

Abstract
Inflammatory bowel disease (IBD) is a chronic debilitating disease resulting from a complex interaction of multiple genetic factors with the environment. To identify modifier genes of IBD, we used an F2 intercross of IBD-resistant C57BL/6J-Il10−/− mice and IBD-susceptible C3H/HeJBir-Il10−/− (C3Bir-Il10−/−) mice. We found a prominent involvement of lymphatic vessels in IBD and applied a scoring system to quantify lymphatic vascular changes. Quantitative trait locus (QTL) analyses revealed a large-effect QTL on chromosome 3, mapping to an interval of 43.6 Mbp. This candidate interval was narrowed by fine mapping to 22 Mbp, and candidate genes were analyzed by a systems genetics approach that included quantitative gene expression profiling, search for functional polymorphisms, and haplotype block analysis. We identified vascular adhesion molecule 1 (Vcam1) as a candidate modifier gene in the interleukin 10-deficient mouse model of IBD. Importantly, VCAM1 protein levels were increased in susceptible C3H/HeJ mice, compared with C57BL/6J mice; systemic blockade of VCAM1 in C3Bir-Il10−/− mice reduced their inflammatory lymphatic vessel changes. These results indicate that genetically determined expression differences of VCAM1 are associated with susceptibility to colon inflammation, which is accompanied by extensive lymphatic vessel changes. VCAM1 is, therefore, a promising therapeutic target for IBD.
Keywords: lymphangiogenesis, inflammation, systems genetics, colitis
Introduction
Inflammatory bowel disease (IBD) is a chronic inflammatory condition that is believed to be caused by an inadequate reaction of a genetically susceptible host to the normal intestinal microflora. Genome-wide association studies of the two major forms of human IBD, Crohn's disease and ulcerative colitis, have identified several possible susceptibility genes, which are involved in innate immunity, autophagy, and immune regulation.1–5 However, IBD pathogenesis likely involves complex interactions of multiple genes with the environment, and the exact molecular mechanisms that lead to IBD have remained elusive.
Previously, vascular changes have been suggested to contribute to IBD pathogenesis.6 As a response to inflammatory cytokines such as tumor necrosis factor, endothelial cells lining the blood vessels become activated and upregulate the expression of cell adhesion molecules, promoting the transmigration of leukocytes into the inflamed tissue.7 In contrast, the function of lymphatic vessels in inflammation became a focus of research only recently. Lymphatic vessels have a major function in the maintenance of tissue fluid homeostasis, immune surveillance, and intestinal lipid absorption.8 Research into the lymphatic system was hampered by the lack of tools to reliably distinguish these vessels from blood vessels. However, the recent discovery of molecular lymphatic vessel markers (for example lymphatic vessel endothelial hyaluronan receptor 1, LYVE1; PROX1; podoplanin) has opened new avenues to detailed investigations of the lymphatic vasculature in inflammation.
