

Abstract
The Entamoeba histolytica upstream regulatory element 3-binding protein (URE3-BP) is a transcription factor that binds DNA in a Ca2+-inhibitable manner. The protein is located in both the nucleus and the cytoplasm but has also been found to be enriched in the plasma membrane of amebic trophozoites. We investigated the reason for the unusual localization of URE3-BP at the amebic plasma membrane. Here we identify and characterize a 22-kDa Ca2+-dependent binding partner of URE3-BP, EhC2A, a novel member of the C2-domain superfamily. Immunoprecipitations of URE3-BP and EhC2A showed that the proteins interact and that such interaction was enhanced in the presence of Ca2+. Recombinant and native EhC2A bound phospholipid liposomes in a Ca2+-dependent manner, with half-maximal binding occurring at 3.4 μM free Ca2+. A direct interaction between EhC2A and URE3-BP was demonstrated by the ability of recombinant EhC2A to recruit recombinant URE3-BP to phospholipid liposomes in a Ca2+-dependent manner. URE3-BP and EhC2A were observed to translocate to the amebic plasma membrane upon an increase in the intracellular Ca2+ concentration of trophozoites, as revealed by subcellular fractionation and immunofluorescent staining. Short hairpin RNA-mediated knockdown of EhC2A protein expression significantly modulated the mRNA levels of URE3-BP-regulated transcripts. Based on these results, we propose a model for EhC2A-mediated regulation of the transcriptional activities of URE3-BP via Ca2+-dependent anchoring of the transcription factor to the amebic plasma membrane.
The parasite Entamoeba histolytica is the causative agent of amebiasis, estimated to be the second leading protozoan cause of morbidity and mortality in humans worldwide (2). The molecular mechanisms that regulate the outcome of infection have yet to be elucidated; however, we hypothesize that transcriptional regulation may play an essential role in determining such outcomes.
The calcium ion (Ca2+) is a universal regulator of many cellular processes in eukaryotic cells, including cell signaling events, gene expression, and cell death. In Entamoeba, Ca2+ has been implicated in many aspects of the parasite's pathophysiology, including cell motility (35), adhesion to fibronectin (6), cytolytic activity (36), transcriptional regulation (16–18), and growth and developmental stage transition (25, 26). In eukaryotes, these processes are generally mediated by proteins possessing Ca2+-binding motifs, such as EF-hands, endonexin folds, and C2 domains, which, in response to changes in intracellular Ca2+ levels, are responsible for the transduction of signaling events, several of which have been identified in Entamoeba (for a review, see reference 4).
The upstream regulatory element 3-binding protein (URE3-BP) is an E. histolytica Ca2+-regulated transcription factor first identified for its roles in the control of the expression of ferredoxin (fdx1) and the heavy subunit of the galactose/N-acetylgalactosamine (Gal/GalNAc)-inhibitable lectin (hgl5). The protein was isolated by a yeast one-hybrid screen using the cognate URE3 DNA motif (17), though the amino acid sequence of URE3-BP revealed no DNA-binding motifs. The protein did, however, possess two canonical and one noncanonical EF-hand motifs. URE3-BP localized to both the nucleus and the cytoplasm of trophozoites and was also found to be enriched in the plasma membrane. Ca2+ induced the release of binding of URE3-BP from the URE3 motif in vitro and in vivo (18). Mutation of the second EF-hand motif at residues critical for Ca2+ coordination abrogated this effect and allowed for the genome-wide identification of targets of URE3-BP in parasites overexpressing the mutant protein (16).
In this study, we explored the reason for the unusual plasma membrane localization of the URE3-BP transcription factor. We discovered a novel URE3-BP binding partner, EhC2A, containing an N-terminal Ca2+-binding C2 domain and a proline-rich C-terminal region. C2 domains are ∼120-residue motifs that confer phospholipid-binding activity on the majority of proteins which possess them (reviewed in references 9, 22, and 37). EhC2A coimmunoprecipitated with URE3-BP in a Ca2+-dependent manner and was shown to be a high-affinity Ca2+-dependent membrane binding protein. In addition, recombinant EhC2A recruited recombinant URE3-BP to phospholipid vesicles in vitro in a Ca2+-dependent manner, indicative of a direct interaction between the binding partners. In amebic trophozoites, EhC2A and URE3-BP translocated to the plasma membrane upon exposure to increased intracellular Ca2+ concentrations, suggesting an important role for EhC2A as a regulator of URE3-BP cellular localization.
MATERIALS AND METHODS
Cultivation and transfection of E. histolytica trophozoites.
E. histolytica strain HM1:IMSS trophozoites were grown at 37°C in TYI-S-33 medium supplemented with penicillin (100 U/ml) and streptomycin sulfate (100 μg/ml) (Gibco/BRL) (13). In some experiments (as indicated below), cells were incubated in medium containing 5 mM EGTA or 1 mM CaCl2 and 1 μM calcium ionophore A23187 (Sigma) for 1 h at 37°C. Stable transfection of trophozoites with plasmids expressing EhC2A short hairpin RNA (shRNA) comprising nucleotides 363 to 391 [EhC2A (363-391)] EhC2A (363-391 scrambled) control shRNA was achieved using the lipofection technique as previously described (24, 33). Transfectant trophozoites were maintained under selection with hygromycin (30 μg/ml). Hygromycin selection was gradually increased to 90 μg/ml in order to achieve protein knockdown (24).
Construction of expression vectors and recombinant protein purification.
To generate a glutathione S-transferase (GST)-tagged EhC2A fusion protein expression vector, the full-length open reading frame of EhC2A was amplified by PCR from E. histolytica HM1:IMSS genomic DNA using the following primers, which introduced EcoRI and XhoI restriction sites to the DNA fragment: 5′-CCGGAATTCATGAGAATAGAACTTAAAGTTATTGAAGG-3′ and 5′-CCGCTCGAGTTAATAAAATCCTGGTGGTGGCATCATACC-3′. This fragment was then cloned into pGST-Parallel1 using the corresponding sites (38). The His6-tagged EhC2A fusion protein expression vector was generated by using PCR to amplify the full-length open reading frame of EhC2A with the following primers: 5′-ATGAGAATAGAACTTAAAGTTATTGAAGG-3′ and 5′-TTAATAAAATCCTGGTGGTGGCATCATACC-3′. This fragment was then cloned into the PCRT7/NT TOPO expression vector (Invitrogen).
The expression of His6-tagged EhC2A and GST-tagged EhC2A fusion proteins in Escherichia coli BL21(DE3)/pLysS cells (Invitrogen) was induced with 1 mM isopropyl-β-d-thiogalactopyranoside. The GST-tagged EhC2A fusion protein was purified by affinity chromatography using glutathione-Sepharose (Sigma) according to the manufacturer's directions. The His6-tagged EhC2A fusion protein was purified by affinity chromatography using TALON metal affinity resin (Clontech) as specified by the manufacturer. Recombinant URE3-BP was a kind gift from Eric Smith, Taryn Haffner, and Amy Raymond of Emerald BioStructures.
Construction of short hairpin RNA expression vectors.
