9 occurs first during acute HIV-1 subtype C infection in HLA-B*57+ African populations (Crawford et al. 2009). The processing escape mutation A146P is often observed in the N-terminal flanking position of this epitope and prevents ER aminopeptidase I from cleaving an N-extended peptide to an optimal size for binding to the HLA-B*57 molecule (Draenert et al. 2004). A further escape substitution of Ile for Leu at site 147 has also been described (Draenert et al. 2004; Peters et al. 2008). Figure 2 shows strong evidence in support of the epitope model at position 146 and positive evidence at position 147. The two-state phylo-HMM captures both escape mutations with sharp peaks in the posterior probability of the epitope state at sites 146 and 147 in figure 3. Viterbi decoding of the phylo-HMMwD+M correctly predicts the ISW9 epitope.
Two escape sites at positions 242 and 247 within the TW10 epitope are identified by large Bayes factors in figure 2 and high posterior probabilities in figure 3. The former position corresponds to the well-known T242N mutation observed in almost all HLA-B*57+ individuals (Brumme et al. 2008), whereas a mutation from Ile to Val at the latter position appears to be specific to HLA-B*57+ individuals infected with HIV-1 subtype C (Crawford et al. 2009). The T242N mutation disrupts critical interactions with the host protein cyclophilin A (CypA) resulting in a 10-fold reduction in viral replicative capacity (Borghans et al. 2007). This fitness loss may be partially restored by compensatory substitutions, particularly in or near the CypA-binding loop upstream of the TW10 epitope (Brockman et al. 2007). Indeed, figure 3 indicates some, albeit weaker, evidence in favor of the epitope state at sites corresponding to the previously described H219Q and M228I/L compensatory mutations within this region.
The KF11 epitope is immunodominant during chronic infection (Leslie et al. 2005). Escape mutations have been described at positions 163 and 165 within this epitope, although their prevalence depends on the HIV-1 subtype and the specific HLA-B*57 allele, with more frequent escape observed in clade C sequences in the context of HLA-B*5703 (Yu et al. 2007). It is therefore not surprising that our two-state phylo-HMM predicts both these mutations. Indeed, the Bayes factors too favor the epitope model at these positions. A mutation from Ala to Gly at position 163 occurs first in HLA-B*57+ individuals and inhibits intracellular processing of the epitope precursor but compromises viral fitness substantially (Leslie et al. 2005). This fitness cost is partially restored by a compensatory substitution of Ser for Asn at position 165, which may explain the lower Bayes factor and posterior escape probability at this site relative to position 163.
Finally, a further B*57-restricted epitope QW9 (308QATQDVKNW316) has also been mapped to the p24 capsid protein (Stewart-Jones et al. 2005). Two mutations within this epitope, namely S310T and E312D, have been described (Navis et al. 2007). However, these mutations represent the most common variant of QW9 in HIV-1 subtype C and do not appear to be the result of CTL escape (Currier et al. 2005). Correspondingly, we too do not find any evidence of escape at these or any other sites within the QW9 epitope.
Epitope Predictions for Other HLA Alleles
We used our phylo-HMMs to predict epitopes for a further 31 HLA alleles that were observed at relatively high frequencies and for which anchor motifs were available in the Los Alamos HIV Molecular Immunology Database. For each HLA allele, the phylo-HMMwD+M was defined to accommodate epitopes of 8 to 11 amino acids in length with an N-terminal flanking region of 3 amino acids. Full plots of the epitopes predicted in HIV-1 Gag p17 and p24 are provided as Supplementary figures S1 and S2, Supplementary Material online, for all the HLA molecules considered. The anchor motifs used for each allele are given in supplementary table S1, Supplementary Material online.
Overall, Viterbi decoding revealed 81 and 65 epitopes across all HLA alleles in the phylo-HMMwD and phylo-HMMwD+M, respectively, compared with 191 nonoverlapping epitopes identified by simply counting the number of segments with appropriately spaced anchor residues present in at least 30% of sequences (see supplementary table S2, Supplementary Material online, for details). The results clearly indicate that the number of potential epitopes based solely on anchor motifs is much larger than that predicted by either phylo-HMM considering evolutionary information. Indeed, the phylo-HMMs limit the set of predictions to only those epitopes, which exhibit HLA allele-dependent immune escape. We also examined the proportion of predicted epitopes, which overlap with known epitopes documented in Yusim et al. (2009) for each of the three methods but did not find any notable differences (anchor motifs only, 0.33; phylo-HMMwD, 0.26; and phylo-HMMwD+M, 0.37). However, predicted epitopes that are unknown are not necessarily false positives, and, in fact, these are more likely to be valid epitopes under the phylo-HMMwD+M where predictions satisfy anchor motifs and exhibit immune-mediated escape patterns. Moreover, we envisage that our model be combined with additional sequence and structural information so as to further reduce the number of false positives that may occur.
