

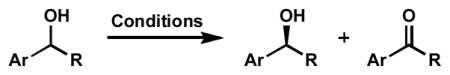
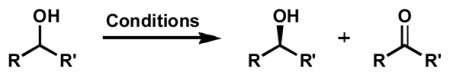
Abstract
The first palladium-catalyzed enantioselective oxidation of secondary alcohols has been developed, utilizing the readily available diamine (−)-sparteine as chiral ligand and molecular oxygen as the stoichiometric oxidant. Mechanistic insights regarding the role of base and hydrogen bond donors have resulted in several improvements to the original system. Namely, addition of cesium carbonate and tert-butyl alcohol greatly enhances reaction rates, promoting rapid resolutions. The use of chloroform as solvent allows the use of ambient air as the terminal oxidant at 23 °C, resulting in enhanced catalyst selectivity. These improved reaction conditions have permitted the successful kinetic resolution of benzylic, allylic, and cyclopropyl secondary alcohols to high enantiomeric excess with good to excellent selectivity factors. This catalyst system has also been applied to the desymmetrization of meso-diols, providing high yields of enantioenriched hydroxyketones.
Keywords: alcohols, asymmetric catalysis, oxidation, palladium, synthetic methods
Introduction
The oxidation of alcohols to carbonyl compounds is one of the most fundamental reactions in organic chemistry.[1] Many different systems have been developed using a wide variety of oxidants.[2] Until recently, however, catalytic enantioselective variants have been largely unexplored.[3,4] The limited number of these alcohol oxidations is somewhat understandable, since this process is inherently complexity-minimizing.[5] While many enantioselective oxidative transformations involve the selective creation of stereogenicity from prochiral starting materials by transfer of a heteroatom to the organic substrate (e.g., epoxidation, dihydroxylation, sulfide oxidation),[6] enantioselective alcohol oxidation requires the selective destruction of a stereocenter in a stereoablative kinetic resolution process.[7,8] Kinetic resolutions have the ability to provide compounds of high enantiomeric excess for even modestly selective processes at higher levels of conversion. Furthermore, the ready availability of a wide range of racemic alcohols, the prevalence of chiral alcohols in organic synthesis, and the potential for product recycling by a simple reduction could make an oxidative kinetic resolution of alcohols a synthetically useful process for the production of enantioenriched materials.
As part of a general program directed toward enantioselective oxidation, we chose to pursue palladium(II) as a catalytic metal for this process. Not only is palladium catalysis prevalent in a variety of enantioselective transformations,[9] but a number of systems involving palladium have been applied to the aerobic oxidation of alcohols.[10,11] Particularly intriguing was a report by Uemura of racemic alcohol oxidation utilizing a palladium(II) catalyst and pyridine in toluene (Scheme 1).[12] Employing molecular oxygen as the sole stoichiometric oxidant, high yields of aldehydes and ketones were obtained for a variety of alcohols. These conditions were attractive for a number of reasons. Molecular oxygen, essential for cellular respiration in all aerobic organisms, is an inexpensive, abundant, and environmentally benign oxidant. The lack of additional co-oxidants simplified reaction complexity, producing water as the sole byproduct. Also, pyridine was found to be critical to the reaction both as ligand and base. Uemura reported no catalytic activity in the absence of pyridine, indicating a strong ligand acceleration effect. We anticipated that the use of chiral ligands in the place of pyridine could lead to significant enantiodiscrimination in the oxidation, while the ligand acceleration could minimize racemic background oxidation by other Pd(II) species that could be present in the reaction. Finally, the non-coordinating nature of toluene could limit solvent displacement of a chiral ligand from the palladium center.
Scheme 1.

Uemura’s oxidation of alcohols.
The proposed mechanism for the oxidation involves alcohol substitution and deprotonation of a palladium(II) complex to generate intermediate palladium alkoxide 1 (Scheme 2). Subsequent β-hydride elimination from this complex forms the product ketone and palladium hydride 2, which then reacts with oxygen and another equivalent of alcohol to reform 1. Efforts by a number of researchers have further clarified this mechanism.[13–15]
Scheme 2.

Proposed mechanism for the palladium-catalyzed alcohol oxidation.
Based on this previous report of alcohol oxidation, we recently developed a palladium-catalyzed oxidative kinetic resolution of secondary alcohols.[16, 17] Utilizing [Pd(nbd)Cl2] (nbd = norbornadiene) and the commercially available diamine (−)-sparteine, a variety of alcohols were resolved to high enantiomeric excesses, often with good corresponding catalyst selectivity. In this article, we describe in detail the development of this catalytic enantioselective oxidation, modification of these conditions leading to dramatic improvements in reaction rate and selectivity, our extensive substrate scope investigation, and applications, including the desymmetrization of meso-diols.
Results and Discussion
Reaction Development
Our initial exploration of the aerobic kinetic resolution of secondary alcohols with catalytic Pd focused on substituting an appropriate chiral ligand for pyridine in the oxidation.[16] While many of the compounds evaluated as ligands led to little or no alcohol oxidation, some provided moderately reactive Pd complexes. However, (−)-sparteine rapidly emerged as a uniquely effective ligand for this transformation, providing modest levels of selectivity[18] in the oxidation of (±)-1-phenylethanol ((±)-3, Table 1).[19]
Table 1.
Initial ligand screen.
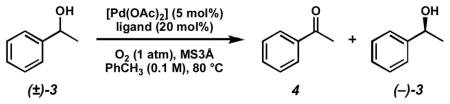 | |||||
|---|---|---|---|---|---|
| entry | ligand[a] | time | conversion[b] | alcohol ee[c] | s |
| 1 | (S,S)-Ph-PYBOX | 72 h | 2% | – | – |
| 2 | (S)-t-Bu-BOX | 24 h | 3% | – | – |
| 3 | cinchonidine | 72 h | 2% | – | – |
| 4 | quinine | 24 h | 0% | – | – |
| 5 | (R,R)-Jacobsen ligand | 24 h | 3% | – | – |
| 6 | (−)-isopinocampheylamine | 24 h | 0% | – | – |
| 7 | (R)-BINAP | 24 h | 29.0% | 0% | 1.0 |
| 8 | brucine | 24 h | 77.0% | 0% | 1.0 |
| 9 | (DHQ)2PHAL | 24 h | 31.6% | 8.7% | 1.6 |
| 10 | (−)-sparteine | 24 h | 15.1% | 13.7% | 8.8 |
See Figure 1.
Measured by GC.
Measured by chiral HPLC. See Supporting Information for details.
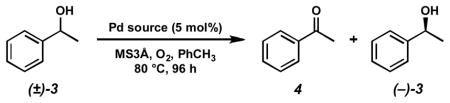
Reexamination of the general mechanism proposed by Uemura was critical for optimization of the reaction. Notably, an acetate is coordinated to the palladium center throughout the catalytic cycle, indicating the possible importance of the Pd(II) counterion in the resolution. Investigation of a number of palladium sources permitted counterion modification, resulting in greatly improved reactivity and selectivity in the oxidation employing palladium chloride complexes. [Pd(nbd)Cl2] proved to be the most effective precatalyst, providing good selectivity in the oxidation of a number of benzylic alcohols (Scheme 3).
Scheme 3.

Initial conditions for the Pd-catalyzed enantioselective oxidation of alcohols.
Although useful in principle, a major limitation of this resolution is the sluggish reaction rates. Usually, over 4 days are required to provide alcohols in high enantiomeric excess. The high operating temperature (80 °C) further demonstrates the low activity of the system. During the course of our studies to improve the reaction rate, we prepared the discrete complex [Pd(sparteine)Cl2] (5). The reactivity of this complex was substantially decreased relative to the complex generated in situ with a 4:1 sparteine: Pd loading (Table 2).[17a] Interestingly, reactivity could be restored by adding 3 equivalents of sparteine relative to complex 5. We hypothesized that the excess sparteine acts as a general base for the hydrogen chloride byproduct generated as a result of palladium alkoxide formation. Indeed, kinetic studies have confirmed the role of sparteine as base.[15a] Without this added sparteine, palladium alkoxide formation is much less favorable. Thus, it was anticipated that the addition of a stoichiometric base would promote palladium alkoxide formation, facilitating alcohol oxidation.
Table 2.
Effect of excess sparteine on the resolution for in situ and preformed catalysts.
 | ||||
|---|---|---|---|---|
| entry | catalyst[a] | conversion[b] | alcohol ee[c] | s |
| 1 | [Pd(nbd)Cl2] (−)-sparteine (20 mol%) |
59.9% | 98.7% | 23.1 |
| 2 | [Pd(sparteine)Cl2] (5) | 4.6% | 3.4% | 6.0 |
| 3 | [Pd(sparteine)Cl2] (5) (−)-sparteine (15 mol%) |
60.6% | 99.2% | 23.8 |
1 atm O2, 500 mg MS3Å/mmol substrate, 0.1 M substrate in PhCH3.
Measured by GC.
Measured by chiral HPLC. See Supporting Information for details.
Early studies to supplant sparteine as a base centered on the addition of amines (Table 3).[20] Initially, triethylamine seemed promising (entry 3), but rapid catalyst deactivation was ultimately observed. Additional equivalents proved more detrimental (entries 5 and 6). The more nucleophilic base 1,4-diazobicyclo[2.2.2]octane (DABCO) was even poorer. The loss of reactivity with these bases is presumably due to competition between the amine and sparteine for coordination to the palladium center.[21] However, carbonate bases were effective at accelerating the oxidations. Cesium carbonate was found to be optimal, leading to the greatest reactivity enhancement.[22] Surprisingly, excess sparteine relative to palladium was still required.
Table 3.
Added base in the kinetic resolution.
 | |||||
|---|---|---|---|---|---|
| entry | additive | time | conversion[b] | alcohol ee[c] | s |
| 1 | sparteine (0.3 equiv) | 13 h | 29% | 34% | 15 |
| 2 | none | 13 h | 2% | <2% | - - |
| 3 | Et3N (0.4 equiv) | 13 h | 26% | 31% | 22 |
| 4 | Et3N (0.4 equiv) | 26 h | 29% | 33% | 13 |
| 5 | Et3N (2.0 equiv) | 13 h | 19% | 19% | 11 |
| 6 | Et3N (4.0 equiv) | 13 h | 14% | 11% | 6 |
| 7 | DABCO (0.4 equiv) | 13 h | 7% | 8% | 20 |
| 8 | Na2CO3 (1.0 equiv) | 13 h | 27% | 31% | 15 |
| 9 | K2CO3 (1.0 equiv) | 13 h | 56% | 84% | 13 |
| 10 | Cs2CO3 (1.0 equiv) | 13 h | 68% | 99% | 13 |
10 mol% [Pd(nbd)Cl2], 10 mol% (−)-sparteine, 1 atm O2, 500 mg MS3Å/mmol substrate, 0.1 M substrate in PhCH3.
Measured by 1H NMR.
Measured by chiral HPLC. See Supporting Information for details.
Several observations suggest cesium carbonate is acting as a heterogeneous base. The solubility of cesium carbonate in toluene is minimal, even at elevated temperatures. Also, finely milled cesium carbonate performs much better than the granular base of lower surface area.[23] Thus, sparteine, which is much more soluble in toluene, may act as a better kinetic base than the heterogeneous cesium carbonate for neutralizing hydrogen chloride generated in the formation of the palladium alkoxide. While the pKa values of sparteine·HCl[24] and cesium bicarbonate[25] are similar, the excess carbonate base could minimize sparteine·HCl concentration,[26] inhibiting protonation of the palladium alkoxide and accelerating the reaction (Scheme 4).[27]
Scheme 4.