Construction of the shRNA-expressing vectors was performed as previously described (24). Briefly, the RNA polymerase III promoter of the E. histolytica U6 gene was amplified beginning at position −333 from the transcription start site of the U6 small nuclear RNA gene, and the shRNA-encoding DNA was added by PCR at the transcription start site. The resulting U6 promoter-shRNA constructs were cloned into pGIR310 modified to contain a short polylinker. The shRNAs were designed to have a 29-nucleotide complementary stem with a 9-nucleotide loop. The oligonucleotides used for the construct expressing an shRNA corresponding to nucleotides 363 to 391 of the gene encoding EhC2A were C2AR1 (5′-TCTCTTGAATCATGCCTGGTTGCATTGGTGGAACCATTGGGCCCAATTTTATTTTTCTTTTTATCC-3′) and C2AR2 (5′-TCGATCGCGGCCGCAAAAAATGGTTCCACCAATGCAACCAGGCATGATCTCTTGAA-3′) The oligonucleotides used for the construct expressing a scrambled shRNA control with the same nucleotide composition in a random order were C2ASR1 (5′-TCTCTTGAAATCTGGAACGGTCTGGATTGTCTAGCCTTGGGCCCAATTTTATTTTTCTTTTTATCC-3′) and C2ASR2 (5′-TCGATCGCGGCCGCAAAAAAGGCTAGACAATCCAGACCGTTCCAGATTCTCTTGA A-3′).
Antibodies.
A polyclonal antiserum was raised against EhC2A by immunizing New Zealand White rabbits with purified GST-tagged EhC2A fusion protein (Cocalico Biologicals Co.). For the preabsorption experiments to determine antiserum specificity, EhC2A antiserum (1:2,000) was preabsorbed with 500 μg of GST alone or GST-EhC2A for 6 h at room temperature prior to probing polyvinylidene fluoride (PVDF) membranes. The mouse anti-URE3-BP monoclonal antibodies (MAbs) 4D6 and 3E6 have been previously described (18). The mouse anti-light subunit of the E. histolytica Gal/GalNAc-inhibitable lectin (LGL), MAb 1D4, has also been previously characterized (27). Rabbit polyclonal antibody against actin was purchased from Santa Cruz Biotechnology. Goat anti-mouse IgG and goat anti-rabbit IgG were purchased from Sigma. Goat anti-rabbit IgG Alexa Fluor 488 and goat anti-mouse IgG Alexa Fluor 546 were purchased from Invitrogen.
Preparation of cellular extracts, immunoprecipitation, and Western blotting.
E. histolytica trophozoites were washed once with phosphate-buffered saline (PBS) and lysed in lysis buffer (150 mM NaCl, 50 mM Tris, pH 8.3, 1% Nonidet P-40, 1.8 mM E-64, supplemented with protease inhibitor cocktail I [Sigma] according to the manufacturer's instructions). The protein concentrations of extracts were measured using a bicinchoninic acid (BCA) protein assay kit according to the manufacturer's protocol (Pierce Biotechnology), and samples were normalized for protein concentration. To detect EhC2A, whole-cell lysates (50 μg total protein/well) or recombinant His6-tagged EhC2A was separated by 15% polyacrylamide gel electrophoresis and transferred to PVDF membranes.
For immunoprecipitations, lysates (supplemented with 5 mM EGTA, 5 mM EDTA or 5 mM CaCl2, as indicated in Fig. 1) were preincubated for 1 h with 50 μl of protein G-Dynabeads (Invitrogen) to reduce nonspecific protein binding. For immunoprecipitations to detect URE3-BP-interacting proteins, lysates were incubated overnight at 4°C with 50 μl of protein G-beads to which URE3-BP MAb 4D6 or 3E6 or control mouse IgG had been covalently attached as specified by the manufacturer. For immunoprecipitations to determine the URE3-BP–EhC2A interaction, lysates supplemented with 5 mM EDTA or 0.1 mM CaCl2 (as indicated in Fig. 3) were incubated at 4°C for 3 h with the antibodies indicated below and further incubated with protein G-beads for 1 h. The beads were magnetically concentrated as directed by manufacturer and washed two times with lysis buffer containing 0.5% Nonidet P-40 and once with PBS. Proteins bound to the beads were eluted in Laemmli sample buffer and boiled for 5 min. The proteins were separated by 15% polyacrylamide gel electrophoresis and transferred to PVDF membranes, which were blocked with 5% milk. For far-Western blot analysis, the membranes were overlaid with amebic whole-cell lysate at a final concentration of 100 μg/ml, enriched with 5 mM EDTA or 5 mM CaCl2. Membranes were stained using URE3-BP MAb 4D6 (5 μg/ml), preimmune serum, or EhC2A anti-serum (1:500) and horseradish peroxidase-conjugated goat anti-mouse or horseradish peroxidase-conjugated goat anti-rabbit antibody. Pierce ECL Western blotting substrate (Thermo Scientific) was used for visualization.
Protein band sequencing and analysis.
Protein sequencing was performed at the Keck Center for Biomedical Research at the University of Virginia. The polyacrylamide gel piece was transferred to a siliconized tube and washed and destained in 200 μl of 50% methanol overnight. The gel pieces were dehydrated in acetonitrile, rehydrated in 30 μl of 10 mM dithiothreitol (DTT) in 0.1 M ammonium bicarbonate, and reduced at room temperature for 30 min. The DTT solution was removed, and the sample alkylated in 30 μl of 50 mM iodoacetamide in 0.1 M ammonium bicarbonate at room temperature for 30 min. The reagent was removed, and the gel pieces dehydrated in 100 μl of acetonitrile. The acetonitrile was removed, and the gel pieces rehydrated in 100 μl of 0.1 M ammonium bicarbonate. The gel pieces were dehydrated in 100 μl of acetonitrile, the acetonitrile removed, and the pieces completely dried by vacuum centrifugation. The gel pieces were rehydrated in 20 ng/μl trypsin in 50 mM ammonium bicarbonate on ice for 10 min. Any excess trypsin solution was removed, and 20 μl of 50 mM ammonium bicarbonate added. The sample was digested overnight at 37°C, and the resulting peptides were extracted from the polyacrylamide in two 30-μl aliquots of 50% acetonitrile–5% formic acid. These extracts were combined and evaporated to 25 μl for mass spectrometry (MS) analysis.
The liquid chromatography (LC)-MS system consisted of a Finnigan LCQ ion trap mass spectrometer system with a Protana nanospray ion source interfaced to a self-packed 8-cm by 75-μm (inner diameter) Phenomenex Jupiter 10-μm C18 reversed-phase capillary column. Volumes of 0.5 to 5 μl of the extract were injected, and the peptides eluted from the column by an acetonitrile–0.1 M acetic acid gradient at a flow rate of 0.25 μl/min. The nanospray ion source was operated at 2.8 kV. The digest was analyzed using the double play capability of the instrument, acquiring full-scan mass spectra to determine peptide molecular weights and product ion spectra to determine amino acid sequences in sequential scans. This mode of analysis produces approximately 400 collision-activated dissociation (CAD) spectra of ions ranging in abundance over several orders of magnitude, though not all CAD spectra are derived from peptides. The data were analyzed by database searching using the Sequest search algorithm (14). Peptides that were not matched by this algorithm were interpreted manually and searched versus the expressed sequence tag databases using the Sequest algorithm.
Liposome binding assays.
For quantitative liposome binding assays, 3H-labeled liposomes were prepared by drying a mixture of 0.5 mg of l-α-phosphatidylserine, 1.5 mg of l-α-phosphatidylcholine (both at 10 mg/ml in chloroform; Avanti Polar Lipids), and 20 μl of cholesterol [7-3H(N)] (1 mCi/ml with specific activity of 20 Ci/mmol; American Radiolabeled Chemicals) under a nitrogen gas flow, followed by overnight vacuum drying to remove residual chloroform. Dried phospholipids were resuspended in 10 ml of buffer A (50 mM HEPES, pH 7.2, 0.1 M NaCl) and sonicated for 30 s using a probe sonicator at 30 kHz. The liposome suspension was centrifuged at 10,000 × g for 10 min, washed two times, stored in the same buffer at 4°C, and used within a week.
