
Figure 9. Proposed mechanism for impaired oxidation of the fully reduced N139L oxidase by oxygen.
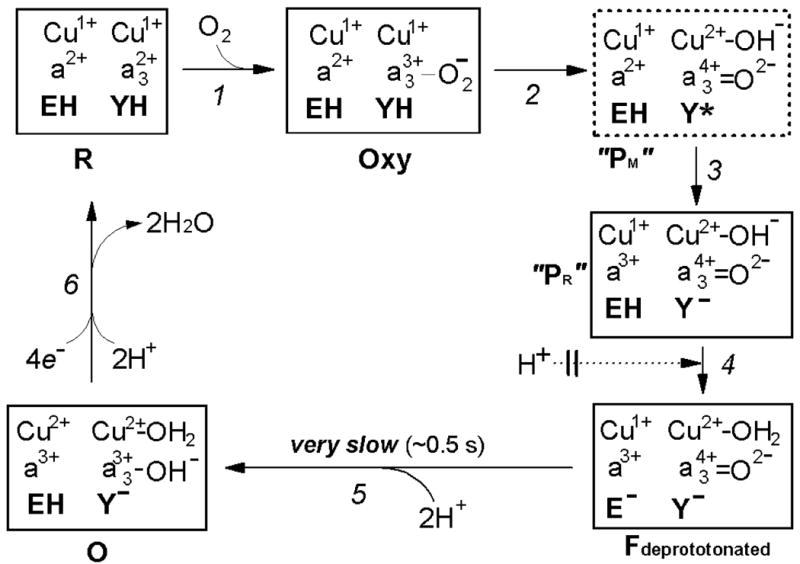
EH and YH denote the protonated glutamic acid E286 and tyrosine Y288, respectively. The transition of the oxycomplex of the fully reduced oxidase (Fe2+-O2 CuB1+) to the initial ferryl-oxene state (Fe4+=O2- Cub2+-OH-) during the reductive splitting of the O-O bond requires extraction of at least one proton from the protein by oxygen. The proton is most likely donated initially by Y288-OH, oxidized to the neutral radical Y288-O• at the O-O-bond breaking step (80, 89-91). In concordance with recent views (84, 85), we assume that the O-O-bond splitting mechanism may be essentially the same for oxidation of the fully reduced and the mixed-valence enzyme, and in either case it results initially in formation of a peroxidase Compound I-type intermediate of the binuclear site (Fe4+=O2- state of heme a3 + amino acid radical, Y288-O•). In this intermediate of cytochrome oxidase, the O-O bond is already broken (92), but it is still labeled here as “Peroxy” state (“PM”) solely to avoid discrepancy with the outdated traditional nomenclature in the literature (see (17, 22) for more consistent possible nomenclature of the oxidase compounds). Insofar, this intermediate has not been directly observed during the oxidation of the fully reduced COX and therefore is shown in a dashed line box. When the reaction starts from the fully reduced state of the oxidase with both CuA and heme a initially reduced, the neutral Y288• radical in the putative “PM” intermediate is rapidly reduced by heme a to tyrosine anion, Y, forming a peroxidase Compound II-type intermediate denoted as “PR” in cytochrome oxidase literature. This designation, although widely used, is misleading because the oxygen-reducing site in “PR” contains 1 reducing equivalent more than “PM” and, actually, its redox state corresponds to the next intermediate F580 analogous to Compound II of peroxidases. For the reasons not yet understood, the “PR” intermediate retains a strong absorption band of heme a3 at 607 nm, typical of the Compound I-type intermediate of cytochrome oxidase (“PM”), and hence may be denoted as F607. In the wild type enzyme, the F607 (“PR”) intermediate converts to the “normal” F580 state with a maximum at ∼580 nm upon H+ uptake by the enzyme. The proton converting F607 (“PR”) to F580 is believed to be provided by E286-COOH that is then rapidly reprotonated via the D-channel. Where exactly the proton goes, has not been established yet with certainity. Originally, it was believed that the proton goes to the Y288 anion. However, according to the recent data (26), Y288 is likely to remain deprotonated in F580 at pH >8. Alternatively, the H+ may be accepted by CuB-bound hydroxide (cf. ref. (85)) as depicted provisionally in Figure 9, or by some other group (e.g. water) in the surroundings of the oxygen reducing site. In the N139L or D132(N,A) mutants, in which the entrance to D-channel is blocked, the “chemical” proton can still be extracted from E286-COOH, allowing for transition to the F580 state, but subsequent rapid reprotonation of E286 is no longer possible. This conclusion is corroborated by the fact that there is no proton uptake coupled to the “PR”→F580 transition in the D132A mutant oxidase of R. sphaeroides (70); moreover, deprotonated state of E286 (E278Pd) has been directly demonstrated for D124N mutant oxidase from P. denitrificans by ATR-FTIR spectroscopy (62). Accordingly, cytochrome oxidase gets stuck at a specific intermediate denoted as Fdeprotonated (Figures 8, 9) that is 1 proton deficient relative to the normal F state. At this unusual state, the proton deficiency is shown to reside on E286 anion, but actually it may be shared among E286 –COOH/E286-COO-and [Y288-OH/Y288-O- ---CuB-H2O/OH-] proton acceptors. Subsequent reduction of Fdeprotonated to the oxidized state by the fourth electron stored within the equilibrating heme a/CuA pair is rate limited (k ∼ 2 s-1) by delivery of at least one, but probably two protons required to protonate the reduced oxygen at heme a3 and reprotonate E286. Only one of the 2 protons is delivered electrogenically from the N-phase (see the text).