Potential role of excess sparteine and base.
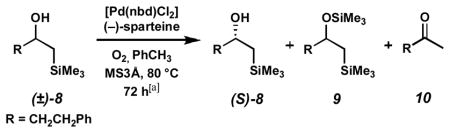
Another key development arose from our early investigations of the substrate scope. Generally, viable resolution substrates featured a π-system that has the capacity to overlap with a cation at the alcohol carbon (Scheme 5).[15b] Because the transition state for β-hydride elimination involves some cationic character at this carbon, we speculated that methods of cation stabilization other than adjacent aromatic rings could lower the energy barrier for alcohol oxidation, increasing reaction rate. Based on this hypothesis, β-silyl alcohol (±)-8 was prepared and exposed to our palladium-catalyzed oxidation conditions. This alcohol could potentially benefit from β-silicon stabilization by hyperconjugation in the transition state.[28] Indeed, (±)-8 proved to be a moderately active substrate for oxidative kinetic resolution, providing partially resolved alcohol in 16% ee (Table 4, entry 1). However, a problematic side reaction complicated analysis of this resolution. The partially enantioenriched alcohol underwent silyl transfer from the α-silyl ketone to provide oxidation-resistant silyl ether 9 and desilylated ketone 10. In order to suppress formation of side product 9, a primary alcohol was added to the reaction. This unactivated alcohol would not be readily oxidized under our resolution conditions, but could act as a sacrificial alcohol to react with the silyl ketone. As seen in entries 2–4, increasing amounts of n-BuOH led to decreased formation of silyl ether 9. Fortuitously, reaction rates also increased with up to 2.1 equivalents of n-BuOH. Rate enhancement was not limited to the oxidation of silyl alcohol (±)-8, as increased rates were also observed in the resolution of other activated alcohols.
Scheme 5.

Stabilization of the β-hydride elimination transition state by resonance and a β-silicon.
Table 4.
n-BuOH as additive in the kinetic resolution.
 | ||||
|---|---|---|---|---|
| entry | n-BuOH equiv | % (S)-8[b] (ee)[c] | % 9[b] | % 10[b] |
| 1 | 0.0 | 75 (16) | 15 | 10 |
| 2 | 0.7 | 60 (34) | 10 | 30 |
| 3 | 2.1 | 35 (81) | 5 | 60 |
| 4 | 4.2 | 45 (61) | <5 | 50 |
5 mol% [Pd(nbd)Cl2], 20 mol% (−)-sparteine, 1 atm O2, 500 mg MS3Å/mmol substrate, 0.25 M substrate in PhCH3.
Measured by 1H NMR.
Measured by chiral HPLC. See Supporting Information for details.
Having observed a significant dependence on the rate of oxidative kinetic resolution with the inclusion of n-BuOH, a variety of alcohol additives were evaluated for their ability to enhance the reaction rate when coupled with cesium carbonate in the resolution of alcohol (±)-6 (Table 5). Addition of 1 equivalent of either 1-butanol or trifluoroethanol led to enhanced reactivity (entries 2 and 5). Increasing equivalents led to decreased selectivity (entries 3 and 6). Large excess of either alcohol impeded the reaction (entries 4 and 7). This trend of initial rate acceleration followed by rate inhibition and selectivity erosion at high concentrations was observed across a wide variety of primary alcohol additives.
Table 5.
Various non-oxidizing alcohols as additives.
 | |||||
|---|---|---|---|---|---|
| entry | additive | time | conversion[b] | alcohol ee[c] | s |
| 1 | none | 5.5 h | 31% | 35% | 12 |
| 2 | n-BuOH (1.0 equiv) | 5.5 h | 66% | 98% | 12 |
| 3 | n-BuOH (2.0 equiv) | 5.5 h | 67% | 95% | 10 |
| 4 | n-BuOH (8.0 equiv) | 19 h | 48% | 56% | 7 |
| 5 | CF3CH2OH (1.0 equiv) | 5.5 h | 60% | 89% | 11 |
| 6 | CF3CH2OH (2.0 equiv) | 5.5 h | 42% | 48% | 8 |
| 7 | CF3CH2OH (8.0 equiv) | 19 h | 19% | 16% | 6 |
| 8 | t-BuOH (1.0 equiv) | 22.5 h[d] | 57% | 90% | 16 |
| 9 | t-BuOH (4.0 equiv) | 11.5 h[d] | 57% | 94% | 20 |
| 10 | t-BuOH (8.0 equiv) | 19 h[e] | 57% | 90% | 16 |
10 mol% [Pd(nbd)Cl2], 10 mol% (−)-sparteine, 10 mol% Cs2CO3, 1 atm O2, 500 mg MS3Å/mmol substrate, 0.1 M substrate in PhCH3.
Measured by 1H NMR.
Measured by chiral HPLC. See Supporting Information for details.
Conducted at 50 °C.
Conducted at 45 °C.
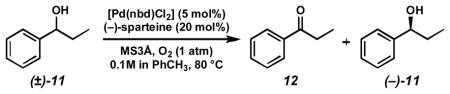
Interestingly, inclusion of tert-butyl alcohol proved to be an excellent exogenous alcohol additive, providing the highest and most reliable levels of selectivity across a wide range of concentrations (entries 8–10).[29] After optimizing exogenous base and tert-butyl alcohol equivalents, the dramatic effect of these additives on the resolution of (±)-1-phenyl-1-propanol ((±)-11) was realized (Table 6). This resolution provided highly enantioenriched (−)-11 in only 4.5 h instead of 192 h without cesium carbonate and tert-butyl alcohol. Importantly, selectivity is maintained in these dramatically rate accelerated conditions. Further selectivity improvements could be obtained by decreasing the temperature to 60 °C.[30]
Table 6.
Direct comparison of original and rate accelerated oxidative kinetic resolution conditions.
 | ||||
|---|---|---|---|---|
| additives | time | conversion[a] | alcohol ee[b] | s |
| none | 192 h | 59% | 93% | 15 |
| Cs2CO3 (1.2 equiv) t-BuOH (4.0 equiv) |
4.5 h | 63% | 98% | 16 |
Measured by 1H NMR.
Measured by chiral HPLC. See Supporting Information for details.
While the purpose of the carbonate base fit well with our mechanistic model for the oxidative kinetic resolution, the role of tert-butyl alcohol remained more subtle. One possibility was that the modified reaction conditions were transforming [Pd(sparteine)Cl2] into a more reactive complex.[31] In order to verify this theory, 5 was exposed to 1 equivalent each of tert-butyl alcohol and cesium carbonate. After 4 d at 60 °C, carbonate 13 was isolated in nearly quantitative yield (Scheme 6). Surprisingly, 13 displayed neither catalytic nor even stoichiometric activity under kinetic resolution conditions. This finding suggests carbonate 13 is a catalyst deactivation product. While these additives provide dramatic rate enhancement, they could also detrimentally affect catalyst longevity.[32]
Scheme 6.