Quantitative liposome binding assays were performed using 50 μg of GST-tagged EhC2A bound to 20 μl (wet volume) of MagneGST beads (Promega). Beads were prewashed with buffer A and resuspended in 0.1 ml of buffer A containing 3H-labeled liposomes and the respective additions of CaCl2 and EGTA to attain the indicated level of free Ca2+ as calculated using the WEBMAXC extended version (available at http://maxchelator/stanford.edu). The mixture was incubated at room temperature for 30 min. Beads were magnetically concentrated as specified by the manufacturer and washed three times with 1 ml of the same buffered Ca2+ solution without liposomes. Liposome binding was then quantified by liquid scintillation counting of the beads. Curve fitting and binding constant calculations were performed using Origin software (OriginLab Corp.). All Ca2+-containing buffers were prepared using a 0.1 M CaCl2 standard solution (Thermo Electron Corp.). All experimental results were confirmed with at least three independent experiments. Statistical analysis was performed using Student's t test.
Liposome sedimentation assays were performed essentially as previously described (40). Briefly, liposomes were prepared as described above without cholesterol [7-3H(N)]. Amebic trophozoites were lysed by pulse sonication in buffer A, followed by centrifugation at 10,000 × g for 10 min. Fifty micrograms of the resulting supernatant (S10) was resuspended with 200 μg of liposomes in 50 μl of buffer A supplemented with 5 mM EGTA or 50 μM CaCl2 for 30 min at room temperature. Sedimentation controls were performed in the absence of liposomes. Mixtures were centrifuged at 10,000 × g for 10 min, and lipid pellets analyzed by Western blotting for EhC2A.
For liposome recruitment assays, 0.5 μg of recombinant URE3-BP and, separately, 0.1 μg, 0.25 μg, or 0.5 μg of GST or GST-tagged EhC2A in 10 μl of buffer B (120 mM KCl, 20 mM histidine, pH 6.4) containing either 5 mM EGTA or 10 μM CaCl2 were incubated at room temperature for 30 min. The solutions were spun at 10,000 × g to remove background protein sedimentation. Precleared recombinant URE3-BP and GST or GST-EhC2A were resuspended with 200 μg of liposomes and incubated at room temperature for 1 h. Sedimentation controls were performed in the absence of liposomes. Mixtures were centrifuged at 10,000 × g for 10 min, and liposome-containing pellets and supernatants analyzed by Western blotting for EhC2A and URE3-BP.
Subcellular fractionation.
Subcellular fractionation was performed by lysing the amebic trophozoites by pulse sonication in 50 mM Tris-HCl, pH 7.4, 10 mM NaCl, 1 mM DTT, and protease inhibitors (as described above), followed by centrifugation at 100,000 × g (Beckman MLA-130 rotor) for 60 min at 4°C, producing particulate (P100) or soluble (S100) fractions. Pellets were resuspended in lysis buffer with 1% Triton X-100. Samples were analyzed by Western blotting for URE3-BP, EhC2A, LGL, and actin content.
Confocal microscopy.
E. histolytica trophozoites were bound to glass coverslips for 1 h at 37°C in TYI-S-33 medium. Where indicated below, adherent cells were then incubated for 1 h with medium supplemented with 5 mM EGTA or 1 mM CaCl2 and 1 μM calcium ionophore A23187. Cells were then immediately fixed in 4% formaldehyde for 30 min at room temperature. Next, cells were permeabilized with 0.1% Triton X-100 in PBS for 2 min. Nonspecific binding was blocked by incubation with 10% horse serum in PBS for 30 min at room temperature. Detection of URE3-BP was performed by incubation with anti-URE3-BP MAb 4D6 diluted at 5 μg/ml in PBS containing 10% horse serum for 1 h. Detection of EhC2A was performed by incubation with anti-EhC2A rabbit polyclonal antibody diluted at 1:500 in PBS containing 10% horse serum for 1 h. Cells were incubated with goat anti-rabbit IgG Alexa Fluor 488 and goat anti-mouse IgG Alexa Fluor 546 for 1 h at room temperature (all at 1:1,000 dilution in 10% horse serum in PBS). The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) before mounting. Cells were viewed with a Zeiss LSM 510 laser scanning confocal microscope using a 40× oil immersion lens and LSM Image Browser software (Zeiss).
RNA isolation and real-time qRT-PCR.
Quantitative real-time reverse transcription-PCR (real-time qRT-PCR) was used to independently measure the mRNA abundance of trophozoites transfected with the EhC2A (363–391) shRNA- and EhC2A (363–391 scrambled) shRNA-expressing plasmids. Approximately 2 × 106 E. histolytica trophozoites were lysed by the addition of 1.0 ml of TRIzol reagent (Invitrogen), and total RNA was isolated according to the manufacturer's directions. RNA that was more than 200 nucleotides in length was isolated from total RNA using an RNeasy kit (Qiagen). On-column DNA digestion using an RNase-free DNase set (Qiagen) was performed to remove contaminating genomic DNA. RNA was retrotranscribed using SuperScript II reverse transcriptase (Invitrogen) and oligo(dT) primers. The cDNA was subjected to 40 amplification cycles with HotStar Taq (Qiagen). All primers used for qRT-PCR were designed to amplify <300-bp fragments of the genes being studied (see Table S2 in the supplemental material). Each oligonucleotide pair was validated against the E. histolytica genome database (http://www.amoebadb.org for specificity, with the exception of actin, which was designed to amplify all actin family members (3). Amplified cDNA was detected using the fluorescent dye SYBR green I (Molecular Probes). Continuous SYBR green I monitoring during amplification was performed using an MJ Research Opticon II DNA Engine (Bio-Rad) according to the manufacturer's recommendations. All amplifications were performed in triplicate, and the resulting fluorescence values averaged. Cycle threshold (CT) values (the cycle number at which fluorescence exceeds the threshold value) were linked to the quantity of initial DNA after calibration of the effectiveness of the amplifying primer pair (19). Statistical analysis was performed using Student's t test (two tailed).
RESULTS
Detection of a Ca2+-dependent URE3-BP-interacting protein.
We were interested in identifying proteins that might be involved in the regulation of the transcriptional activity and cellular localization of URE3-BP. To search for URE3-BP-interacting proteins, URE3-BP was immunoprecipitated with the anti-URE3-BP MAbs 4D6 and 3E6 from amebic lysates enriched (with CaCl2) or depleted (with EGTA or EDTA) of Ca2+. A silver-stained SDS-PAGE gel revealed a protein of 22 kDa that coimmunoprecipitated with URE3-BP in the presence of Ca2+ but not in the presence of the divalent ion chelator EDTA or the Ca2+ chelator EGTA (Fig. 1A), indicating that we had potentially identified a Ca2+-dependent binding partner. Incidentally, immunoprecipitation of URE3-BP was enhanced in the presence of Ca2+ (Fig. 1B).
Fig. 1.