There is growing evidence that lymphatic vessels actively participate in the inflammatory response. Pronounced lymphangiogenesis has been found in mouse models of chronic airway inflammation, rheumatoid arthritis, and psoriasis.9–11 Activation of lymphatic vessels might support inflammation by promoting inflammatory cell transport to draining lymph nodes.12 This process was shown to involve active interaction with lymphatic vessels and upregulation of intercellular adhesion molecule 1 and vascular adhesion molecule 1 (VCAM1) on lymphatic endothelium in vivo.13 On the other hand, the resolution of chronic inflammation might be promoted by lymphatic vessel drainage, and thereby removal of accumulated fluid, immune cells, and inflammatory cytokines from the sites of inflammation.14,15 Indeed, impairment of the lymphatic vascular network and flow can exacerbate inflammation, and blockade of lymphangiogenic factors in inflammation results in delayed antigen clearance and inflammation resolution.16–19
Genetic variation among mouse inbred strains most commonly results in quantitative traits, measurable continuous phenotypic variations.20 Quantitative traits are influenced by multiple genes and environmental factors and their analysis in mice is being successfully used to gain insight into the genetics of complex human diseases such as IBD.21 To identify genes that determine susceptibility to IBD, we used interleukin 10 (IL10)-deficient (Il10−/−) mice. Il10−/− mice develop a form of colitis that is similar to human IBD, because of impaired regulation of the immune response to microbial antigens in the colon.22 Importantly, the extent of the IBD phenotype in Il10−/− mice is highly influenced by the genetic background: C57BL/6J-Il10−/− (B6-Il10−/−) mice are relatively resistant to colitis, whereas C3H/HeJBir-Il10−/− (C3Bir-Il10−/−) mice are highly susceptible (for full designation of the strains, see Materials and methods).23
In a pilot study, we found that in C3Bir-Il10−/− mice, colitis is accompanied by increased numbers of enlarged and tortuous lymphatic vessels and by pronounced submucosal edema. Moreover, it has been reported that obstruction of lymphatic vessels in the intestine leads to Crohn's disease-like symptoms in experimental animals.24,25 A higher density of lymphatic vessels was also observed in the colons of human IBD patients.26 Moreover, a number of eminent pathologists during the early 20th century pointed to the important function of the lymphatic vasculature in the etiology of Crohn's disease.27 On the basis of these findings, we developed and applied a scoring system to quantify lymphatic vessel traits in an F2 intercross between B6-Il10−/− and C3Bir-Il10−/− mice. This cross was earlier established for the purpose of mapping susceptibility loci for colon mucosal inflammation.28 Quantitative trait locus (QTL) analyses revealed a large-effect QTL on chromosome 3 (Chr 3), mapping to an interval of 43.6 Mbp. This candidate interval was narrowed by fine mapping and by a systems genetics approach that included quantitative gene expression profiling of the QTL genes and functional polymorphisms and haplotype block analyses. These studies identified Vcam1 as a candidate modifier gene in the Il10−/− model of IBD. We found that VCAM1 protein levels were increased in susceptible C3H/HeJ mice, compared with C57BL/6J mice. Systemic blockade of VCAM1 in C3Bir-Il10−/− mice reduced their inflammatory lymphatic vessel changes and the distal colon colitis. Collectively, our results indicate that the higher expression level of VCAM1 in C3Bir-Il10−/− mice represents a genetic component of their susceptibility to colon inflammation, accompanied by extensive lymphatic vessel changes. VCAM1 might, therefore, be a useful therapeutic target in patients with IBD.
Results
Pronounced lymphatic vessel abnormalities in the Il10−/− mouse model of IBD
Il10−/− mice spontaneously develop inflammation of the colon, caused by their inability to control immune responses against the normal bacterial flora.22,29 In line with earlier reports,22,29 histological examination of tissue sections from 7-week-old C3Bir-Il10−/− mice revealed epithelial hyperplasia, focal erosions and ulceration, infiltration by leukocytes, and enlarged vessels (Supplementary Figure S1). Importantly, the degree of inflammation was much more severe in C3Bir-Il10−/− mice than in age-matched B6-Il10−/− mice (Supplementary Figure S1). To further investigate the vascular reaction in Il10−/− mice colons, we used antibodies for the panendothelial marker CD31 and for the lymphatic vessel-specific marker LYVE1.30 Unexpectedly, we found that lymphatic vessels were prominently enlarged in the submucosal area of inflamed colons and that they invaded the lamina propria (Figure 1a; Supplementary Figures S2 and S7). These changes were much more pronounced in C3Bir mice than in C57BL/6J mice. Although there was a dramatic increase in the area covered by lymphatic vessel endothelium in Il10−/− mouse colons compared with wild-type mouse colons, there was no significant change in the colon area covered by blood vessel endothelium in inflammation (Supplementary Figure S2).
Figure 1.