Formation of inactive Pd complex 13.
Alternatively, the observed rate enhancement could be due to the hydrogen bonding potential of tert-butyl alcohol. While hydrogen bonding plays an essential and well-documented role in the catalysis of a number of biological[33] and synthetic processes,[34] less is known about its role in organometallic transformations. A hydrogen-bond donor may enhance reactivity by stabilizing and solubilizing polar or charged intermediates in the nonpolar solvent toluene. In particular, cationic palladium complexes generated as the immediate products of alcohol coordination and β-hydride elimination result in the formation of chloride anions, which could be solubilized by hydrogen-bond donors.[15b,d]
Based on this hypothesis, a more rigorous solvent screen was performed with an emphasis on solvents capable of hydrogen-bond donation and solubilization of polar or charged intermediates (Table 7).[35] Surprisingly, while common polar solvents led to little oxidation (entries 12, 15, 16, 19, and 20), halogenated solvents that could act as weak hydrogen-bond donors provided rapid reactions. Oxidations conducted in the hydrogen-bond donating solvent CH2Cl2 were the fastest (entry 1), but catalyst selectivity in kinetic resolutions suffered.[36] Chloroform, on the other hand, emerged as an outstanding nonflammable solvent for rapid and selective oxidation, even at 23 °C (entry 2). Strikingly, chlorinated solvents lacking the ability to donate a hydrogen bond (entries 13, 14, 17, and 21), were less effective. Some other factors do not appear to explain the observed trends. Oxygen solubility is fairly similar in many of the solvents.[37] Also, there is no clear trend between reaction rate and dielectric constant, a measure of solvent polarity.[38]
Table 7.
Impact of solvent on reaction rate.
 | |||
|---|---|---|---|
| entry | solvent | dielectric constant | conversion[a] |
| 1 | CH2Cl2 | 8.9 | 83% |
| 2 | CHCl3 | 4.8 | 74% |
| 3 | CH2Br2 | 7.8 | 73% |
| 4 | CHCl3/1 equiv t-BuOH | 4.8/12.5 | 72% |
| 5 | CHBr3 | 4.4 | 68% |
| 6 | ClCH2CH2Cl | 10.4 | 46% |
| 7 | PhCH3/1 equiv t-BuOH | 2.4/12.5 | 39% |
| 8 | PhCH3/t-BuOH (1:1) | 2.4/12.5 | 29% |
| 9 | PhCH3 | 2.4 | 23% |
| 10 | pinacolone | 12.7 | 21% |
| 11 | t-amyl alcohol | 5.8 | 21% |
| 12 | THF | 7.5 | 14% |
| 13 | Cl2C=CHCl | 3.4 | 10% |
| 14 | Cl3CCH3 | 7.2 | 9% |
| 15 | EtOAc | 6.1 | 8% |
| 16 | 2-propanol | 20.2 | 7% |
| 17 | Cl2C=CCl2 | 2.3 | 6% |
| 18 | H2O/2-propanol | 80.1/20.2 | 3% |
| 19 | CH3CN | 36.6 | 3% |
| 20 | CH3NO2 | 37.3 | 2% |
| 21 | CCl4 | 2.2 | 2% |
Measured by GC relative to internal standard (tridecane). See Supporting Information for details.
Spectroscopic evidence for hydrogen-bond formation between chloroform and catalytic species is found in IR spectra of CDCl3 (Table 8).[39] A significant shift in the C-D stretching frequency of CDCl3 occurred in the presence of either (−)-sparteine (entry 2) or [Pd(sparteine)Cl2] (5) (entry 3).[40,41] The observed decrease in λmax corresponds to a lower energy C-D stretching frequency due to a weaker C-D bond over free CDCl3 when hydrogen-bond accepting species are present. Notably, no shift in the C-D stretch was observed with the catalytically inactive carbonate 13, suggesting little or no hydrogen bonding occurs.
Table 8.
C-D stretch of CDCl3 in the presence of potential H-bond acceptors.
| entry | concentration | CDCl3 solution | λmax (cm−1) | predicted D-X bond |
|---|---|---|---|---|
| 1 | neat | 2258 | – | |
| 2 | 0.25 M |  |
2175 | 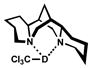 |
| 3 | 0.25 M |  |
2230 | 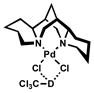 |
| 4 | 0.10 M[a] |  |
2258 | – |
Near saturation concentration at 23 °C.
The change in solvent allows decreased amounts of (−)-sparteine and cesium carbonate to be used with little effect on rate. Performing the reactions at 23 °C also provides a dramatic increase in the catalyst selectivity. Furthermore, as little as 5% O2 atmosphere is sufficient for oxidation, permitting the use of ambient air as the terminal oxidant. These developments greatly improve the safety of the oxidative kinetic resolution, avoiding the use of flammable solvents at elevated temperatures under an oxygen atmosphere.[42]
Concurrent with studies centered on the development of improved conditions for oxidation, efforts were undertaken to understand the mechanism of the reaction. We were especially interested in elucidating the steric and electronic influences of the sparteine ligand on the outcome of the kinetic resolution. To this end, a number of palladium complexes with sparteine were prepared.[15c] Subsequent X-ray analysis of these crystalline complexes, coupled with theoretical calculations,[15d] allowed us to develop a model for selectivity in the oxidation (Scheme 7). Coordination of alcohol to complex 5 leads to one of two diastereomeric alkoxides, 14 or 15. Alkoxide 14, formed from the fast reacting enantiomer of alcohol, proceeds through a four-membered β-hydride elimination transition state to afford product ketone. Complex 15, on the other hand, has a higher energy barrier to β-hydride elimination due to developing unfavorable steric interactions with the departing chloride counterion. Confronted with this high-energy transition state, protonation regenerates the observed enantiomer of alcohol.
Scheme 7.