URE3-BP coimmunoprecipitation with a 22-kDa protein is Ca2+ dependent. (A) Trophozoites were lysed in the presence of 5 mM EDTA, 5 mM EGTA, or 5 mM CaCl2 as described in Materials and Methods. Whole-cell extracts were immunoprecipitated (IP) with anti-URE3-BP MAb 4D6 (IgG2B) or 3E6 (IgG2A) covalently coupled to protein G-Dynabeads. Aliquots of immunoprecipitated proteins were separated by polyacrylamide gel electrophoresis using comparable amounts of the relevant samples and analyzed by silver stain. An asterisk indicates a 22-kDa coimmunoprecipitated protein. M, size ladder; α, anti. (B) Relative abundance of URE3-BP in the immunoprecipitants was determined by Western blotting using anti-URE3-BP MAb 4D6. (C) Far-Western blot of proteins immunoprecipitated with anti-URE3-BP monoclonal antibody 4D6 from amebic lysate enriched with 5 mM CaCl2 and probed with amebic cell lysate supplemented with either 5 mM CaCl2 or 5 mM EDTA in order to detect URE3-BP binding partners. Bound URE3-BP was detected by Western blotting using anti-URE3-BP MAb 4D6. Asterisk indicates a 22-kDa protein band bound by URE3-BP.
To determine if the coimmunoprecipitating 22-kDa protein band truly interacted with URE3-BP in a Ca2+-dependent manner, a combined immunoprecipitation–far-Western blot analysis was performed. URE3-BP was immunoprecipitated in the presence of Ca2+, and the immunoprecipitate was subjected to SDS-PAGE, transferred onto PVDF membranes, and then overlaid with E. histolytica whole-cell lysates in the presence or absence of Ca2+. Probing the membrane with URE3-BP MAb 4D6 revealed the interaction of URE3-BP with a 22-kDa protein band only in the presence of CaCl2, indicating that the URE3-BP-22-kDa protein interaction was Ca2+ dependent (Fig. 1C).
Mass spectrometry analysis of the coimmunoprecipitating 22-kDa band revealed a C2 domain-containing protein.
In order to determine the identity of the 22-kDa coimmunoprecipitating band, the gel slice containing this band was excised and subjected to mass spectrometry peptide sequence analysis. The sequences obtained were analyzed against the E. histolytica genome database (http://www.amoebadb.org) using the Sequest algorithm (14). Peptides corresponding to 31 different proteins were identified in the excised gel band (see Table S1 in the supplemental material). We focused on one of the most abundant and size-appropriate proteins identified in the MS analysis, EhC2A.
The amino acid sequence of EhC2A predicted a protein mass of 20.7 kDa with an N-terminal C2 domain, identified in silico using InterProScan (48), as well as a proline-rich C-terminal tail (Fig. 2A). When the sequence of EhC2A was queried against the E. histolytica genome database using BLASTP (NCBI), several additional C2 domain-containing proteins with similar domain architecture were identified, including EhC2B (GenBank accession no. XM_647805), a protein with a predicted mass of 22.1 kDa that has significant sequence identity (63%) to EhC2A. No EhC2B-specific peptides were identified in the 22-kDa coimmunoprecipitated band (Fig. 2B).
Fig. 2.

Sequence and domain structure of EhC2A (GenBank accession no. XM_650207). (A) Schematic representation of EhC2A. The C2 domain starts at amino acid 2 and terminates at amino acid 118. (B) Amino acid sequence alignment of EhC2A and EhC2B. The C2A sequences corresponding to the four peptides (P1 to P4) identified by mass spectrometry are indicated. Asterisks show identical amino acids, double dots show conserved amino acids, and dots show semiconserved amino acids.
C2 domains are known to confer the ability to bind phospholipids and target proteins to specific membranes within eukaryotic cells. Given that URE3-BP was enriched in the amebic plasma membrane (17), we were interested in studying EhC2A as a potential binding partner responsible for this association.
Coimmunoprecipitation of URE3-BP with EhC2A.
Rabbit antiserum developed against an E. coli-expressed GST-tagged EhC2A fusion protein recognized a 22-kDa protein in whole-cell extracts of E. histolytica, as well as recombinant amino-terminally His6-tagged EhC2A (Fig. 3A). A second band of 16 kDa was also recognized by the antiserum in the whole-cell extract. The 22-kDa and 16-kDa protein bands disappeared when the antiserum was preabsorbed with recombinant GST-EhC2A but not when the antiserum was preabsorbed with recombinant GST alone (see Fig. S1 in the supplemental material). This suggests that the 16-kDa band shared antigenic epitopes with the 22-kDa band and was likely a product of EhC2A processing.
Fig. 3.

Identification of the 22-kDa URE3-BP-interacting protein as EhC2A. (A) Western blots were performed on amebic whole-cell lysates or recombinant purified His6-tagged EhC2A using anti-EhC2A rabbit serum developed with recombinant purified GST-tagged EhC2A. Preimmune serum lanes are indicated. α, anti. (B) Trophozoites were lysed in the presence of 5 mM EGTA (E) and immunoprecipitated (IP) with anti-EhC2A rabbit serum (EhC2A) or control rabbit serum (Control). Coimmunoprecipitated proteins were detected by Western blotting (WB). (C) Trophozoites were lysed in the presence of 5 mM EGTA (E) or 0.1 mM CaCl2 (C) and immunoprecipitated (IP) with anti-URE3-BP MAb 4D6 (URE3-BP) or control mouse MAb (Control). Coimmunoprecipitated proteins were detected by Western blotting (WB).
In order to confirm that EhC2A was a binding partner of URE3-BP, we performed a combined immunoprecipitation-Western blot analysis. URE3-BP was detected when EhC2A was immunoprecipitated using anti-EhC2A rabbit polyclonal antiserum in the presence of EGTA (Fig. 3B). Similarly, EhC2A was detected in the immunoprecipitation using URE3-BP MAb 4D6 (Fig. 3C). The 16-kDa protein detected by immunoblotting of whole-cell lysates was not detected when immunoprecipitations were performed using the EhC2A antiserum and was not consistently detected when immunoprecipitations were performed using URE3-BP MAb 4D6. The amount of URE3-BP coimmunoprecipitating with EhC2A was increased in the presence of CaCl2 (Fig. 3C), consistent with the Ca2+ dependence of the EhC2A–URE3-BP interaction. We concluded that the 22-kDa (and not the 16 kDa) presumed isoform of EhC2A interacted with URE3-BP.
Characterization of the Ca2+-dependent phospholipid-binding activity of EhC2A.
Structural and functional studies have facilitated the prediction of which putative residues in C2 domains are required for Ca2+ coordination and membrane binding (10, 28, 44). Alignment of the C2 domains of EhC2A and EhC2B to the C2 domain of human protein kinase C-α identified these conserved residues, as well as the predicted calcium-binding loops (Fig. 4A). The conserved Asp-17, Asp-69, and Asp-71 residues of EhC2A have been mutated in other C2 domains, and those mutations abolished the domain's ability to bind phospholipid (10). Ca2+-coordinating residues of EhC2A were located between the first and second β-strands and between the fifth and sixth β-strands, the same organization as was observed for the type II topology C2 domains of the human nonclassical protein kinase C isoforms and phospholipase C isoforms (29). Therefore, we predicted that the C2 domains of EhC2A and EhC2B were of type II topology.
Fig. 4.

Ca2+-dependent phospholipid binding by purified recombinant GST-EhC2A. (A) Alignment of the C2 domains of EhC2A and EhC2B with the C2 domain of human PKC-α using ClustalW2 (23). The β-sheets and calcium-binding loops (CBL) for PKC-α (boxes) are assigned according to the designation of its crystal structure (44). The predicted positioning of the β-sheets for EhC2A and EhC2B are as determined by PORTER (34). The residues in gray boxes are conserved calcium-coordinating aspartyl residues. (B) GST or GST-EhC2A bound to the MagneGST beads were incubated with 3H-cholesterol-labeled phosphatidylcholine/phosphatidylserine liposomes (3:1 [wt/wt]) in the absence (black) or presence of CaCl2 (0.1 mM) (gray). Liposome binding to absorbed beads was determined by scintillation counting. Error bars indicate the standard deviations (n = 3). **, P < 0.0001. (C) Binding of 3H-labeled liposomes to GST-EhC2A bound to MagneGST beads as a function of the free Ca2+ concentration. Error bars indicate standard deviations (n = 3). CPM, counts per minute.