Many well-known epitopes do not conform to the documented HLA-binding motifs and will be missed by the phylo-HMMwD+M. For example, none of the known HLA-B*5802-restricted epitopes in HIV-1 Gag p17 and p24 documented in Yusim et al. (2009) contain the anchor motif X[ST]XXX[R]XX[F] suitable for binding to an HLA-B*5802 molecule (Marsh et al. 2000; Schuler et al. 2007). The two-state epitope model and phylo-HMMwD are likely to be more useful in such cases where there may be uncertainty regarding the binding motif. For example, the posterior probabilities of the two-state model for HLA-C*04 clearly indicate strong immune pressure within the known HLA-C*04-restricted epitope QW9 (308QATQDVKNW316), even though this epitope is undetected by the phylo-HMMwD+M, which specifies a Tyr, Phe, or Pro B-pocket anchor residue and a Leu, Phe, or Met F-pocket anchor residue.
Discussion
Pathogens, such as HIV-1, adapt to common immune responses in host populations (Kawashima et al. 2009) and to the immune repertoire of specific infected hosts (Bhattacharya et al. 2007). This phenomenon has attracted substantial interest in the HIV-1 field, partly because it provides insights into the future trajectory of the pandemic but also because the interplay between the immune response and the evolving virus is an essential component of HIV-1 disease dynamics. In this work, we have developed the first mutation–selection model of CTL escape, incorporating host HLA information explicitly into the evolutionary model. We can then make use of model comparison techniques to identify sites within the virus that are correlated with host HLA alleles. Amino acid positions that support the model of epitope evolution are consistent with viral polymorphisms that have been reported as associated with the host HLA genotype. Because CTLs recognize linear epitopes, we can incorporate this model into a phylo-HMM to predict epitopes on the basis of patterns of evolution at contiguous amino acid positions. Indeed, the possibility of combining evidence from evolutionary models over contiguous sites makes the problem of predicting linear epitopes a natural application of phylo-HMMs.
Current methods for predicting T-cell epitopes are based on the characteristics of individual amino acid sequences that affect peptide processing in the endogenous pathway or binding by HLA molecules. As described above, epitopes are also associated with characteristic patterns of evolution and we demonstrate here that the phylo-HMM framework allows us to integrate this evolutionary evidence into an existing sequence-based strategy to enhance epitope prediction. In doing so, we shift the focus from the prediction of epitopes in individual sequences to the prediction of epitopes that tend to occur in a set of viruses circulating in a host population for which HLA data are available. Our approach is unlikely to identify all the potential epitopes in a given sequence; rather, it is designed to identify epitopes that are associated with consistent patterns of immune escape and reversion. This is the case for many well-studied and important epitopes in HIV-1. Epitopes that exhibit consistent patterns of immune escape and reversion are significant because they frequently correspond to immune responses for which there are a limited number of possible escape pathways and for which escape mutations incur a fitness cost, such that they tend to revert upon transmission to a host lacking the required HLA allele.
The evolutionary models are fitted separately for each HLA allele, followed by decoding of the phylo-HMM to predict the corresponding epitopes. However, HLA alleles do not occur independently of one another. Instead, it is possible for different HLA alleles to cooccur more often than would be expected by chance, either as a result of linkage disequilibrium or population structure. To identify HLA alleles that cooccur, we carried out Bonferroni-corrected Fisher's exact tests (supplementary table S3, Supplementary Material online) and subsequently examined the epitope predictions of the associated alleles to see if we could identify false positives resulting from linkage. In several cases, common peaks in the posterior probabilities of the epitope state based on the two-state epitope model are observed for HLA alleles, which exhibit significant linkage disequilibrium (see, e.g., the p17 region in Supplementary figure S1, Supplementary Material online, for HLA-B*4201 and HLA-C*1701). Undocumented epitopes predicted by the phylo-HMMwD+M and presented by such HLA molecules were screened and highlighted as potentially spurious where appropriate. However, in most cases, the specification of HLA specific–binding motifs in the phylo-HMMwD+M eliminates these potential false positives.
The characteristic pattern of HLA-associated evolution provides a basis for epitope prediction, which is orthogonal to the characteristics of individual sequences. As a result, it is possible to combine this with any existing sequence- or structure-based epitope prediction strategy. We demonstrated this using the example of motif-based epitope prediction. Because the sequence motifs associated with HLA binding have a low-information content, an epitope prediction method that considers motifs exclusively has a very high false positive rate. Combining the phylo-HMM with motif-based prediction, we were able to substantially reduce the number of predicted epitopes. We argue that the resulting predictions are the most relevant because they are associated with a shared pattern of immune escape and reversion across different infections. The combination of the phylo-HMM with motif-based prediction also provided a fully probabilistic approach to epitope prediction and is easily interpretable. For example, we can calculate the posterior probability of an epitope at each amino acid site using the forward–backward algorithm, which is accurate given the explicit assumptions of the model. Consequently, we present the phylo-HMM as a general epitope prediction framework, which may be readily adapted to combine evolutionary evidence of an epitope with any other epitope prediction method.