Model for kinetic resolution selectivity.
As a result of our exploration of the oxidative kinetic resolution of secondary alcohols, four distinct sets of conditions have been developed (Table 9): the original conditions (A) in toluene with no exogenous base, the rate enhanced conditions (B) that take advantage of cesium carbonate and tert-butyl alcohol additives, and the chloroform conditions at 23 °C under an atmosphere of either molecular oxygen (C) or ambient air (D). In general, resolutions performed without added carbonate base have slower rates but greater catalyst longevity. The rate enhanced (B) conditions are the fastest, often achieving highly enantioenriched alcohol in a small fraction of the time required for the original (A) conditions. Reactions performed in chloroform at 23 °C (C and D) are the most selective, nearly doubling the s factor for the resolution of some alcohols. Typically, both molecular oxygen and ambient air can be used in oxidations in chloroform with similar rates and selectivities. The development of four distinct sets of conditions provides the opportunity to resolve the widest range of alcohol substrates in order to maximize the selectivity of the process while maintaining reactivity and minimizing side reactions. The benefit of this flexibility is evident in the broad scope of this system.
Table 9.
Variety of conditions for the resolution of secondary alcohols.
 | |||||
|---|---|---|---|---|---|
| entry | conditions | time | conversion[a] | ee[b] | s |
| A | [Pd(nbd)Cl2] (5 mol%) (−)-sparteine (20 mol%) PhCH3 (0.1 M), O2 MS3Å, 80 °C |
96 h | 67% | 96% | 12 |
| B | [Pd(nbd)Cl2] (5 mol%) (−)-sparteine (20 mol%) Cs2CO3 (1.2 equiv) t-BuOH (4.0 equiv) PhCH3 (0.25 M), O2 MS3Å, 60 °C |
9.5 h | 67% | 99% | 15 |
| C | [Pd(nbd)Cl2] (5 mol%) (−)-sparteine (12 mol%) Cs2CO3 (0.4 equiv) CHCl3 (0.25 M), O2 MS3Å, 23 °C |
48 h | 63% | 99% | 27 |
| D | [Pd(nbd)Cl2] (5 mol%) (−)-sparteine (12 mol%) Cs2CO3 (0.4 equiv) CHCl3 (0.25 M), Air MS3Å, 23 °C |
24 h | 62% | 99% | 25 |
Measured by GC relative to internal standard (tridecane).
Measured by chiral HPLC. See Supporting Information for details.
Substrate Scope[43]
Our early studies focused on benzylic alcohols, as the racemates are commercially available or easily prepared. The resolution is quite general for this class of substrates, providing a wide range of 1-arylethanols in excellent enantiomeric excess (Table 10). Substitution on the aryl ring at the 3- and 4-positions (entries 1–8, 12–14) is well tolerated. Ortho-substitution leads to much slower rates of oxidation (entry 9), although reactivity improves if the substituent is constrained in a ring (entries 10 and 11). Some heteroaromatic substrates (entries 18 and 19) can also be resolved to high enantiomeric excesses.
Table 10.
Resolution of 1-arylethanols.
 | ||||||
|---|---|---|---|---|---|---|
| entry | alcohol, major enantiomer | conditions[a] | time | conv.[b] | alcohol ee[c] | s |
| 1 | R = H | B | 12.5 h | 64% | 99% | 20 |
| 2 | C | 48 h | 60% | 99% | 31 | |
| 3 |
 R = OMe R = OMeR = F |
B | 9.5 h | 67% | 99% | 15 |
| 4 | C | 45 h | 63% | 99% | 27 | |
| 5 | D | 24 h | 62% | 99% | 25 | |
| 6 | A | 54 h | 63% | 97% | 14 | |
| 7 | C | 48 h | 59% | 98% | 23 | |
| 8 | D | 24 h | 57% | 93% | 20 | |
| 9 |  |
C | 164 h | 60% | 89% | 12 |
| 10 | 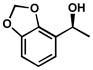 |
B | 15 h | 57% | 99% | 47 |
| 11 | C[d] | 12 h | 55% | 95% | 29 | |
| 12 | 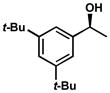 |
A | 28 h | 52% | 93% | 44 |
| 13 | C | 24 h | 54% | 98% | 47 | |
| 14 | D | 22 h | 54% | 98% | 54 | |
| 15 | 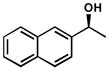 |
A | 112 h | 55% | 99% | 47 |
| 16 | C | 48 h | 60% | 99% | 31 | |
| 17 | D | 24 h | 56% | 98% | 37 | |
| 18 | 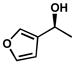 |
A | 120 h | 67% | 94% | 8.3 |
| 19 | 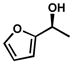 |
A | 120 h | 67% | 94% | 8.8 |
Conditions A: [Pd(nbd)Cl2] (5 mol%), (−)-sparteine (20 mol%), O2 (1 atm), MS3Å (500 mg/mmol substrate), PhCH3 (0.1 M substrate), 80 °C. Conditions B: [Pd(nbd)Cl2] (5 mol%), (−)-sparteine (20 mol%), O2 (1 atm), Cs2CO3 (0.5 equiv), t-BuOH (1.5 equiv), MS3Å (500 mg/mmol substrate), PhCH3 (0.25 M substrate), 60 °C. Conditions C: [Pd(nbd)Cl2] (5 mol%), (−)-sparteine (12 mol%), O2 (1 atm), Cs2CO3 (0.4 equiv), MS3Å (500 mg/mmol substrate), CHCl3 (0.25 M substrate), 23 °C. Conditions D: [Pd(nbd)Cl2] (5 mol%), (−)-sparteine (12 mol%), Cs2CO3 (0.4 equiv), MS3Å (500 mg/mmol substrate), CHCl3 (0.25 M substrate), 23 °C, open to ambient air under a short tube of Drierite.
Conversion measured by GC or NMR.
Measured by chiral HPLC or chiral GC. See Supporting Information for details.
Conducted at 40 °C.
Other structural variations are also tolerated by the catalyst system (Table 11). Cyclic benzylic alcohols are able to be resolved successfully. 1-Indanol (entries 1 and 2) is oxidized rapidly, albeit with decreased selectivity, compared to 1-tetralol (entries 3–5). Other functional groups on the alcohol substrate, including protected amines (entries 9 and 10), a tertiary alcohol (entry 11), and even aryl bromides (entries 11 and 12), are tolerated under the reaction conditions.[44]
Table 11.
Resolution of other benzylic alcohols.
 | ||||||
|---|---|---|---|---|---|---|
| entry | alcohol, major enantiomer | conditions[a] | time | conv.[b] | alcohol ee[c] | s |
| 1 | 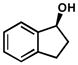 |
B[d] | 12 h | 74% | 99% | 10 |
| 2 | C | 23 h | 70% | 98% | 10 | |
| 3 | 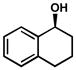 |
B[d] | 12 h | 62% | 99% | 21 |
| 4 | C | 24 h | 58% | 98% | 28 | |
| 5 | D | 16 h | 60% | 99% | 28 | |
| 6 | B[e] | 4.5 h | 63% | 98% | 16 | |
| 7 | C | 72 h | 63% | 98% | 24 | |
| 8 | D | 48 h | 57% | 95% | 22 | |
| 9 |
R = Boc |
A | 14.5 h | 70% | 97% | 9.0 |
| 10 | A | 24 h | 58% | 93% | 18 | |
| 11 | 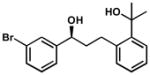 |
B | 4.5 h | 71% | 99% | 15 |
| 12 | 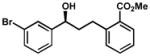 |
B | 4.5 h | 63% | 93% | 11 |
| 13 | 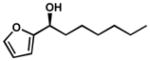 |
C | 122 h | 55% | 75% | 9.0 |
The broad utility of chiral allylic alcohols in organic synthesis led us to investigate this important class of molecules next. Conditions in chloroform are particularly effective for these substrates, providing enhanced selectivity over the other methods (Table 12, cf. entries 11 and 12). While acyclic allylic alcohols are challenging, often leading to oxidation with low selectivity, many cyclic allylic alcohols are resolved successfully. Of particular note are vinyl bromides (entries 1, 2, and 5), which rapidly decompose with darkening of the reaction mixture at elevated temperatures, indicating the formation of aggregated Pd(0). While 2-bromocyclopent-2-enol (entry 5) oxidizes fast enough to allow moderate resolution at 60 °C with cesium carbonate and tert-butyl alcohol, 2-bromocyclohex-2-enol (entries 1 and 2) is cleanly resolved with no decomposition only at lower temperatures. Alkyl enol ethers are stable in the reactions and are resolved to high enantiomeric excess (entries 4, 7, and 8), providing access to enantioenriched α-hydroxyketone derivatives. The catalyst is also tolerant of a variety of alkene substitution patterns. Cyclopentenols are usually oxidized more quickly, though less selectively, than cyclohexenols (cf. entries 4 and 7).