Not all C2 domains bind phospholipids or Ca2+, and sequence homology by itself is not necessarily predictive of the ability of a particular C2 domain to bind phospholipids (9, 22). To determine if EhC2A was a functional phospholipid-binding protein, E. coli-expressed EhC2A was tested for its ability to bind liposomes in a Ca2+-dependent manner. GST alone or GST-EhC2A was immobilized on MagneGST beads and mixed with 3H-cholesterol-labeled phosphatidylcholine/phosphatidylserine (PC/PS) liposomes (3:1 [wt/wt]) in the presence or absence of Ca2+ (EGTA), and the amount of protein-bound liposomes quantitated by liquid scintillation counting. GST-EhC2A protein but not GST alone bound liposomes in a Ca2+-dependent manner (Fig. 4B). Half-maximal binding activity was determined to occur at ∼3.4 μM free Ca2+ (Fig. 4C), which is similar to other determinations observed for the human C2 domain-containing proteins DOC2B (5 μM) (21) and cPLA2-α (4.7 μM) (31). Native EhC2A from amebic lysates was confirmed to bind phospholipids in a Ca2+-dependent manner, utilizing liposome sedimentation assays (see Fig. S2 in the supplemental material). Only the 22-kDa full-length EhC2A and not the 16-kDa presumed isoform recognized by the EhC2A antiserum bound liposomes.
Ca2+-dependent recruitment of URE3-BP to phospholipids.
Given the ability of EhC2A to bind liposomes in a Ca2+-dependent manner and its Ca2+-dependent association with URE3-BP, we hypothesized that EhC2A might function by recruiting URE3-BP to phospholipids. To determine if EhC2A was able to recruit URE3-BP to phospholipids in vitro, liposome recruitment assays were performed using recombinant URE3-BP and GST-tagged EhC2A. Recombinant URE3-BP was found to associate with PC/PS liposomes in the presence of GST-tagged EhC2A but not in the presence of the control GST alone (Fig. 5). The recruitment of URE3-BP to PC/PS liposomes required Ca2+, as recombinant URE3-BP and GST-tagged EhC2A only bound liposomes in the presence of Ca2+. The proteins were not observed to bind liposomes in the presence of the Ca2+ chelator EGTA. We concluded that the interaction between URE3-BP and EhC2A was direct and that EhC2A was able to recruit the transcription factor to phospholipids in a Ca2+-dependent manner.
Fig. 5.

EhC2A-mediated recruitment of URE3-BP to phospholipid vesicles is Ca2+ dependent. GST or GST-EhC2A (0.1 μg, 0.1 μg, 0.25 μg, or 0.5 μg) was incubated with recombinant URE3-BP (0.5 μg) and phosphatidylcholine/phosphatidylserine liposomes (3:1 [wt/wt]) in the presence of 5 mM EGTA or 10 μM CaCl2. Liposomes were precipitated by centrifugation, and liposome-bound and unbound (Supernatants) proteins were detected by Western blotting using EhC2A antiserum (EhC2A) or anti-URE3-BP MAb 4D6 (URE3-BP). Sedimentation controls were performed in the absence of liposomes (not shown).
Translocation of URE3-BP and EhC2A is induced by increased intracellular Ca2+.
URE3-BP was previously observed to be enriched in the inner leaflet of the amebic plasma membrane (17). The finding of Ca2+-dependent sequestration of URE3-BP to phospholipid liposomes mediated by EhC2A in vitro led us to examine the role of Ca2+ and EhC2A in the cellular localization of URE3-BP. The distribution of EhC2A and URE3-BP was examined in trophozoites incubated under conditions that elevated or depleted intracellular Ca2+ (3). When trophozoites were incubated in medium depleted of Ca2+ using EGTA, URE3-BP and EhC2A were recovered in the soluble (S100) fractions, with some protein observed in the crude membrane fractions of trophozoites (Fig. 6). However, when trophozoites were incubated in medium supplemented with ionophore A21387 and 1 mM CaCl2 to induce an increase in intracellular calcium, URE3-BP and EhC2A were recovered in the crude membrane fraction (P100) of trophozoites. We concluded that the Ca2+-dependent redistribution of EhC2A and URE3-BP was consistent with EhC2A having a direct role in anchoring URE3-BP to amebic membrane.
Fig. 6.
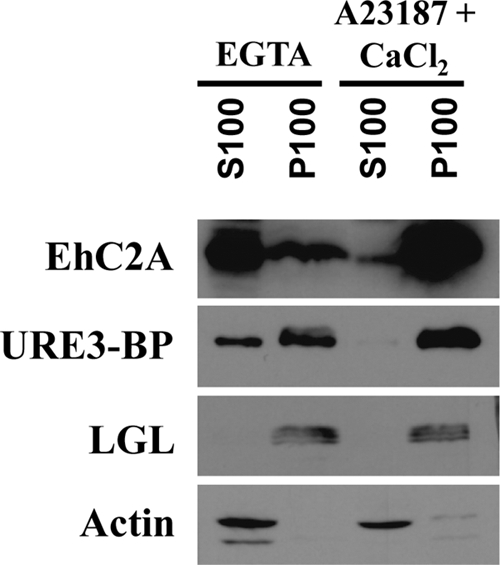
Ca2+-induced association of EhC2A and URE3-BP with crude membranes of trophozoites. E. histolytica trophozoites were incubated at 37°C for 1 h in TYI-S-33 medium with 5 mM EGTA or 1 mM CaCl2 and 1 μM A23187, lysed, and separated into particulate (P100) and soluble (S100) fractions by high-speed centrifugation. Fifty micrograms of each fraction was separated by polyacrylamide gel electrophoresis and immunoblotted with EhC2A antiserum (EhC2A), anti-URE3-BP MAb 4D6 (URE3-BP), anti-LGL MAb 1D4 (LGL) as a membrane fraction control, or actin antiserum (Actin) as a soluble fraction control.
To confirm the subcellular localization of EhC2A and URE3-BP in trophozoites under conditions that elevate or deplete intracellular Ca2+, double-immunofluorescence staining using anti-URE3-BP MAb 4D6 and anti-EhC2A rabbit polyclonal serum was performed and the results analyzed by confocal laser scanning microscopy. EhC2A and URE3-BP were homogenously distributed in the cytoplasm of the cells when trophozoites were incubated in medium lacking Ca2+ (Fig. 7A). URE3-BP was also observed in the nucleus under these conditions. In contrast, when trophozoites were incubated in calcium-containing medium, elevating intracellular Ca2+, EhC2A and URE3-BP showed a clearly distinct distribution characterized by intense plasma membrane-associated fluorescence (Fig. 7B). These findings suggest the involvement of EhC2A in the translocation of URE3-BP to the amebic plasma membrane.
Fig. 7.

Ca2+-induced translocation of URE3-BP and EhC2A to the plasma membrane of amebic trophozoites. E. histolytica trophozoites were incubated at 37°C for 1 h in TYI-S-33 medium with 5 mM EGTA (A) or 1 mM CaCl2 and 1 μM A23187 (B). Fixed permeabilized cells were probed with rabbit anti-EhC2A polyclonal antibody, Alexa Fluor 488 (green) goat anti-rabbit IgG (EhC2A), mouse anti-URE3-BP MAb 4D6, and Alexa Fluor 546 (red) goat anti-mouse IgG (URE3-BP). Nuclei were stained blue with 4′,6-diamidino-2-phenylindole (DAPI). Merged images demonstrate colocalization of the two proteins.