Lymphatic vessels are enlarged and tortuous in colitis; the phenotype differs between C3Bir- and B6-Il10−/− mice. (a) Representative images of wild-type (WT) and Il10−/− C3Bir or B6 mouse colon sections at different time points stained for LYVE1 (red) and counterstained with hematoxylin. The age of mice is indicated (w = weeks). Whereas WT mice had normal lymphatic vasculature, 4-week-old C3Bir-Il10−/− mice showed signs of inflammation and a slightly denser lymphatic vasculature. At 6 and 8 weeks of age, lymphatics were dramatically enlarged and dense. In B6-Il10−/− mice, the lymphatic vasculature first showed changes at 6 weeks, whereas at 8 weeks, changes were less pronounced than in age-matched C3Bir-Il10−/− mice. Scale bars: 100 mm. (b) Quantification of the lymphatic vascular phenotype in the two parental Il10−/− strains at different time points. Summary scores of the lymphatic vascular phenotype (y axis) are shown versus age (x axis). At 6 weeks, there was the most significant difference between the strains (Po0.001). Data are expressed as mean ± s.e.m. (N = 3–5 per strain and time point). The percentage of area covered by lymphatic vessels (c) and the average size of lymphatic vessels (d) in colon hot spots are normally distributed in F2 generation Il10−/− mice.
In C3Bir-Il10−/− mice, the first inflammatory changes in the colon were visible at 4 weeks of age, along with marked lymphangiogenesis and lymphatic vessel enlargement (from a normal average size of approximately 500 μm2 to >45000 μm2), whereas no such changes were observed in B6-Il10−/− mice (Figure 1a). At 6 weeks, C3Bir-Il10−/− mice had further increases in the number of enlarged lymphatic vessels in the submucosal area of inflamed colons, whereas B6-Il10−/− mice had only mild colitis without any major lymphatic abnormalities. The extent of colitis was severe at 8 weeks of age in C3Bir-Il10−/− mice and included a dramatic enlargement of the lymphatic vessels and their invasion into the lamina propria (Figure 1a; Supplementary Figure S7). In B6-Il10−/− mice, the changes were milder with only moderately enlarged lymphatic vessels (Figure 1a). Quantitative analyses of five lymphatic vessel traits (Supplementary Table S1) revealed a much greater lymphatic vessel phenotype score of C3Bir-Il10−/− mice than that of B6-Il10−/− mice at all time points, most notably at 6 weeks (P = 0.0003; Figure 1b). Thus, all further analyses were performed in 6-week-old mice.
QTL analysis identifies a candidate region for modifier genes of lymphatic inflammatory changes on Chr 3
We next performed a quantitative analysis of the lymphatic vessel involvement in colitis (described in Supplementary Table S1) using LYVE1-immunostained colon sections from 243 F2 mice from reciprocal crosses of B6-Il10−/− and C3Bir-Il10−/− mice. Quantitative lymphatic vessel traits were approximately normally distributed (Figures 1c and d; Supplementary Figure S3). All F2 mice were genotyped for 110 polymorphic microsatellite markers that provided complete coverage of the genome (Supplementary Table S2). Genome-wide interval mapping identified major QTLs on Chr 3 for all five lymphatic vessel traits (Figures 2a and b). The mapped QTLs had high logarithm of odds (LOD) scores (4.4–11.7; Table 1), indicating the strength of evidence for a modifier gene effect. The combined Bayesian credible interval (CI) (95% significance) for the five QTLs spanned between 39.7 and 61.8 cM (83.1–126.7 Mbp) on the distal part of Chr 3 (Figure 2b). The Chr 3 QTL did not show any interaction with other loci, as determined by two-dimensional genetic mapping; however, large interaction LOD scores were found between loci on chromosome pairs X:18 and X:19 (Figure 2c; Supplementary Figure S5). An investigation of the interacting loci on these chromosomes revealed a potentially interesting region on Chr 18, previously mapped as a QTL that affects corneal angiogenesis in response to fibroblast growth factor-2 (Angfq4: 74.8–84.8 Mbp).31 As the growth of new vessels was shown to have an effect on IBD, this locus might contribute to the severity of the colitis phenotype. In earlier studies, colitis was quantified by measuring several aspects of mucosal inflammation in Il10−/− F2 mice.28,29 We found a high degree of correlation (correlation coefficients: 0.52–0.78) between the colitis traits quantified in the earlier studies and the lymphatic vessel traits analyzed here (Supplementary Figure S6).