Table 12.
Resolution of allylic alcohols.
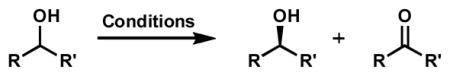 | ||||||
|---|---|---|---|---|---|---|
| entry | alcohol, major enantiomer | conditions[a] | time | conv.[b] | alcohol ee[c] | s |
| 1 |
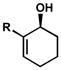 R = Br R = BrR = iPr R = OiBu |
C | 33 h | 64% | 96% | 12 |
| 2 | D | 25 h | 55% | 86% | 16 | |
| 3 | C | 31 h | 51% | 74% | 13 | |
| 4 | C | 74 h | 52% | 86% | 23 | |
| 5 |
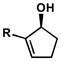 R = Br R = BrR = iPr R = OiBu |
B | 4 h | 75% | 97% | 7.0 |
| 6 | B | 7 h | 66% | 93% | 9.2 | |
| 7 | C | 45 h | 65% | 99% | 15 | |
| 8 | D | 24 h | 61% | 97% | 17 | |
| 9 | 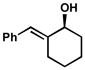 |
C | 27 h | 61% | 96% | 16 |
| 10 | D | 24 h | 63% | 98% | 17 | |
| 11 | 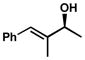 |
B | 12 h | 65% | 88% | 7.5 |
| 12 | C | 48 h | 63% | 99% | 18 | |
| 13 | D | 44 h | 65% | 99% | 16 | |
| 14 | 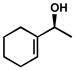 |
C | 43 h | 64% | 97% | 13 |
| 15 | D | 43 h | 61% | 91% | 12 | |
| 16 | 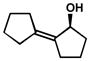 |
C | 75 h | 57% | 94% | 19 |
See Table 10 footnotes.
Our success with benzylic alcohols led us to explore aryl substituents on cyclic allylic alcohols. These substrates are readily prepared via Suzuki coupling of arylboronate esters and iodoenones[45] followed by ketone reduction.[46] To our delight, subjection of these alcohols to any of our developed kinetic resolution conditions affords highly enantioenriched allylic alcohols with short reaction times (Table 13).[47] Both electron rich (entries 7–9) and electron poor (entries 13–15) 2-aryl substituents lead to excellent selectivity. The lack of an electronic influence on these resolutions suggests selectivity is primarily due to steric factors, as elucidated by our selectivity model (Scheme 7). Even heteroaromatic substitution (entries 22–24) and a larger ring size (entries 25–27) are tolerated, albeit with somewhat longer reaction times.
Table 13.
Resolution of 2-aryl allylic alcohols.
 | ||||||
|---|---|---|---|---|---|---|
| entry | alcohol major enantiomer | conditions[a] | time | conv.[b] | alcohol ee[c] | s |
| 1 | R = H | A | 7 h | 57% | 99% | 35 |
| 2 | B | 1.5 h | 53% | 99% | 89 | |
| 3 | D | 10 h | 56% | 99% | 47 | |
| 4 | R = Me | B | 3.5 h | 58% | 99% | 31 |
| 5 | C | 11 h | 58% | 99% | 28 | |
| 6 |
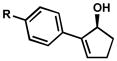 R = OMe R = OMe |
D | 10 h | 56% | 99% | 39 |
| 7 | A | 31 h | 58% | 99% | 30 | |
| 8 | B | 1.5 h | 54% | 97% | 46 | |
| 9 | C | 24 h | 55% | 92% | 23 | |
| 10 | R = F | A | 10 h | 57% | 99% | 33 |
| 11 | B | 3.5 h | 60% | 99% | 24 | |
| 12 | C | 11 h | 58% | 99% | 30 | |
| 13 | R = CF3 | A | 10 h | 59% | 99% | 26 |
| 14 | C | 10 h | 57% | 94% | 21 | |
| 15 | D | 10 h | 59% | 99% | 26 | |
| 16 | 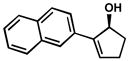 |
A | 10 h | 59% | 99% | 28 |
| 17 | B | 1.5 h | 53% | 94% | 36 | |
| 18 | D | 10 h | 56% | 99% | 45 | |
| 19 | 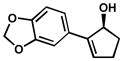 |
A | 24 h | 54% | 99% | 59 |
| 20 | C | 10 h | 59% | 99% | 25 | |
| 21 | D | 6 h | 57% | 97% | 27 | |
| 22 | 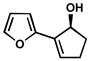 |
B | 10 h | 64% | 99% | 19 |
| 23 | C | 72 h | 57% | 96% | 23 | |
| 24 | D | 42.5 h | 57% | 98% | 27 | |
| 25 | 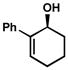 |
A | 17 h | 52% | 99% | 122 |
| 26 | B | 3 h | 57% | 97% | 27 | |
| 27 | C | 24 h | 55% | 97% | 33 | |
See Table 10 footnotes.
Substitution at the 3-position of cyclic allylic alcohols was also explored (Table 14). Again, allylic alcohols with 2-aryl substituents are oxidized rapidly, and with exceptionally high selectivity (entries 1–6). These resolutions have some of the highest selectivities seen with this catalyst system. 3-Substituted alcohols with 2-alkyl substituents are resolved to high enantiomeric excess as well (entries 7–13). Interestingly, analogous 2-alkyl allylic alcohols unsubstituted at the 3-position oxidize with poor selectivity.[48]
Table 14.
Resolution of 3-substituted cyclic allylic alcohols.
 | ||||||
|---|---|---|---|---|---|---|
| entry | alcohol, major enantiomer | conditions[a] | time | conv.[b] | alcohol ee[c] | s |
| 1 | 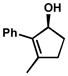 |
A | 4 h | 53% | 99% | 83 |
| 2 | B | 1 h | 53% | 98% | 63 | |
| 3 | D | 6 h | 55% | 94% | 26 | |
| 4 | 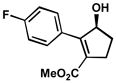 |
A | 4 h | 56% | 99% | 50 |
| 5 | B | 1 h | 56% | 99% | 51 | |
| 6 | C | 9 h | 51% | 95% | 83 | |
| 7 | 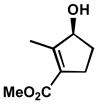 |
A | 8 h | 73% | 98% | 8.6 |
| 8 | B | 1 h | 61% | 85% | 9.0 | |
| 9 | C | 10 h | 78% | 99% | 7.5 | |
| 10 |
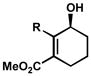 R = Me R = MeR = Bn |
B | 8 h | 65% | 99% | 15 |
| 11 | C | 27 h | 58% | 94% | 19 | |
| 12 | D | 43 h | 64% | 97% | 13 | |
| 13 | C | 72 h | 61% | 91% | 11 | |
See Table 10 footnotes.
Other forms of activation of the racemic alcohols were also explored, such as α-cyclopropyl substituents. Several substrates were exposed to our oxidative kinetic resolution conditions (Table 15). Again, the chloroform conditions are especially effective in providing highly selective oxidation (cf. entries 1 and 2, entries 4 and 6). Even 1-cyclopropylethanol (entries 1–3), with relatively little steric differentiation between alcohol substituents, is able to be resolved to high enantiomeric excess. These resolutions also produce alcohols with three contiguous stereocenters (entries 4–11), including a quaternary stereocenter (entries 7 and 8). Furthermore, for entries 4–11, the product ketones are also enantioenriched (Figure 2). Importantly, these molecules have the opposite configuration at C(3) and C(4) relative to the resolved alcohol, opening the door to enantiodivergent opportunities in synthesis.
Table 15.
Resolution of cyclopropylcarbinyl alcohols.
 | ||||||
|---|---|---|---|---|---|---|
| entry | alcohol, major enantiomer | conditions[a] | time | conv.[b] | alcohol ee[c] | s |
| 1 | 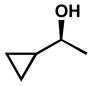 |
B | 22 h | 76% | 96% | 6.1 |
| 2 | C | 72 h | 67% | 99% | 13 | |
| 3 | D | 68 h | 66% | 96% | 10 | |
| 4 | 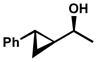 |
A | 23 h | 69% | 99% | 12 |
| 5 | B | 3 h | 59% | 91% | 14 | |
| 6 | C | 25 h | 59% | 99% | 28 | |
| 7 | 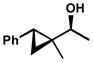 |
C | 71 h | 60% | 99% | 25 |
| 8 | D | 38 h | 57% | 89% | 15 | |
| 9 | 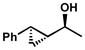 |
A | 17 h | 66% | 99% | 15 |
| 10 | B | 9 h | 71% | 99% | 10 | |
| 11 | C | 24 h | 51% | 76% | 15 | |
See Table 10 footnotes.
Figure 2.