Knockdown of EhC2A modulates transcription of URE3 motif-regulated genes.
The overexpression of a calcium- insensitive dominant-positive mutant of URE3-BP [EF(2)mutURE3-BP] was used to profile the transcriptome of URE3-BP (16). This led to the identification of several genes with URE3 motifs in their promoters that were modulated when URE3-BP was overexpressed, including the genes coding for acyl-coenzyme A (CoA) synthetase (GenBank accession no. XM_645995) and a predicted membrane protein (GenBank accession no. XM_644369). Ca2+-inhibitable URE3-BP binding to the promoter of the predicted membrane protein in vivo has been determined by chromatin immunoprecipitation accompanied by real time RT-PCR (C. A. Gilchrist, unpublished data).
Short hairpin RNA (shRNA)-mediated knockdown of protein expression in E. histolytica has recently been described (5, 24). The system is based on the stable episomal transfection of amebic trophozoites with a vector expressing an RNA hairpin consisting of a 29-base-pair target-gene-specific sequence, a 9-base-pair loop, and the reverse complementary 29-base-pair sequence, all under the regulation of the E. histolytica RNA polymerase III U6 promoter. Stable transfection of trophozoites with a construct expressing shRNA specifically targeting EhC2A resulted in significant knockdown of EhC2A protein (97%) compared to the results with a scrambled-shRNA control transfectant (24). These trophozoites were tested by qRT-PCR to compare the mRNA transcript levels of the URE3 motif-regulated genes encoding acyl-CoA synthetase and the predicted membrane protein. In EhC2A shRNA transfectants, the transcripts for acyl-CoA synthetase and the membrane protein were significantly modulated (Fig. 8). The URE3-BP transcript levels were not significantly changed. A 3-fold increase in the transcript level of acyl-CoA synthetase (P = 0.0396) and a 7-fold decrease in the transcript level of membrane protein (P = 0.0496) were observed when the RNA from amebae transfected with EhC2A shRNA and amebae transfected with scrambled-shRNA control plasmids were compared. Interestingly, the direction of transcript modulation of these two genes correlated with the direction of transcript modulation previously observed for trophozoites overexpressing the dominant-positive URE3-BP (16). These findings are suggestive of a regulatory role for EhC2A in the transcriptional activities of URE3-BP.
Fig. 8.

EhC2A shRNA protein knockdown transfectants modulate URE3 motif-regulated transcripts. mRNA levels of URE3-BP, acyl-CoA synthetase, and membrane protein from E. histolytica trophozoites transfected with EhC2A (363–391) (shRNA) or EhC2A (363–391 scrambled) (control) as measured by qRT-PCR (mean ± standard error). Average results for three biological replicates are expressed as the percentages of the EhC2A (363–391 scrambled) level (control), standardized for each transcript with plasmid control mRNA levels and normalized to the actin level. Statistical analysis was performed using Student's t test. **, P < 0.05.
DISCUSSION
In this study, we identified and characterized the association of URE3-BP, a Ca2+-regulated transcription factor of the pathogenic protozoan E. histolytica, with a second Ca2+-regulated protein, the phospholipid-binding EhC2A. URE3-BP regulates the transcription of several virulence factors in Entamoeba in a very unique manner: Ca2+ binding directly regulates the association of the protein to its DNA-binding motif in gene promoters via its EF-hand motifs. Only one other protein has been demonstrated to utilize this mechanism, the human EF-hand-containing protein DREAM (7). Our results demonstrate an added level of Ca2+-mediated regulation of URE3-BP, as we showed that Ca2+ also regulates the translocation of URE3-BP to the amebic plasma membrane and its association with the plasma membrane binding protein EhC2A. Hence, we propose a tripartite system by which changes in intracellular calcium directly regulate gene expression in the parasite (Fig. 9). In the absence of an intracellular calcium signaling event, URE3-BP binds to its cognate URE3 element on the promoters of the genes it regulates. Upon an increase in intracellular Ca2+, URE3-BP releases the URE3 element and binds EhC2A. Furthermore, Ca2+ also induces the association of the EhC2A–URE3-BP protein complex with the plasma membrane of trophozoites, resulting in the inhibition of the transcriptional activities of URE3-BP.
Fig. 9.

Proposed model for regulation of URE3-BP induced by Ca2+. Under basal intracellular Ca2+ conditions (left), the EF-hand-containing transcription factor URE3-BP binds to the URE3 motif in promoters, thereby activating or repressing gene transcription. Upon an increase in intracellular Ca2+, URE3-BP is released from DNA promoters and is sequestered to the plasma membrane by EhC2A (right), resulting in an inhibition of its transcriptional activity.
The evidence that EhC2A is a binding partner of URE3-BP involved in its translocation to the amebic plasma membrane in a Ca2+-dependent manner is comprised of the following: (i) URE3-BP and EhC2A immunoprecipitated with one another; (ii) the amount of EhC2A that coimmunoprecipitated with URE3-BP increased in the presence of Ca2+; (iii) recombinant EhC2A recruited recombinant URE3-BP binding to phosphatidylcholine/phosphatidylserine liposomes only in the presence of Ca2+; (iv) EhC2A and URE3-BP colocalized in amebic trophozoites in the presence of Ca2+, as determined by immunofluorescence confocal microscopy; and (v) EhC2A and URE3-BP translocated to the amebic plasma membrane when the intracellular Ca2+ concentration was increased in trophozoites. Several examples of C2 domain proteins functioning as molecular scaffolds that mediate the association of proteins with membranes have been reported (39, 42). The C2 domain-containing copines interact with several proteins, including transcription factors, and were proposed to target such proteins to membrane surfaces (41). The Ca2+-induced mobilization of URE3-BP represents a novel mechanism in which a C2 domain protein is involved in the anchoring of a transcription factor to the plasma membrane.
C2 domain-containing proteins have extensive functional diversity, participating in a variety of Ca2+-regulated processes, including signal transduction, membrane trafficking, lipid second-messenger generation, and others. Our finding of conserved C2 domain proteins in this evolutionarily divergent parasite is evidence of the importance of Ca2+ signaling in regulating the parasite's activities. EhC2A is the first C2 domain-containing protein that has been characterized in E. histolytica. The C2 domains of EhC2A and EhC2B are remarkably conserved, with 75% identity across this region and similar predicted molecular masses. Despite this conservation, we only identified peptides corresponding to EhC2A when the URE3-BP coimmunoprecipitating band was sequenced. EhC2A and EhC2B therefore might have nonredundant roles, as only EhC2A appears to interact with URE3-BP. It is interesting to speculate that EhC2B may also be involved in membrane anchoring of associated proteins.
Intracellular Ca2+ fluxes have been observed in E. histolytica upon contact with fibronectin (6) and increased extracellular Ca2+ concentrations (18). The precise Ca2+ concentration of fluxes induced by these extracellular cues has not been determined. It is interesting to note that similar concentrations of Ca2+ were required for EhC2A membrane binding (∼3.4 μM), EhC2A-mediated recruitment of URE3-BP to PC/PS liposomes (≤10 μM), and URE3-BP dissociation from the URE3 DNA motif (≤6.7 μM) (18). However, it is possible that the effective Ca2+ concentration required for membrane binding of EhC2A in vivo is much lower than the concentration we observed utilizing the PC/PS liposomes (3:1 [wt/wt]). The Ca2+ affinity of C2 domains has been observed to increase when the cognate target membrane lipid composition is recreated for in vitro determinations (11). Unfortunately, attempts to define the exact lipid composition of the Entamoeba plasma membrane, the target of EhC2A, have been limited (1, 43).