Figure 2.

Mapping of a large-effect QTL for lymphatic vessel inflammatory changes to chromosome 3. (a) The result of interval mapping for five lymphatic vessel subphenotypes (x axis: chromosomes and marker positions, y axis: LOD score). A single locus exceeding the genome-wide 5% significance levels gave a high peak on Chr 3 for all five traits (purple dashed line indicates the significance threshold determined by permutation test). (b) Detailed view of mapped QTL on Chr 3 showing LOD score curves for the five traits, with horizontal lines of matching color indicating the span of 95% approximate Bayesian credible intervals (x axis: marker position in cM). (c) Two-dimensional genome scan performed with 243 F2 mice and 110 microsatellite markers for the total score for lymphatic vessel changes revealed no interaction of the Chr 3 QTL with other loci and a high LOD score interaction between chromosome pairs X:19 and X:18. The upper left triangle contains the interaction model LOD scores, and the lower right triangle contains the conditional-interactive model LOD scores. The color bar indicates the LOD scores for the left and right triangle, respectively.
Table 1.
Results of genome-wide interval mapping and Chr 3 QTL interval fine mapping
| Trait | Initial genome scan |
Fine mapping the Chr 3 QTL interval |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LOD score | Peak Sanger | Position (cM) | Position (bp) | LOD score | Peak marker | Position (bp) | Variance explained (%) | P-value (χ2-test) | P-value (F-test) | |
| Percentage of colon with large and tortuous lymphatics | 12.0 | D3Mit348 | 61.8 | 126 744 151 | 24.4 | rs3687177 | 120 471 745 | 24.3 | 0 | 0 |
| Percentage of area covered by lymphatics in colon hot spots | 6.9 | D3Mit189 | 49.7 | 100 779 519 | 15.1 | rs13477318 | 107 918 926 | 14.5 | 1.92E-14 | 4.31E–09 |
| Average size of lymphatics in colon hot spots | 6.4 | D3Mit189 | 49.7 | 100 779 519 | 8.7 | rs3720182 | 123 406 475 | 9.4 | 2.45E-09 | 2.85E–09 |
| Proliferation of lymphatics into lamina propria | 4.4 | D3Mit189 | 49.7 | 100 779 519 | 13.9 | rs13477315 | 106 767 998 | 14.3 | 3.30E-14 | 4.16E–14 |
| Total score for lymphatic vascular changes in colon wall area | 10.0 | D3Mit189 | 49.7 | 100 779 519 | 22.4 | rs3687177 | 120 471 745 | 22.6 | 0 | 0 |
Abbreviations: Chr 3, chromosome 3; LOD, logarithm of odds; QTL, quantitative trait locus.
Names and positions of the peak marker or closest to the peak markers are shown. The percent of the lymphatic vessel phenotype variance explained by the single QTL on Chr 3 is shown along with the P-values for χ2 test and F-test.
Fine mapping narrows the QTL interval on Chr 3
To increase the resolution of genetic mapping, we increased the number of F2 histological samples from 243 to 402, and we fine mapped the Chr 3 QTL region by genotyping 18 single nucleotide polymorphism (SNP) markers spanning across the QTL interval in all 402 F2 mice (Figure 3a; Supplementary Table S3). Interval mapping using these additional data yielded considerably higher LOD scores (8.7–24.3) for all five lymphatic vessel traits (Figures 3b and c; Table 1). The combined Bayesian 95% CI was narrowed from the earlier 43.6 Mbp to only 22 Mbp (104.5–126.5 Mbp). The narrowed Chr 3 interval contained approximately 220 genes. C3Bir allele homozygosity at rs3687177, rs13477318, and rs3720182 markers was associated with the highest scores for multiple lymphatic vessel subphenotypes (Supplementary Figure S4). The percent of lymphatic phenotype variance explained by the single QTL on Chr 3 ranged from 9.4 to 24.3% for the five subphenotypes; this conferred the major genetic contribution of the Chr 3 QTL (Table 1).