Enantioenriched ketones obtained from the kinetic resolution.
Though a broad range of secondary alcohols is successfully resolved with this system, limitations to the methodology exist. A number of alcohols display limited rates of oxidation, preventing their resolution (Figure 3). Benzylic alcohols with ortho-substituents (e.g. (±)-18 and (±)-19) and sterically hindered alcohols such as (±)-20 and (±)-21 have dramatically decreased oxidation rates. The presence of vicinal heteroatoms (e.g. (±)-22 and (±)-23) impedes the oxidation, presumably through catalyst coordination and deactivation.[49] Finally, unactivated alcohols (e.g. (±)-24 and (±)-25), particularly primary alcohols, are slow to oxidize under any of our developed conditions.[50]
Figure 3.

Examples of alcohols displaying poor reactivity.
In addition to unreactive alcohols, certain classes of alcohols are resolved with poor selectivity (Figure 4). In some cases, the steric difference between the two alcohol substituents seems too small for the catalyst to adequately distinguish between enantiomers (e.g. (±)-26, (±)-27, and (±)-28). Substrates with electron-poor aromatic substituents are much less selectively resolved than their electron-rich counterparts (cf. (±)-29 and (±)-30 with Table 10 entries 4 and 12, respectively). At least in the case of benzylic alcohols, steric effects alone do not fully account for these selectivity differences. Saturated alcohols, such as (±)-31 also tend to display lower selectivity factors in the resolution.
Figure 4.

Examples of alcohols oxidized with poor selectivity.
Applications
The wide use of alcohols in synthesis provides numerous applications for our kinetic resolution.[51] A number of alcohols successfully resolved are intermediates in the synthesis of a variety of pharmaceuticals (Scheme 8).[44, 52] Boc-protected γ-aminoalcohol (−)-32, resolved with good selectivity, is an intermediate in the synthesis of a number of related antidepressants, including fluoxetine·HCl (33). Tertiary alcohol (−)-34 and a related ester were transformed to a known intermediate in the synthesis of the leukotriene receptor antagonist montelukast sodium (35). Finally, allylic alcohol (−)-36 is an intermediate in the enantioselective synthesis of human neurokinin-1 (hNK-1) receptor antagonist 37.
Scheme 8.

Secondary alcohols as drug intermediates.
To further highlight the utility of the method, we have explored the Claisen rearrangement of resolved cyclic allylic alcohols such as (−)-38 (Scheme 9).[47] Vinyl ether formation and treatment with DIBAL-H at low temperature induces Claisen rearrangement and subsequent reduction to form primary alcohol (+)-39. Importantly, because our kinetic resolution is able to produce alcohol (−)-38 in high enantiomeric excess, the product alcohol is also highly enantioenriched. Furthermore, this alcohol can undergo a second palladium-catalyzed oxidative process developed in our laboratories[53] to form highly enantioenriched tetrahydrofuran (−)-40 containing vicinal, fully substituted stereocenters.
Scheme 9.

Conversion of a resolved alcohol to a functionalized tetrahydrofuran.
In addition to kinetic resolution, our catalyst system is well suited for selective oxidation of meso-diols to hydroxyketones.[17a,c,d] These reactions have the potential to provide highly enantioenriched products in greater than 50% yield. One example of this process was demonstrated with our initially developed resolution conditions (Scheme 10).[16] Selective oxidation of diol 41 provides ketoalcohol (+)-42 in 72% yield and 95% ee.
Scheme 10.

Desymmetrization of diol 41.
Further efforts in the area of meso-diol desymmetrization were inspired by the abundance of complex polyol- and polyether-containing natural products, such as polymethoxydienes 43a-e (Figure 5).[54] We envisioned access to these molecules in enantioenriched form by late stage desymmetrization of a meso-diol stereochemical framework. This polyether array could be prepared via diastereoselective bidirectional chain synthesis from a simple symmetric precursor.[55,56]
Figure 5.

Polyether natural products potentially accessible by meso-diol desymmetrization.
To demonstrate the utility of our palladium-catalyzed oxidation system in this area, we developed a unified synthetic approach to several meso-diol stereochemical arrays. Synthesis of these meso-diols commenced from readily available alcohol 44 (Scheme 11).[57] Alcohol protection as a benzyl ether and palladium-catalyzed diene oxidation provides anti-tris-benzyl ether 45.[58] Alkene dihydroxylation and oxidative cleavage with [Pb(OAc)4] affords meso-dialdehyde 46. No epimerization of the sensitive α-benzyloxy stereocenter is observed with this two-step procedure. Diastereoselective chelation-controlled addition of aryl Grignard reagents produces meso-diols 47a and 47b. The syn-ether array was generated by a similar sequence. Again starting from alcohol 44, TBS ether formation followed by [4+2] cycloaddition with singlet oxygen, reductive opening, and benzylation of the resulting diol affords syn-bis-benzylether 48.[59] Alkene cleavage and diastereoselective addition of aryl nucleophiles produces diols 50a and 50b.
Scheme 11.

Preparation of meso-diols from a common starting material.
Having generated the desired meso-diol substrates, we looked to apply our enantioselective oxidation conditions. Exposure of the meso-diols to catalytic quantities of [Pd(sparteine)Cl2] (5) and excess (−)-sparteine under a balloon of oxygen in chloroform provides highly enantioenriched hydroxyketones in excellent yields (Scheme 12). These reactions establish the absolute configuration of four stereocenters in a single catalytic asymmetric operation. Based on these methods, we have begun to construct even more complex polyether structural motifs toward the synthesis of a variety of natural product frameworks.
Scheme 12.

Oxidative desymmetrization of meso-diols.
Conclusion
Enantioselective oxidation with Pd(II) catalysts is a powerful method for the preparation of enantioenriched secondary alcohols. Research in this area has led to dramatic improvements in reaction rate, selectivity, and operational simplicity. The development of a number of distinct methods has allowed the kinetic resolution of a wide range of substrates. Benzylic, allylic, and α-cyclopropyl alcohols can be resolved to high enantiomeric excesses, in many cases with excellent selectivity. A variety of applications, including the desymmetrization of meso-diols, have demonstrated the utility of this oxidation in synthesis. Ongoing efforts to enhance reactivity, to use other chiral ligands in place of sparteine,[60] and to apply this kinetic resolution to natural product synthesis will be reported in due course.
Experimental Section
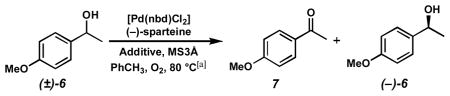
General Oxidative Kinetic Resolution Conditions
Kinetic Resolution Conditions A
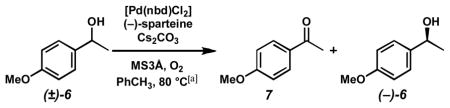
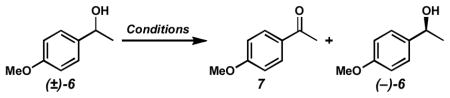
To an oven dried reaction tube with stir bar was added 3Å molecular sieves (250 mg). After cooling, [Pd(nbd)Cl2] (6.7 mg, 0.025 mmol, 0.05 equiv) followed by toluene (2.5 mL) and then (−)-sparteine (23.0 μL, 23.4 mg, 0.10 mmol, 0.20 equiv) were added. The reaction tube was then cooled to −78 °C, then vacuum evacuated and purged with O2 (3x). Then, the tube was heated to 80 °C with vigorous stirring under O2 atmosphere (1 atm, balloon) for 20 min. A solution of (±)-6 (70.5 μL, 76.1 mg, 0.50 mmol, 1.0 equiv) and tridecane (36.6 μL, 27.7 mg, 0.15 mmol, 0.30 equiv) in toluene (2.5 mL) was added, and the reaction was allowed to proceed under O2 atmosphere at 80°C. Aliquots were filtered through a small plug of silica gel (Et2O eluent), evaporated, and analyzed by GC for conversion and chiral HPLC for alcohol ee. Purification of 4-methoxyacetophenone (7) and (−)-6 was accomplished by direct chromatography of the crude reaction mixture.
Kinetic Resolution Conditions B
To an oven dried reaction tube with stir bar was added 3Å molecular sieves (500 mg). After cooling, [Pd(nbd)Cl2] (13.5 mg, 0.050 mmol, 0.05 equiv), followed by toluene (2 mL) and then (−)-sparteine (46.0 μL, 46.9 mg, 0.20 mmol, 0.20 equiv) were added. The reaction tube was cooled to −78 °C, then vacuum evacuated and purged with O2 (3x). The tube was heated to 60 °C with vigorous stirring under O2 atmosphere (1 atm, balloon) for 20 min. Finely powdered Cs2CO3 (163 mg, 0.50 mmol, 0.50 equiv) was added, followed by a solution of (±)-6 (141 μL, 152 mg, 1.0 mmol, 1.0 equiv), anhydrous t-BuOH (143 μL, 111 mg, 1.5 mmol, 1.5 equiv), and tridecane (73.2 μL, 55.3 mg, 0.30 mmol, 0.30 equiv) in toluene (2 mL). The reaction was allowed to proceed under O2 atmosphere at 60 °C. Aliquots were filtered through a small plug of silica gel (Et2O eluent), evaporated, and analyzed by GC for conversion and chiral HPLC for alcohol ee. Purification of 7 and (−)-6 was accomplished by direct chromatography of the crude reaction mixture.
Kinetic Resolution Conditions C
To an oven dried reaction tube with stir bar was added 3Å molecular sieves (500 mg). After cooling, [Pd(nbd)Cl2] (13.5 mg, 0.050 mmol, 0.05 equiv), followed by chloroform (2 mL, ACS reagent grade, stabilized with amylenes) and then (−)-sparteine (27.6 μL, 28.1 mg, 0.12 mmol, 0.12 equiv) were added. The reaction tube was cooled to −78 °C, then vacuum evacuated and purged with O2 (3x). The reaction was allowed to warm to 23 °C and stirred vigorously under O2 atmosphere (1 atm, balloon) for 15 min. Finely powdered Cs2CO3 (130 mg, 0.40 mmol, 0.40 equiv) was added, followed by a solution of (±)-6 (141 μL, 152 mg, 1.0 mmol, 1.0 equiv) and tridecane (73.2 μL, 55.3 mg, 0.30 mmol, 0.30 equiv) in chloroform (2 mL). The reaction was allowed to proceed under O2 atmosphere at 23 °C. Aliquots were filtered through a small plug of silica gel (Et2O eluent), evaporated, and analyzed by GC for conversion and chiral HPLC for alcohol ee. Purification of 7 and (−)-6 was accomplished by direct chromatography of the crude reaction mixture.
Kinetic Resolution Conditions D
To an oven dried reaction tube with stir bar was added 3Å molecular sieves (500 mg). After cooling, [Pd(nbd)Cl2] (13.5 mg, 0.050 mmol, 0.05 equiv), followed by chloroform (2 mL, ACS reagent grade, stabilized with amylenes) and then (−)-sparteine (27.6 μL, 28.1 mg, 0.12 mmol, 0.12 equiv) were added. A short tube containing Drierite was attached to the reaction tube. The reaction was stirred vigorously at 23°C for 15 min. Finely powdered Cs2CO3 (130 mg, 0.40 mmol, 0.40 equiv) was added, followed by a solution of (±)-6 (141 μL, 152 mg, 1.0 mmol, 1.0 equiv) and tridecane (73.2 μL, 55.3 mg, 0.30 mmol, 0.30 equiv) in chloroform (2 mL). The reaction was allowed to proceed under an ambient air atmosphere at 23 °C. Aliquots were filtered through a small plug of silica gel (Et2O eluent), evaporated, and analyzed by GC for conversion and chiral HPLC for alcohol ee. Purification of 7 and (−)-6 was accomplished by direct chromatography of the crude reaction mixture.
Supplementary Material
Figure 1.