Supplementary Material
Supplementary figures S1 and S2 and tables S1, S2, and S3 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Supplementary Material
Acknowledgments
The authors wish to thank Sergei L. Kosakovsky Pond for use of his Linux cluster and assistance with HyPhy.
References
- Bhattacharya T, Daniels M, Heckerman D, et al. (20 co-authors) Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science. 2007;315:1583–1586. doi: 10.1126/science.1131528. [DOI] [PubMed] [Google Scholar]
- Borghans JAM, Mølgaard A, Boer RJD, Keşmir C. HLA alleles associated with slow progression to AIDS truly prefer to present HIV-1 p24. PLoS One. 2007 doi: 10.1371/journal.pone.0000920. 2:e920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman MA, Schneidewind A, Lahaie M, et al. (14 co-authors) Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin. A. J Virol. 2007;81:12608–12618. doi: 10.1128/JVI.01369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumme ZL, Brumme CJ, Carlson J, et al. (21 co-authors) Marked epitope- and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J Virol. 2008;82:9216–9227. doi: 10.1128/JVI.01041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusic V, Bajic VB, Petrovsky N. Computational methods for prediction of T-cell epitopes—a framework for modelling, testing, and applications. Methods. 2004;34:436–443. doi: 10.1016/j.ymeth.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Cao K, Moormann AM, Lyke KE, et al. (18 co-authors) Differentiation between African populations is evidenced by the diversity of alleles and haplotypes of HLA class I loci. Tissue Antigens. 2004;63:293–325. doi: 10.1111/j.0001-2815.2004.00192.x. [DOI] [PubMed] [Google Scholar]
- Carlson JM, Brumme ZL. HIV evolution in response to HLA-restricted CTL selection pressures: a population-based perspective. Microbes Infect. 2008;10:455–461. doi: 10.1016/j.micinf.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Chopera DR, Woodman Z, Mlisana K, et al. (12 co-authors) Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog. 2008 doi: 10.1371/journal.ppat.1000033. 4:e1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford H, Lumm W, Leslie A, et al. (18 co-authors) Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J Exp Med. 2009;206:909–921. doi: 10.1084/jem.20081984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currier JR, Harris ME, Cox JH, McCutchan FE, Birx DL, Maayan S, Ferrari G. Immunodominance and cross-reactivity of B5703-restricted CD8 T lymphocytes from HIV type 1 subtype C-infected Ethiopians. AIDS Res Hum Retroviruses. 2005;21:239–245. doi: 10.1089/aid.2005.21.239. [DOI] [PubMed] [Google Scholar]
- Draenert R, Le Gall S, Pfafferott KJ, et al. (12 co-authors) Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J Exp Med. 2004;199:905–915. doi: 10.1084/jem.20031982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Flower DR. Towards in silico prediction of immunogenic epitopes. Trends Immunol. 2003;24:667–674. doi: 10.1016/j.it.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Goonetilleke N, Liu MK, Salazar-Gonzalez JF, et al. (24 co-authors) The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206:1253–1272. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Halpern AL, Bruno WJ. Evolutionary distances for protein-coding sequences: modeling site-specific residue frequencies. Mol Biol Evol. 1998;15:910–917. doi: 10.1093/oxfordjournals.molbev.a025995. [DOI] [PubMed] [Google Scholar]
- Heckerman D, Kadie C, Listgarten J. Leveraging information across HLA alleles/supertypes improves epitope prediction. J Comput Biol. 2006;14:736–746. doi: 10.1089/cmb.2007.R013. [DOI] [PubMed] [Google Scholar]
- Kass RE, Raftery AE. Bayes factors. J Am Stat Assoc. 1995;90:773–795. [Google Scholar]
- Kawashima Y, Pfafferott K, Frater J, et al. (43 co-authors) Adaptation of HIV-1 to human leukocyte antigen class I. Nature. 2009;458:641–645. doi: 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Muse SV. Site-to-site variation of synonymous substitution rates. Mol Biol Evol. 2005;22:2375–2385. doi: 10.1093/molbev/msi232. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SDW. GARD: a genetic algorithm for recombination detection. Bioinformatics. 2006;22:3096–3098. doi: 10.1093/bioinformatics/btl474. [DOI] [PubMed] [Google Scholar]
- Larsen MV, Lundegaard C, Lamberth K, Buus S, Brunak S, Lund O, Nielsen M. An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol. 2005;35:2295–2303. doi: 10.1002/eji.200425811. [DOI] [PubMed] [Google Scholar]