EhC2A was recently found to be associated with the amebic phagosome (32), which may be indicative of intracellular Ca2+ fluxes induced by amebic phagocytosis of host cells. Interestingly, the copine homolog of the social ameba Dictyostelium discoideum, CpnA, contains a Ca2+-dependent phospholipid-binding C2 domain that also associates with the amebic phagosome (12).
URE3-BP contains two canonical EF-hand motifs, associated with the ability to bind Ca2+ and induce conformational changes upon Ca2+ binding or release in several proteins (46). C2 domain-containing proteins have also been found to undergo conformational changes that extend beyond the motif's Ca2+-binding region upon Ca2+ binding (8, 30, 45). It is therefore possible that one or both proteins undergo Ca2+-induced conformational changes necessary for the protein association to occur. There is precedent for direct interaction of EF-hand motifs with C2 domains. A Ca2+-dependent interaction between the EF-hand motifs of the human major vault protein and the C2 domain of the tumor suppressor PTEN was identified (47), and intramolecular interactions between the C2 and EF-hand domains of phospholipase C-δ1 have been revealed by structural analysis (15, 20). It would therefore be interesting to determine if the interaction between URE3-BP and EhC2A occurs via similar interactions.
The translocation of URE3-BP and EhC2A to the plasma membrane in response to increased intracellular Ca2+ and the Ca2+-dependent nature of their interaction are perhaps indicative of an enhanced regulatory mechanism. Using trophozoites transfected with plasmids that express shRNA targeting EhC2A, we have knocked down the protein expression levels of EhC2A (24). We examined the transcript levels of two genes, encoding acyl-CoA synthetase and a predicted membrane protein, that were previously identified as being modulated when the dominant-positive EF(2)mutURE3-BP protein was overexpressed in amebae (16). The mRNA levels of these two genes were modulated in the same direction as in cells overexpressing dominant-positive URE3-BP. This finding is in agreement with our hypothesis that by sequestering URE3-BP to the amebic plasma membrane, EhC2A regulates the ability of the transcription factor to bind its cognate DNA element. Future studies will examine the extent to which the interaction with EhC2A controls the nuclear availability and transcriptional activities of URE3-BP and consequently regulates amebic virulence.
Supplementary Material
ACKNOWLEDGMENTS
We thank Carl E. Creutz for valuable advice on membrane binding assays. We also thank Nicholas E. Sherman of the W. M. Keck Biomedical Mass Spectrometry Laboratory at the University of Virginia for his technical support of the MS analysis. Recombinant URE3-BP protein was provided by Eric Smith, Taryn Haffner, and Amy Raymond from Emerald BioStructures, as part of the Seattle Structural Genomics Center for Infectious Disease, which is funded by NIAID contract no. HHSN272200700057C.
This work was supported by NIH grant AI-37941.
Footnotes
Supplemental material for this article may be found at http://ec.asm.org/.
Published ahead of print on 18 December 2009.
REFERENCES
- 1.Aley S. B., Scott W. A., Cohn Z. A. 1980. Plasma membrane of Entamoeba histolytica. J. Exp. Med. 152:391–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anonymous. 1997. Amoebiasis. Wkly. Epidemiol. Rec. 72:97–99 [PubMed] [Google Scholar]
- 3.Beck D. L., Boettner D. R., Dragulev B., Ready K., Nozaki T., Petri W. A., Jr 2005. Identification and gene expression analysis of a large family of transmembrane kinases related to the Gal/GalNAc lectin in Entamoeba histolytica. Eukaryot. Cell 4:722–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharya A., Padhan N., Jain R., Bhattacharya S. 2006. Calcium-binding proteins of Entamoeba histolytica. Arch. Med. Res. 37:221–225 [DOI] [PubMed] [Google Scholar]
- 5.Boettner D. R., Huston C. D., Linford A. S., Buss S. N., Houpt E., Sherman N. E., Petri W. A., Jr 2008. Entamoeba histolytica phagocytosis of human erythrocytes involves PATMK, a member of the transmembrane kinase family. PLoS Pathog. 4:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carbajal M. E., Manning-Cela R., Pina A., Franco E., Meza I. 1996. Fibronectin-induced intracellular calcium rise in Entamoeba histolytica trophozoites: effect on adhesion and the actin cytoskeleton. Exp. Parasitol. 82:11–20 [DOI] [PubMed] [Google Scholar]
- 7.Carrion A. M., Link W. A., Ledo F., Mellstrom B., Naranjo J. R. 1999. DREAM is a Ca2+-regulated transcriptional repressor. Nature 398:80–84 [DOI] [PubMed] [Google Scholar]
- 8.Chapman E. R., Davis A. F. 1998. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J. Biol. Chem. 273:13995–14001 [DOI] [PubMed] [Google Scholar]
- 9.Cho W., Stahelin R. V. 2006. Membrane binding and subcellular targeting of C2 domains. Biochim. Biophys. Acta 1761:838–849 [DOI] [PubMed] [Google Scholar]
- 10.Corbalan-Garcia S., Rodriguez-Alfaro J. A., Gomez-Fernandez J. C. 1999. Determination of the calcium-binding sites of the C2 domain of protein kinase Calpha that are critical for its translocation to the plasma membrane. Biochem. J. 337(Pt. 3):513–521 [PMC free article] [PubMed] [Google Scholar]
- 11.Corbin J. A., Evans J. H., Landgraf K. E., Falke J. J. 2007. Mechanism of specific membrane targeting by C2 domains: localized pools of target lipids enhance Ca2+ affinity. Biochemistry 46:4322–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damer C. K., Bayeva M., Hahn E. S., Rivera J., Socec C. I. 2005. Copine A, a calcium-dependent membrane-binding protein, transiently localizes to the plasma membrane and intracellular vacuoles in Dictyostelium. BMC Cell Biol. 6:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diamond L. S., Harlow D. R., Cunnick C. C. 1978. A new medium for the axenic cultivation of Entamoeba histolytica and other Entamoeba. Trans. R. Soc. Trop. Med. Hyg. 72:431–432 [DOI] [PubMed] [Google Scholar]
- 14.Eng J. K., McCormack A. L., Yates J. R. 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5:976–989 [DOI] [PubMed] [Google Scholar]
- 15.Essen L. O., Perisic O., Cheung R., Katan M., Williams R. L. 1996. Crystal structure of a mammalian phosphoinositide-specific phospholipase C delta. Nature 380:595–602 [DOI] [PubMed] [Google Scholar]
- 16.Gilchrist C. A., Baba D. J., Zhang Y., Crasta O., Evans C., Caler E., Sobral B. W. S., Bousquet C. B., Leo M., Hochreiter A., Connell S. K., Mann B. J., Petri W. A., Jr 2008. Targets of the Entamoeba histolytica transcription factor URE3-BP. PLoS Negl. Trop. Dis. 2:e282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilchrist C. A., Holm C. F., Hughes M. A., Schaenman J. M., Mann B. J., Petri W. A., Jr 2001. Identification and characterization of an Entamoeba histolytica upstream regulatory element 3 sequence-specific DNA-binding protein containing EF-hand motifs. J. Biol. Chem. 276:11838–11843 [DOI] [PubMed] [Google Scholar]