Ligands evaluated in Table 1.
Acknowledgments
The authors are grateful to the NDSEG (predoctoral fellowship to D.C.E.), the NSF (predoctoral fellowships to D.C.E. and E.M.F.), the University of California TRDRP (predoctoral fellowship to J.T.B.), Bristol-Myers Squibb Company (predoctoral fellowship to E.M.F.), the American Chemical Society Division of Organic Chemistry and Bristol-Myers Squibb Foundation (predoctoral fellowship to R.M.T.), Eli Lilly (predoctoral fellowships to D.D.C. and R.M.M.), the NIH-NIGMS (R01 GM65961-01), King Abdullah University of Science and Technology (KAUST, Award No. KUS-I1-006-02), California Institute of Technology, A. P. Sloan Foundation, the Dreyfus Foundation, Research Corporation, Abbott, Amgen, AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Johnson and Johnson, Eli Lilly, Merck, Novartis, Pfizer, and Roche for generous funding.
Footnotes
[Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.a) Hudlicky M. Oxidations in Organic Chemistry, ACS Monograph Series. American Chemical Society; Washington, DC: 1990. [Google Scholar]; b) Tidwell TT. Org React. 1990;39:297–572. [Google Scholar]; c) Trost BM, Fleming I, editors. Comprehensive Organic Synthesis. Pergamon; Oxford, U.K.: 1991. [Google Scholar]; d) Luzzio FA. Org React. 1998;53:1–221. [Google Scholar]
- 2.Larock RC. Comprehensive Organic Transformations. Wiley & Sons; New York: 1999. pp. 1234–1248. [Google Scholar]
- 3.a) Ohkubo K, Hirata K, Yoshinaga K, Okada M. Chem Lett. 1976:183–184. [Google Scholar]; b) Berti C, Perkins MJ. Angew Chem. 1979;91:923–924. [Google Scholar]; Angew Chem Int Ed Engl. 1979;18:864–865. [Google Scholar]; c) Ishii Y, Suzuki K, Ikariya T, Saburi M, Yoshikawa S. J Org Chem. 1986;51:2822–2824. [Google Scholar]; d) Ma Z, Huang Q, Bobbitt JM. J Org Chem. 1993;58:4837–4843. [Google Scholar]; e) Rychnovsky SD, McLernon TL, Rajapakse H. J Org Chem. 1996;61:1194–1195. [Google Scholar]; f) Kashiwagi Y, Yanagisawa Y, Kurashima F, Anzai J-i, Osa T, Bobbitt JM. Chem Commun. 1996:2745–2746. doi: 10.1039/b209871g. [DOI] [PubMed] [Google Scholar]; g) Hashiguchi S, Fujii A, Haack KJ, Matsumura K, Ikariya T, Noyori R. Angew Chem. 1997;109:300–303. [Google Scholar]; Angew Chem Int Ed Engl. 1997;36:288–289. [Google Scholar]; h) Hamada T, Irie R, Mihara J, Hamachi K, Katsuki T. Tetrahedron. 1998;54:10017–10028. [Google Scholar]; i) Nishibayashi Y, Takei I, Uemura S, Hidai M. Organometallics. 1999;18:2291–2293. [Google Scholar]; j) Gross Z, Ini S. Org Lett. 1999;1:2077–2080. [Google Scholar]; k) Kashiwagi Y, Kurashima F, Kikuchi C, Anzai J-i, Osa T, Bobbitt JM. Tetrahedron Lett. 1999;40:6469–6472. [Google Scholar]; l) Masutani K, Uchida T, Irie R, Katsuki T. Tetrahedron Lett. 2000;41:5119–5123. [Google Scholar]; m) Kuroboshi M, Yoshihisa H, Cortona MN, Kawakami Y, Gao Z, Tanaka H. Tetrahedron Lett. 2000;41:8131–8135. [Google Scholar]
- 4.Subsequent to our initial report in this area, a number of further studies were reported, see: Shimizu H, Nakata K, Katsuki T. Chem Lett. 2002:1080–1081.Sun W, Wang H, Xia C, Li J, Zhao P. Angew Chem. 2003;115:1072–1074. doi: 10.1002/anie.200390268.Angew Chem Int Ed. 2003;42:1042–1044. doi: 10.1002/anie.200390268.Nishibayashi Y, Yamauchi A, Onodera G, Uemura S. J Org Chem. 2003;68:5875–5880. doi: 10.1021/jo0345087.Graetz B, Rychnovsky S, Leu WH, Farmer P, Lin R. Tetrahedron: Asymmetry. 2005;16:3584–3598.Radosevich AT, Musich C, Toste FD. J Am Chem Soc. 2005;127:1090–1091. doi: 10.1021/ja0433424.Li Z, Tang ZH, Hu XX, Xia CG. Chem Eur J. 2005;11:1210–1216. doi: 10.1002/chem.200400818.Weng SS, Shen MW, Kao JQ, Munot YS, Chen CT. Proc Natl Acad Sci USA. 2006;103:3522–3527. doi: 10.1073/pnas.0511021103.Pawar VD, Bettigeri S, Weng SS, Kao JQ, Chen CT. J Am Chem Soc. 2006;128:6308–6309. doi: 10.1021/ja060639o.Li YY, Zhang XQ, Dong ZR, Shen WY, Chen G, Gao JX. Org Lett. 2006;8:5565–5567. doi: 10.1021/ol062244f.Sun W, Wu X, Xia C. Helv Chim Acta. 2007;90:623–626.Chen T, Jiang JJ, Xu Q, Shi M. Org Lett. 2007;9:865–868. doi: 10.1021/ol063061w.Kantam ML, Ramani T, Chakrapani L, Choudary BM. J Mol Catal A: Chem. 2007;274:11–15.Kureshy RI, Ahmad I, Pathak K, Khan N-uH, Abdi SHR, Prathap JK, Jasra RV. Chirality. 2007;19:352–357. doi: 10.1002/chir.20387.Nakamura Y, Egami H, Matsumoto K, Uchida T, Katsuki T. Tetrahedron. 2007;63:6383–6387.Pathak K, Ahmad I, Abdi SHR, Kureshy RI, Khan N-uH, Jasra RV. J Mol Catal A: Chem. 2007;274:120–126.Onomura O, Arimoto H, Matsumura Y, Demizu Y. Tetrahedron Lett. 2007;48:8668–8672.Arita S, Koike T, Kayaki Y, Ikariya T. Angew Chem. 2008;120:2481–2483. doi: 10.1002/anie.200705875.Angew Chem Int Ed. 2008;47:2447–2449. doi: 10.1002/anie.200705875.Tomizawa M, Shibuya M, Iwabuchi Y. Org Lett. 2009;11:1829–1831. doi: 10.1021/ol900441f.
- 5.Stoltz BM. Chem Lett. 2004;33:362–367. [Google Scholar]
- 6.a) Johnson RA, Sharpless KB. In: Catalytic Asymmetric Synthesis. 2. Ojima I, editor. Chapter 6A. Wiley & Sons; New York: 2000. pp. 231–280. [Google Scholar]; b) Wen X, Ojima I. In: Catalytic Asymmetric Synthesis. 2. Ojima I, editor. Chapter 6A. Wiley & Sons; New York: 2000. pp. 281–286. [Google Scholar]; c) Katsuki T. In: Catalytic Asymmetric Synthesis. 2. Ojima I, editor. Chapter 6B. Wiley & Sons; New York: 2000. pp. 287–326. [Google Scholar]; d) Kagan HB. In: Catalytic Asymmetric Synthesis. 2. Ojima I, editor. Chapter 6C. Wiley & Sons; New York: 2000. pp. 327–356. [Google Scholar]; e) Johnson RA, Sharpless KB. In: Catalytic Asymmetric Synthesis. 2. Ojima I, editor. Chapter 6D. Wiley & Sons; New York: 2000. pp. 357–398. [Google Scholar]; f) Bolm C, Hildebrand JP, Muñiz K. In: Catalytic Asymmetric Synthesis. 2. Ojima I, editor. Chapter 6E. Wiley & Sons; New York: 2000. pp. 399–428. [Google Scholar]; g) Katsuki T, editor. Practical Approach in Chemistry Series. Oxford Press; New York: 2001. Asymmetric Oxidation Reactions. [Google Scholar]
- 7.Mohr JT, Ebner DC, Stoltz BM. Org Biomol Chem. 2007;5:3571–3576. doi: 10.1039/b711159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For discussions on kinetic resolution, see: Kagan HB, Fiaud JC. In: Topics in Stereochemistry. Eliel EL, editor. Vol. 18. Wiley & Sons; New York: 1988. pp. 249–330.Keith JM, Larrow JF, Jacobsen EN. Adv Synth Catal. 2001;343:5–26.Vedejs E, Jure M. Angew Chem. 2005;117:4040–4069. doi: 10.1002/anie.200460842.Angew Chem Int Ed. 2005;44:3974–4001. doi: 10.1002/anie.200460842.
- 9.a) Trost BM, Van Vranken DL. Chem Rev. 1996;96:395–422. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; b) Sodeoka M, Shibasaki M. Pure Appl Chem. 1998;70:411–414. [Google Scholar]; c) Hayashi T. J Organomet Chem. 1999;576:195–202. [Google Scholar]; d) Helmchen G. J Organomet Chem. 1999;576:203–214. [Google Scholar]; e) Trost BM. Chem Pharm Bull. 2002;50:1–14. doi: 10.1248/cpb.50.1. [DOI] [PubMed] [Google Scholar]; f) RajanBabu TV. Chem Rev. 2003;103:2845–2860. doi: 10.1021/cr020040g. [DOI] [PubMed] [Google Scholar]; g) Graening T, Schmalz H-G. Angew Chem. 2003;115:2684–2688. doi: 10.1002/anie.200301644. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:2580–2584. doi: 10.1002/anie.200301644. [DOI] [PubMed] [Google Scholar]; h) Dounay AB, Overman LE. Chem Rev. 2003;103:2945–2963. doi: 10.1021/cr020039h. [DOI] [PubMed] [Google Scholar]; i) Tietze LF, Ila H, Bell HP. Chem Rev. 2004;104:3453–3516. doi: 10.1021/cr030700x. [DOI] [PubMed] [Google Scholar]; j) Trost BM. J Org Chem. 2004;69:5813–5837. doi: 10.1021/jo0491004. [DOI] [PubMed] [Google Scholar]; k) Sodeoka M, Hamashima Y. Bull Chem Soc Jpn. 2005;78:941–956. [Google Scholar]; l) Hii KK. Pure Appl Chem. 2006;78:341–349. [Google Scholar]; m) Sodeoka M, Hamashima Y. Pure Appl Chem. 2006;78:477–494. [Google Scholar]; n) Hamashima Y, Sodeoka M. Synlett. 2006:1467–1478. [Google Scholar]
- 10.a) Lloyd WG. J Org Chem. 1967;32:2816–2819. [Google Scholar]; b) Blackburn TF, Schwartz J. J Chem Soc, Chem Commun. 1977:157–158. [Google Scholar]; c) Gómez-Bengoa E, Noheda P, Echavarren AM. Tetrahedron Lett. 1994;35:7097–7098. [Google Scholar]; d) Aït-Mohand S, Hénin F, Muzart J. Tetrahedron Lett. 1995;36:2473–2476. [Google Scholar]; e) Peterson KP, Larock RC. J Org Chem. 1998;63:3185–3189. [Google Scholar]; f) ten Brink G-J, Arends IWCE, Sheldon RA. Science. 2000;287:1636–1639. doi: 10.1126/science.287.5458.1636. [DOI] [PubMed] [Google Scholar]; g) Hallman K, Moberg C. Adv Synth Catal. 2001;343:260–263. [Google Scholar]; h) Sheldon RA, Arends IWCE, ten Brink G-J, Dijksman A. Acc Chem Res. 2002;35:774–781. doi: 10.1021/ar010075n. [DOI] [PubMed] [Google Scholar]; i) Schultz MJ, Park CC, Sigman MS. Chem Commun. 2002:3034–3035. doi: 10.1039/b209344h. [DOI] [PubMed] [Google Scholar]; j) ten Brink G-J, Arends IWCE, Hoogenraad M, Verspui G, Sheldon RA. Adv Synth Catal. 2003;345:497–505. [Google Scholar]; k) Jensen DR, Schultz MJ, Mueller JA, Sigman MS. Angew Chem. 2003;115:3940–3943. doi: 10.1002/anie.200351997. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:3810–3813. doi: 10.1002/anie.200351997. [DOI] [PubMed] [Google Scholar]; l) ten Brink G-J, Arends IWCE, Hoogenraad M, Verspui G, Sheldon RA. Adv Synth Catal. 2003;345:1341–1352. [Google Scholar]; m) Paavola S, Zetterberg K, Privalov T, Csöregh I, Moberg C. Adv Synth Catal. 2004;346:237–244. [Google Scholar]; n) Iwasawa T, Tokunaga M, Obora Y, Tsuji Y. J Am Chem Soc. 2004;126:6554–6555. doi: 10.1021/ja031936l. [DOI] [PubMed] [Google Scholar]
- 11.For reviews of palladium-catalyzed aerobic oxidations, see: Muzart J. Tetrahedron. 2003;59:5789–5816.Stahl SS. Angew Chem. 2004;116:3480–3501.Angew Chem Int Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630.Sigman MS, Schultz MJ. Org Biomol Chem. 2004;2:2551–2554. doi: 10.1039/B409127M.d) Ref [5].
- 12.a) Nishimura T, Onoue T, Ohe K, Uemura S. Tetrahedron Lett. 1998;39:6011–6014. [Google Scholar]; b) Nishimura T, Onoue T, Ohe K, Uemura S. J Org Chem. 1999;64:6750–6755. doi: 10.1021/jo9906734. [DOI] [PubMed] [Google Scholar]; c) Nishimura T, Maeda Y, Kakiuchi N, Uemura S. J Chem Soc, Perkin Trans 1. 2000:4301–4305. [Google Scholar]; d) Nishimura T, Uemura S. Synlett. 2004:201–216. [Google Scholar]