Figure 3.

Fine mapping of the Chr 3 QTL interval narrows the candidate region. (a) Genetic map showing the position of the previously genotyped microsatellite markers and the newly genotyped 18 SNP markers on Chr 3 used for fine mapping (y axis: cM marker positions). (b) Genetic mapping with an increased number of markers and F2 mice provided higher resolution peaks on Chr 3 for all five traits. (c) Detailed view of mapped QTL on Chr 3 showing LOD score curves for the five traits, with horizontal lines indicating the span of 95% approximate Bayesian credible intervals (95% significance) (x axis: marker position in cM).
Gene expression profiling identifies candidate genes that modify IBD
To investigate the possible differences in steady-state gene expression levels between the two parental Il10−/− mouse strains, we isolated RNA from the colons of 10-week-old B6-Il10−/− and C3Bir-Il10−/− mice that were maintained in an SPF facility without enteric microflora needed to trigger the colitis. These mice did not show any macroscopic signs of colon inflammation or upregulation of inflammatory genes such as Tnf, S100a9, or Ifng (data not shown). We simultaneously quantified the expression of 87 genes from the Chr 3 QTL interval by high-throughput real-time RT-PCR, using custom-made low-density arrays (Supplementary Table S4). Four genes with significant differences in expression levels between strains were identified (corrected P≤0.05): ferric-chelate reductase 1 (Frrs1), G-protein-coupled receptor 88 (Gpr88), and Vcam1 were expressed at higher levels in C3Bir-Il10−/− mice whereas glutathione S-transferase, mu 1 (Gstm1) was expressed at higher levels in B6-Il10−/− mice (Figure 4; Supplementary Table S4).
Figure 4.
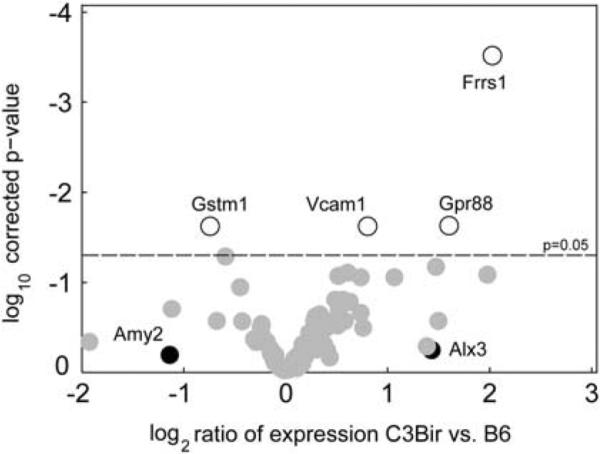
Gene expression levels in colitis-resistant and -susceptible mouse strains, assessed by high-throughput quantitative PCR. Each circle represents a gene from the Chr 3 QTL interval. The log2 ratio of expression in C3Bir mice, compared with B6 strain is shown on the x axis and log10 of Benjamini-Hochberg corrected P-values are shown on the y axis. Genes that had a statistically significant change between the two strains are shown with open circles. The genes that were expressed at levels below the threshold are shown in black. Genes that were expressed at significantly higher levels in the C3H strain are shown on the right, whereas genes that were expressed at lower levels in the C3H strain are shown on the left (N per strain = 5).
In silico analysis enables haplotype block-based reduction of the QTL interval
As a complementary systems genetics approach, we performed in silico haplotype block analysis to narrow the QTL region on Chr 3. We used the mouse haplotype block viewer tool (http://mouse.perlegen.com/mouse/)32 to align the haplotype blocks of C3H/HeJ and C57BL/6J strains within the QTL 95% CI. As C3H/HeJBir is a recently developed substrain of C3H/HeJ, it was reasonable to use the C3H/HeJ strain for the analysis.33 The blocks in which the C3H/HeJ and C57BL/6J strains appear to be non-identical by descent provide the most probable location for the causative polymorphisms.20 Using this approach, we achieved an approximately 40% reduction of the Chr 3 QTL CI; the narrowed interval contained only 89 genes (Figure 5).