- Leslie A, Kavanagh D, Honeyborne I, et al. (27 co-authors) Transmission and accumulation of CTL escape variants drive negative associations between HIV polymorphisms and HLA. J Exp Med. 2005;201:891–902. doi: 10.1084/jem.20041455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh SGE, Parham P, Barber LD. San Diego (CA): Academic Press; 2000. The HLA factsbook. [Google Scholar]
- Navis M, Matas DE, Rachinger A, Koning FA, van Swieten P, Kootstra NA, Schuitemaker H. Molecular evolution of human immunodeficiency virus type 1 upon transmission between human leukocyte antigen disparate donor-recipient pairs. PLoS One. 2008 doi: 10.1371/journal.pone.0002422. 3:e2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navis M, Schellens I, van Baarle D, Borghans J, van Swieten P, Miedema F, Kootstra N, Schuitemaker H. Viral replication capacity as a correlate of HLA-B57/B5801-associated nonprogressive HIV-1 infection. J Immunol. 2007;179:3133–3143. doi: 10.4049/jimmunol.179.5.3133. [DOI] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters HO, Mendoza MG, Capina RE, et al. (13 co-authors) An integrative bioinformatic approach for studying escape mutations in human immunodeficiency virus type 1 Gag in the Pumwani Sex Worker Cohort. J Virol. 2008;82:1980–1992. doi: 10.1128/JVI.02742-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. Vienna (Austria): R Foundation for Statistical Computing; 2009. R: a language and environment for statistical computing. [Google Scholar]
- Rabiner L. A tutorial on hidden Markov models and selected applications in speech recognition. Proc IEEE. 1989;77:257–286. [Google Scholar]
- Rognan D, Lauemoller SL, Holm A, Buus S, Tschinke V. Predicting binding affinities of protein ligands from three-dimensional models: application to peptide binding to class I major histocompatibility proteins. J Med Chem. 1999;42:4650–4658. doi: 10.1021/jm9910775. [DOI] [PubMed] [Google Scholar]
- Rousseau CM, Daniels MG, Carlson JM, et al. (23 co-authors) HLA class I-driven evolution of human immunodeficiency virus type 1 subtype C proteome: immune escape and viral load. J Virol. 2008;82:6434–6446. doi: 10.1128/JVI.02455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler MM, Nastke M, Stevanovikć S. SYFPEITHI: database for searching and T-cell epitope prediction. Methods Mol Biol. 2007;409:75–93. doi: 10.1007/978-1-60327-118-9_5. [DOI] [PubMed] [Google Scholar]
- Siepel A, Haussler D. Phylogenetic hidden Markov models. In: Nielsen R, editor. Statistical methods in molecular evolution. New York: Springer; 2005. pp. 325–351. [Google Scholar]
- Stewart-Jones GBE, Gillespie G, Overton IM, Kaul R, Roche P, McMichael AJ, Rowland-Jones S, Jones EY. Structures of three HIV-1 HLA-B*5703-peptide complexes and identification of related HLAs potentially associated with long-term nonprogression. J Immunol. 2005;175:2459–2468. doi: 10.4049/jimmunol.175.4.2459. [DOI] [PubMed] [Google Scholar]
- Vyas JM, van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol. 2008;8:607–618. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanover C, Hertz T. Predicting protein-peptide binding affinity by learning peptide-peptide distance functions. In: Miyano S, Mesirov J, Kasif S, Istrail S, Pevzner P, Waterman M, editors. Research in Computational Molecular Biology, 9th Annual International Conference, RECOMB 2005. Cambridge, MA: Springer; 2005. pp. 456–471. [Google Scholar]
- Yu K, Petrovsky N, Schonbach C, Koh JLY, Brusic V. Methods for prediction of peptide binding to MHC molecules: a comparative study. Mol Med. 2002;8:137–148. [PMC free article] [PubMed] [Google Scholar]
- Yu XG, Lichterfeld M, Chetty S, et al. (26 co-authors) Mutually exclusive T-cell receptor induction and differential susceptibility to human immunodeficiency virus type 1 mutational escape associated with a two-amino-acid difference between HLA class I subtypes. J Virol. 2007;81:1619–1631. doi: 10.1128/JVI.01580-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusim K, Korber BTM, Brander C, Haynes BF, Koup R, Moore JP, Walker BD, Watkins DI. Los Alamos National Laboratory, Theoretical Biology and Biophysics. Los Alamos, NM: HIV Molecular Immunology 2009; 2009. [Google Scholar]
- Zhao Y, Pinilla C, Valmori D, Martin R, Simon R. Application of support vector machines for T-cell epitopes prediction. Bioinformatics. 2003;19:1978–1984. doi: 10.1093/bioinformatics/btg255. [DOI] [PubMed] [Google Scholar]
- Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion [PhD dissertation] University of Texas at Austin; 2006. [Austin (TX)] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.