- 13.a) Steinhoff BA, Stahl SS. Org Lett. 2002;4:4179–4181. doi: 10.1021/ol026988e. [DOI] [PubMed] [Google Scholar]; b) Steinhoff BA, Guzei IA, Stahl SS. J Am Chem Soc. 2004;126:11268–11278. doi: 10.1021/ja049962m. [DOI] [PubMed] [Google Scholar]; c) Privalov T, Linde C, Zetterberg K, Moberg C. Organometallics. 2005;24:885–893. [Google Scholar]; d) Schultz MJ, Adler RS, Zierkiewicz W, Privalov T, Sigman MS. J Am Chem Soc. 2005;127:8499–8507. doi: 10.1021/ja050949r. [DOI] [PubMed] [Google Scholar]; e) Steinhoff BA, King AE, Stahl SS. J Org Chem. 2006;71:1861–1868. doi: 10.1021/jo052192s. [DOI] [PubMed] [Google Scholar]
- 14.For mechanistic studies of other non-enantioselective palladium-catalyzed alcohol oxidations, see: Stahl SS, Thorman JL, Nelson RC, Kozee MA. J Am Chem Soc. 2001;123:7188–7189. doi: 10.1021/ja015683c.Steinhoff BA, Fix SR, Stahl SS. J Am Chem Soc. 2002;124:766–767. doi: 10.1021/ja016806w.ten Brink G-J, Arends IWCE, Sheldon RA. Adv Synth Catal. 2002;344:355–369.Mueller JA, Goller CP, Sigman MS. J Am Chem Soc. 2004;126:9724–9734. doi: 10.1021/ja047794s.Konnick MM, Guzei IA, Stahl SS. J Am Chem Soc. 2004;126:10212–10213. doi: 10.1021/ja046884u.Landis CR, Morales CM, Stahl SS. J Am Chem Soc. 2004;126:16302–16303. doi: 10.1021/ja044674b.Zierkiewicz W, Privalov T. Organometallics. 2005;24:6019–6028.Arends IWCE, ten Brink G-J, Sheldon RA. J Mol Catal A: Chem. 2006;251:246–254.Steinhoff BA, Stahl SS. J Am Chem Soc. 2006;128:4348–4355. doi: 10.1021/ja057914b.Nielsen RJ, Goddard WA., III J Am Chem Soc. 2006;128:9651–9660. doi: 10.1021/ja060915z.
- 15.For enantioselective palladium-catalyzed alcohol oxidation mechanistic studies, see: Mueller JA, Jensen DR, Sigman MS. J Am Chem Soc. 2002;124:8202–8203. doi: 10.1021/ja026553m.Mueller JA, Sigman MS. J Am Chem Soc. 2003;125:7005–7013. doi: 10.1021/ja034262n.Trend RM, Stoltz BM. J Am Chem Soc. 2004;126:4482–4483. doi: 10.1021/ja039551q.Nielsen RJ, Keith JM, Stoltz BM, Goddard WA., III J Am Chem Soc. 2004;126:7967–7974. doi: 10.1021/ja031911m.Keith JM, Nielsen RJ, Oxgaard J, Goddard WA., III J Am Chem Soc. 2005;127:13172–13179. doi: 10.1021/ja043094b.Mueller JA, Cowell A, Chandler BD, Sigman MS. J Am Chem Soc. 2005;127:14817–14824. doi: 10.1021/ja053195p.Popp BV, Stahl SS.J Am Chem Soc 20071294410–4422.17371024Keith JM, Goddard WA, III, Oxgaard J. J Am Chem Soc. 2007;129:10361–10369. doi: 10.1021/ja070462d.
- 16.Ferreira EM, Stoltz BM. J Am Chem Soc. 2001;123:7725–7726. doi: 10.1021/ja015791z. [DOI] [PubMed] [Google Scholar]
- 17.Concurrent with our publication, a related system was reported, see: Jensen DR, Pugsley JS, Sigman MS. J Am Chem Soc. 2001;123:7475–7476. doi: 10.1021/ja015827n.Jensen DR, Sigman MS. Org Lett. 2003;5:63–65. doi: 10.1021/ol027190y.Mandal SK, Jensen DR, Pugsley JS, Sigman MS. J Org Chem. 2003;68:4600–4603. doi: 10.1021/jo0269161.Mandal SK, Sigman MS. J Org Chem. 2003;68:7535–7537. doi: 10.1021/jo034717r.Sigman MS, Jensen DR. Acc Chem Res. 2006;39:221–229. doi: 10.1021/ar040243m.
- 18.The selectivity factor (s) was determined using the equation s = kfast/kslow = ln[(1−C)(1−ee)]/ln[(1−C)(1+ee)], where C is conversion and ee is enantiomeric excess of recovered alcohol, see ref [8a].
- 19.Recently, a similar palladium-catalyzed system utilizing a different ligand for enantioselective alcohol oxidation has been developed, see ref [4k].
- 20.Bagdanoff JT, Ferreira EM, Stoltz BM. Org Lett. 2003;5:835–837. doi: 10.1021/ol027463p. [DOI] [PubMed] [Google Scholar]
- 21.Complexes of [PdCl2] with a variety of monodentate amines do not catalyze alcohol oxidation. For details, see ref [12b] and Supporting Information.
- 22.The beneficial role of carbonates as exogenous bases in the kinetic resolution was subsequently reported by Sigman as well, see ref [17d].
- 23.A significant effect of cesium carbonate structure and particle size has been found for palladium-catalyzed aryl halide aminations in toluene, consistent with a heterogeneous process, see: Meyers C, Maes BUW, Loones KTJ, Bal G, Lemiere GLF, Dommisse RA. J Org Chem. 2004;69:6010–6017. doi: 10.1021/jo049774e.
- 24.Crow WD. Aust J Chem. 1959;12:474–482. [Google Scholar]
- 25.Lide DR, Frederikse HPR, editors. CRC Handbook of Chemistry and Physics. 76. Chemical Rubber Company; New York: 1995. p. 43. Section 8. [Google Scholar]
- 26.Addition of sparteine HCl inhibits alcohol oxidation, see ref [15a].
- 27.Sigman has proposed that excess chloride ion in solution may inhibit displacement of chloride by alcohol in [Pd(sparteine)Cl2]. Thus, formation of insoluble CsCl may sequester excess chloride and promote alcohol coordination. For details, see ref [15f].
- 28.Lambert JB, Zhao Y, Emblidge RW, Salvador LA, Liu X, So JH, Chelius EC. Acc Chem Res. 1999;32:183–190. [Google Scholar]
- 29.tert-Butyl alcohol was found to be a competent solvent for this oxidation, see ref [17c].
- 30.Resolutions conducted under an atmosphere of air instead of O2 displayed substantially lower reactivity and selectivity.
- 31.An alternate explanation involves the formation of trace amounts of CsOt-Bu in situ. However, reactions conducted with CsOt-Bu as base led to oxidation but no kinetic resolution, see Supporting Information for details.
- 32.Palladium carbonate complex 13 was also observed on prolonged exposure of 5 to cesium carbonate in chloroform at 23 °C.
- 33.Sinnott M, editor. Comprehensive Biological Catalysis. 1–3. Academic Press; San Diego, CA: 1998. [Google Scholar]
- 34.For a recent review, see: Taylor MS, Jacobsen EN. Angew Chem. 2006;118:1550–1573.Angew Chem Int Ed. 2006;45:1520–1543. doi: 10.1002/anie.200503132.
- 35.Bagdanoff JT, Stoltz BM. Angew Chem. 2004;116:357–361. doi: 10.1002/anie.200352444. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:353–357. [Google Scholar]
- 36.Goddard has proposed that the solubilization of charged intermediates by dichloromethane may be so great as to nearly completely separate the chloride anion from the palladium complex in the β-hydride elimination transition state, improving the oxidation rate but limiting a key interaction for selectivity, see ref [15d].
- 37.Fischer F, Pfleiderer G. Z Anorg Allg Chem. 1922;124:61–69. [Google Scholar]
- 38.For dielectric constant tables, see: CRC Handbook of Chemistry and Physics, 76th ed. (Eds.: D. R. Lide, H. P. R. Frederikse), Chemical Rubber Company, New York, 1995, Section 6, pp. 159–192.
- 39.The IR spectrum of CHCl3 was also investigated. However, the C-H stretch was not well resolved. CDCl3 performs identically to CHCl3 as reaction solvent.
- 40.For a discussion of the hydrogen bonding of chloroform and its effect on IR vibrational frequencies, see: Green RD. Hydrogen Bonding by C–H Groups. John Wiley & Sons; New York: 1974.
- 41.A molecule of chloroform is also within hydrogen bonding distance in the solid-state structure of 5. CCDC 203513 for 5 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 42.While we have never experienced an accident, reactions conducted at elevated temperatures in flammable solvents should be conducted with appropriate caution in a fume hood.
- 43.For a brief discussion of the substrate scope of the oxidative kinetic resolution, see: Stoltz BM, Ebner DC. In: Handbook of C–H Transformation. 2. Dyker G, editor. Wiley-VCH; New York: 2005. pp. 393–401.
- 44.Caspi DD, Ebner DC, Bagdanoff JT, Stoltz BM. Adv Synth Catal. 2004;346:185–189. [Google Scholar]
- 45.Ruel FS, Braun MP, Johnson WS. Collect. X. Wiley & Sons; New York: 2004. Organic Syntheses; pp. 467–471. [Google Scholar]
- 46.Luche JL, Gemal AL. J Chem Soc, Chem Commun. 1978:601–602. [Google Scholar]
- 47.Ebner DC, Novák Z, Stoltz BM. Synlett. 2006:3533–3539. [Google Scholar]
- 48.For example with conditions C, (±)-2-methylcyclopent-2-enol and (±)-2-methylcyclohex-2-enol ((±)-26) are oxidized with selectivity factors of 3.2 and 5.3, respectively.
- 49.Low reactivity was also observed in a related system, see: Sayyed IA, Kumar NSCR, Sudalai A. Indian J Chem, Sect B. 2005;44B:1533–1535.
- 50.For palladium-catalyzed alcohol oxidation conditions that allow the resolution of some saturated alkyl alcohols, see ref [17c].
- 51.For recent applications of our kinetic resolution to natural product total synthesis, see: Tambar UK, Ebner DC, Stoltz BM. J Am Chem Soc. 2006;128:11752–11753. doi: 10.1021/ja0651815.Krishnan S, Bagdanoff JT, Ebner DC, Ramtohul YK, Tambar UK, Stoltz BM. J Am Chem Soc. 2008;130:13745–13754. doi: 10.1021/ja804738b.
- 52.Related oxidative kinetic resolutions en route to pharmaceutical agents have been reported, see: Ali IS, Sudalai A. Tetrahedron Lett. 2002;43:5435–5436.Thakur VV, Sudalai A. Indian J Chem, Sect B. 2005;44B:557–562.
- 53.a) Trend RM, Ramtohul YK, Ferreira EM, Stoltz BM. Angew Chem. 2003;115:2998–3001. doi: 10.1002/anie.200351196. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:2892–2895. doi: 10.1002/anie.200351196. [DOI] [PubMed] [Google Scholar]; b) Trend RM, Ramtohul YK, Stoltz BM. J Am Chem Soc. 2005;127:17778–17788. doi: 10.1021/ja055534k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rao MR, Faulkner JD. J Nat Prod. 2002;65:1201–1203. doi: 10.1021/np020040b. [DOI] [PubMed] [Google Scholar]
- 55.For discussions on synthetic approaches based on bidirectional chain synthesis, see: Poss CS, Schreiber SL. Acc Chem Res. 1994;27:9–17.Schreiber SL, Goulet MT, Schulte G. J Am Chem Soc. 1987;109:4718–4720.
- 56.For examples of bidirectional synthesis/terminus differentiation in natural products synthesis, see: Harada T, Kagamihara Y, Tanaka S, Sakamoto K, Oku A. J Org Chem. 1992;57:1637–1639.Poss CS, Rychnovsky SD, Schreiber SL. J Am Chem Soc. 1993;115:3360–3361.Schreiber SL, Sammakia T, Uehling DE. J Org Chem. 1989;54:15–16.Ikemoto N, Schreiber SL. J Am Chem Soc. 1990;112:9657–9659.
- 57.a) Reingold ID, DiNardo LJ. J Org Chem. 1982;47:3544–3545. [Google Scholar]; b) Reingold ID, Kwong KS, Kahr BE, Menard M, Cummings G, Kowalski JA. Synth Commun. 1993;23:1463–1466. [Google Scholar]
- 58.Bäckvall JE, Vågberg JO. J Org Chem. 1988;53:5695–5699. [Google Scholar]
- 59.Johnson CR, Golebiowski A, McGill TK, Steensma DH. Tetrahedron Lett. 1991;32:2597–2600. [Google Scholar]
- 60.Ebner DC, Trend RM, Genet C, McGrath MJ, O’Brien P, Stoltz BM. Angew Chem. 2008;120:6467–6470. doi: 10.1002/anie.200801865. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:6367–6370. doi: 10.1002/anie.200801865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.