Figure 5.

Haplotype block analysis excludes a large part of QTL credible interval genes. On the basis of the identical-by-descent blocks of SNPs, approximately 40% of CI was excluded from the further analysis. C57BL/6J haplotype blocks are indicated by light grey and non-identical-by-descent C3H/HeJ haplotype blocks are indicated by dark grey. Areas that lack genotype information are shown in black. Sequence coordinates of the Chr 3 CI (in Mbp) are provided at the bottom line of the image.
Analysis of candidate genes polymorphisms
Using the Mouse Phenome Database and Sanger Mouse Genomes Project SNP tool, we found 13 579 SNPs within the Chr 3 QTL CI that differ between C3H/HeJ and C57BL/6J strains. We focused our search on functional SNPs (coding non-synonymous, UTR-miRNA-binding sites, regulatory regions, and splice sites) that reside within the blocks that are non-identical-by-descent (Table 2). Importantly, we found a coding non-synonymous SNP in the Vcam1 gene (rs13477355). The presence of this SNP in exon 9 of the Vcam1 gene in C3Bir strain was verified by sequencing. SNP rs13477355 changes the amino-acid 693 in the extracellular domain of the protein from asparagine (B6-Il10−/−) to aspartic acid (C3Bir-Il10−/−). By using the bioinformatics tool GOR4,34 we determined the effect of this change on the secondary structure of VCAM1 to be the replacement of a random coil by an α-helix structure. Furthermore, there were SNPs in the 3′UTR (rs30131404) and in the 5′upstream region (rs30131638) of Vcam1 that might account for the differences of Vcam1 expression observed in the colons of the two mouse strains (Table 2).
Table 2.
Forty-six genes from the reduced QTL interval on chromosome 3 contain one or more potentially functional SNPs
| Gene symbol | Gene start (Mbp) | Gene end (Mbp) | 3′UTR SNP | Coding non-synonymous SNPs | Locus region SNP |
|---|---|---|---|---|---|
| Adora3 | 105.67 | 105.73 | Yes | ||
| I830077J02Rik | 105.73 | 105.74 | Yes | Yes | |
| Atp5f1 | 105.75 | 105.76 | Yes | ||
| Wdr77 | 105.76 | 105.77 | Yes | ||
| Ovgp1 | 105.78 | 105.79 | Yes | ||
| Dennd2d | 106.29 | 106.30 | Yes | ||
| Kcna10 | 106.99 | 107.00 | Yes | Yes | |
| Gm131 | 107.01 | 107.02 | Yes | ||
| Ntng1 | 109.58 | 109.95 | Yes | ||
| LOC625585 | 111.68 | 111.88 | Yes | ||
| EG229746 | 112.11 | 112.11 | Yes | Yes | |
| EG433643 | 112.86 | 112.86 | Yes | ||
| Col11a1 | 113.73 | 113.92 | Yes | Yes | |
| Olfm3 | 114.61 | 114.83 | Yes | ||
| Edg1 | 115.41 | 115.42 | Yes | ||
| Dph5 | 115.59 | 115.63 | Yes | ||
| Slc30a7 | 115.65 | 115.71 | Yes | ||
| Vcam1 | 115.81 | 115.83 | Yes | Yes | Yes |
| Dbt | 116.22 | 116.25 | Yes | ||
| Lrrc39 | 116.27 | 116.29 | Yes | Yes | Yes |
| Ccdc76 | 116.28 | 116.32 | Yes | Yes | |
| Sass6 | 116.3 | 116.33 | Yes | ||
| Slc35a3 | 116.37 | 116.42 | Yes | ||
| Frrs1 | 116.56 | 116.61 | Yes | Yes | |
| Palmd | 116.56 | 116.61 | Yes | ||
| 1700061I17Rik | 116.77 | 116.78 | Yes | Yes | |