- 18.Gilchrist C. A., Leo M., Line C. G., Mann B. J., Petri W. A., Jr 2003. Calcium modulates promoter occupancy by the Entamoeba histolytica Ca2+-binding transcription factor URE3-BP. J. Biol. Chem. 278:4646–4653 [DOI] [PubMed] [Google Scholar]
- 19.Ginzinger D. G., Godfrey T. E., Nigro J., Moore D. H., II, Suzuki S., Pallavicini M. G., Gray J. W., Jensen R. H. 2000. Measurement of DNA copy number at microsatellite loci using quantitative PCR analysis. Cancer Res. 60:5405–5409 [PubMed] [Google Scholar]
- 20.Grobler J. A., Essen L. O., Williams R. L., Hurley J. H. 1996. C2 domain conformational changes in phospholipase C-delta 1. Nat. Struct. Biol. 3:788–795 [DOI] [PubMed] [Google Scholar]
- 21.Groffen A. J., Brian E. C., Dudok J. J., Kampmeijer J., Toonen R. F., Verhage M. 2004. Ca(2+)-induced recruitment of the secretory vesicle protein DOC2B to the target membrane. J. Biol. Chem. 279:23740–23747 [DOI] [PubMed] [Google Scholar]
- 22.Hurley J. H., Misra S. 2000. Signaling and subcellular targeting by membrane-binding domains. Annu. Rev. Biophys. Biomol. Struct. 29:49–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 24.Linford A., Moreno H., Good K., Zhang H., Singh U., Petri W. 2009. Short hairpin RNA-mediated knockdown of protein expression in Entamoeba histolytica. BMC Microbiol. 9:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makioka A., Kumagai M., Kobayashi S., Takeuchi T. 2002. Possible role of calcium ions, calcium channels and calmodulin in excystation and metacystic development of Entamoeba invadens. Parasitol. Res. 88:837–843 [DOI] [PubMed] [Google Scholar]
- 26.Makioka A., Kumagai M., Ohtomo H., Kobayashi S., Takeuchi T. 2001. Effect of calcium antagonists, calcium channel blockers and calmodulin inhibitors on the growth and encystation of Entamoeba histolytica and E. invadens. Parasitol. Res. 87:833–837 [DOI] [PubMed] [Google Scholar]
- 27.McCoy J. J., Weaver A. M., Petri W. A., Jr 1994. Use of monoclonal anti-light subunit antibodies to study the structure and function of the Entamoeba histolytica Gal/GalNAc adherence lectin. Glycoconj. J. 11:432–436 [DOI] [PubMed] [Google Scholar]
- 28.Medkova M., Cho W. 1998. Mutagenesis of the C2 domain of protein kinase C-alpha. Differential roles of Ca2+ ligands and membrane binding residues. J. Biol. Chem. 273:17544–17552 [DOI] [PubMed] [Google Scholar]
- 29.Nalefski E. A., Falke J. J. 1996. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 5:2375–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nalefski E. A., Slazas M. M., Falke J. J. 1997. Ca2+-signaling cycle of a membrane-docking C2 domain. Biochemistry 36:12011–12018 [DOI] [PubMed] [Google Scholar]
- 31.Nalefski E. A., Wisner M. A., Chen J. Z., Sprang S. R., Fukuda M., Mikoshiba K., Falke J. J. 2001. C2 domains from different Ca2+ signaling pathways display functional and mechanistic diversity. Biochemistry 40:3089–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okada M., Huston C. D., Oue M., Mann B. J., Petri W. A., Jr., Kita K., Nozaki T. 2006. Kinetics and strain variation of phagosome proteins of Entamoeba histolytica by proteomic analysis. Mol. Biochem. Parasitol. 145:171–183 [DOI] [PubMed] [Google Scholar]
- 33.Olvera A., Olvera F., Vines R. R., Recillas-Targa F., Lizardi P. M., Dhar S., Bhattacharya S., Petri W., Jr., Alagon A. 1997. Stable transfection of Entamoeba histolytica trophozoites by lipofection. Arch. Med. Res. 28:S49–S51 [PubMed] [Google Scholar]
- 34.Pollastri G., McLysaght A. 2005. Porter: a new, accurate server for protein secondary structure prediction. Bioinformatics 21:1719–1720 [DOI] [PubMed] [Google Scholar]
- 35.Ravdin J. I., Moreau F., Sullivan J. A., Petri W. A., Jr., Mandell G. L. 1988. Relationship of free intracellular calcium to the cytolytic activity of Entamoeba histolytica. Infect. Immun. 56:1505–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravdin J. I., Murphy C. F., Guerrant R. L., Long-Krug S. A. 1985. Effect of antagonists of calcium and phospholipase A on the cytopathogenicity of Entamoeba histolytica. J. Infect. Dis. 152:542–549 [DOI] [PubMed] [Google Scholar]
- 37.Rizo J., Sudhof T. C. 1998. C2-domains, structure and function of a universal Ca2+-binding domain. J. Biol. Chem. 273:15879–15882 [DOI] [PubMed] [Google Scholar]
- 38.Sheffield P., Garrard S., Derewenda Z. 1999. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 15:34–39 [DOI] [PubMed] [Google Scholar]
- 39.Sundberg C., Thodeti C. K., Kveiborg M., Larsson C., Parker P., Albrechtsen R., Wewer U. M. 2004. Regulation of ADAM12 cell-surface expression by protein kinase C epsilon. J. Biol. Chem. 279:51601–51611 [DOI] [PubMed] [Google Scholar]
- 40.Tomsig J. L., Creutz C. E. 2000. Biochemical characterization of copine: a ubiquitous Ca2+-dependent, phospholipid-binding protein. Biochemistry 39:16163–16175 [DOI] [PubMed] [Google Scholar]
- 41.Tomsig J. L., Snyder S. L., Creutz C. E. 2003. Identification of targets for calcium signaling through the copine family of proteins. Characterization of a coiled-coil copine-binding motif. J. Biol. Chem. 278:10048–10054 [DOI] [PubMed] [Google Scholar]
- 42.Tsuboi T., Fukuda M. 2005. The C2B domain of rabphilin directly interacts with SNAP-25 and regulates the docking step of dense core vesicle exocytosis in PC12 cells. J. Biol. Chem. 280:39253–39259 [DOI] [PubMed] [Google Scholar]
- 43.van Vliet H. H., Spies F., Linnemans W. A., Klepke A., Op den Kamp J. A., van Deenen L. L. 1976. Isolation and characterization of subcellular membranes of Entamoeba invadens. J. Cell Biol. 71:357–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verdaguer N., Corbalan-Garcia S., Ochoa W. F., Fita I., Gomez-Fernandez J. C. 1999. Ca(2+) bridges the C2 membrane-binding domain of protein kinase Calpha directly to phosphatidylserine. EMBO J. 18:6329–6338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu G. Y., McDonagh T., Yu H. A., Nalefski E. A., Clark J. D., Cumming D. A. 1998. Solution structure and membrane interactions of the C2 domain of cytosolic phospholipase A2. J. Mol. Biol. 280:485–500 [DOI] [PubMed] [Google Scholar]
- 46.Yap K. L., Ames J. B., Swindells M. B., Ikura M. 1999. Diversity of conformational states and changes within the EF-hand protein superfamily. Proteins 37:499–507 [DOI] [PubMed] [Google Scholar]
- 47.Yu Z., Fotouhi-Ardakani N., Wu L., Maoui M., Wang S., Banville D., Shen S. H. 2002. PTEN associates with the vault particles in HeLa cells. J. Biol. Chem. 277:40247–40252 [DOI] [PubMed] [Google Scholar]
- 48.Zdobnov E. M., Apweiler R. 2001. InterProScan: an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17:847–848 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.