| D3Bwg0562e | 117.02 | 117.06 | Yes | Yes | |
| Alg14 | 120.99 | 121.06 | Yes | ||
| Cnn3 | 121.13 | 121.16 | Yes | ||
| Slc44a3 | 121.16 | 121.24 | Yes | ||
| LOC100043726 | 122.08 | 122.09 | Yes | ||
| Synpo2 | 122.78 | 122.94 | Yes | ||
| Sec24d | 122.97 | 123.07 | Yes | Yes | Yes |
| G430022H21Rik | 123.07 | 123.09 | Yes | Yes | |
| Prss12 | 123.15 | 123.21 | Yes | ||
| EG668804 | 123.21 | 123.21 | Yes | ||
| Ndst3 | 123.23 | 123.38 | Yes | Yes | Yes |
| EG668809 | 123.99 | 124.00 | Yes | ||
| 1700006A11Rik | 124.1 | 124.13 | Yes | Yes | Yes |
| Ndst4 | 125.14 | 125.43 | Yes | ||
| Ugt8a | 125.57 | 125.64 | Yes | Yes | |
| Arsj | 126.07 | 126.14 | Yes | ||
| Camk2d | 126.3 | 126.55 | Yes | ||
| Ank2 | 126.62 | 126.70 | Yes | Yes | |
| 4930422G04Rik | 127.26 | 127.32 | Yes | Yes | |
| Alpk1 | 127.37 | 127.48 | Yes | Yes | Yes |
Abbreviations: QTL, quantitative trait locus; SNP, single nucleotide polymorphism.
The QTL interval was reduced by fine mapping and by excluding identical-by-descent haplotype blocks. Coding-non-synonymous SNP in Vcam1 (rs13477355) was verified by sequencing. Gene symbols and locations are given according to NCBI mouse build 37.1.
Increased VCAM1 expression in several different colon cell types of the C3H/HeJ strain, compared with the C57BL/6J strain
Since Vcam1 was identified as a candidate gene that had increased mRNA levels in the normal colon of C3Bir-Il10−/− mice, compared with B6-Il10−/− mice, and that also contained several candidate SNPs, we investigated whether there might also be differences in VCAM1 protein levels. We prepared colon cell suspensions from wild-type C3H/HeJ and C57BL/6J mice and performed fluorescence-activated cell sorting (FACS) analysis of VCAM1 levels in total colon cells, lymphatic endothelial cells (CD45−CD31+ podoplanin+), and blood vascular endothelial cells (CD45−CD31+ podoplanin−). We found that regardless of the cell type analyzed, VCAM1 expression was higher in the C3H/HeJ strain (Figure 6). A clear trend in the difference of the mean fluorescence intensity between the two strains was evident (P = 0.06–0.08), confirming the results obtained by real-time RT-PCR.
Figure 6.

VCAM1 is expressed at higher levels in colon cells of C3H/HeJ, compared with C57BL/6J mice. Whole-cell suspensions of colon (a) were used to compare the expression levels of VCAM1 between wild-type C3H/HeJ and C57BL/6J mice. To distinguish between different cell types, 4-color FACS analysis was performed with colon cells. VCAM1 mean fluorescence intensity (MFI) values are shown for colon lymphatic endothelial cells (CD45−CD31+ podoplanin+) (b), and colon blood vascular endothelial cells (CD45− CD31+ podoplanin−) (c). A higher expression level of VCAM1 was observed in all C3H/HeJ cell types. Data are shown from four separate experiments. Each box plot shows the median, upper, and lower quartiles as well as minimum and maximum MFI values. The paired t-test was used to calculate the P-values.
Neutralizing antibody against VCAM1 reduces inflammatory lymphatic vessel changes and distal colon inflammation in C3Bir-Il10−/− mice
By 6 weeks of age, C3Bir-Il10−/− mice develop a form of colitis that resembles IBD (Figures 1a and b). To investigate a functional role for VCAM1 in modifying IBD susceptibility, 6-week-old C3Bir-Il10−/− mice raised in an environment with requisite microflora for colitis development were given injections of a monoclonal antibody that specifically blocks VCAM1. After 2 weeks of treatment, colons of mice that were given antibody and control mice were processed and scored for inflammatory lymphatic vessel changes and for colon mucosal inflammation severity. We observed an overall reduction in the lymphatic vessel score (P = 0.04) and in distal colon inflammation (P = 0.04) in mice given anti-VCAM1 antibody (Figure 7), indicating that higher expression of VCAM1 in Il10−/− mice has an important function in the development of IBD.
Figure 7.

Systemic blockade of VCAM1 reduces lymphatic vessel inflammatory changes and distal colon inflammation in a mouse model of IBD. (a) Representative images showing the extent of inflammatory lymphatic vessel changes in colon sections from C3Bir-Il10−/− mice injected with control (IgG) or anti-VCAM1 antibodies, immunostained for LYVE1 (red). Scale bars: 100 μm. (b) Quantitative lymphatic vessel and colitis analysis was performed on colon sections of anti-VCAM1-treated mice (filled triangles) and control IgG-treated mice (filled squares). A significant reduction of the total score for lymphatic vessel changes in the colon wall area and distal colon inflammation was observed (*P<0.05). Horizontal lines represent the mean values (N mice IgG group = 8, N mice anti-VCAM1 group = 6).
Discussion
We used the Il10−/− mouse model of colon inflammation to identify genes that modify susceptibility to IBD, a disease with rising incidence in developed countries. Genetic mapping, together with lymphatic vessel phenotyping, enabled us to identify a large-effect QTL on Chr 3. We identified VCAM1 as a candidate modifier gene for IBD by combining a systems genetics approach, quantitative analysis of protein expression levels, and an in vivo function-blocking approach.
Several mouse models of IBD have been proposed.1 The Il10−/− model faithfully reflects the human IBD processes in several aspects, namely spontaneous chronic disease development because of immune dysregulation, Th1/Th17 polarized response, and intestinal microflora phenotype dependence.22 Moreover, a SNP near the 3′ UTR of the Il10 gene was consistently replicated in human genome-wide ulcerative colitis association studies and low ileal IL10 was found to be predictive of Crohn's disease recurrence.5,35 The Il10−/− model is also valuable because the phenotype ranges from mild to severe, depending on the genetic background of the mouse.23
A colitis QTL (cytokine deficiency-induced colitis susceptibility; Cdcs1) was previously mapped to the distal part of Chr 3 by scoring colon mucosal inflammation.28,29 We found a high degree of correlation between the mucosal inflammation phenotype and the lymphatic vessel phenotype in colons of F2 mice. Importantly, the Cdcs1 interval spans between 62 and 70 cM with the peak marker at 61.8 cM,28,36 thus overlapping at its proximal end with the interval mapped scoring the lymphatic vessel traits. Further subcongenic analysis revealed high genetic complexity of Cdcs1 with likely more than one gene contributing to the phenotype.37 In this study, we have evaluated the thus-far neglected inflammatory changes that occur in the lymphatic vasculature, in contrast to the earlier investigations of the `classical' colitis symptoms of epithelial damage and inflammatory cell accumulation. This new approach of scoring related, but distinct quantitative traits enabled us to identify Vcam1, within the Cdcs1 interval, as a modifier of lymphatic vessel inflammatory changes, underlining the complexity of Cdcs1.
Lymphatic vessels undergo significant changes during inflammation, and a higher density of lymphatic vessels was also observed in the colons of IBD patients.26 Moreover, when normal and diseased human bowel was studied in Crohn's disease patients, lymphatic failure was found in some apparently unaffected areas, and further examination of these areas confirmed the presence of early IBD.38 Pathologists have also noted that marked edema of the submucosa with enlarged lymphatic capillaries is present without mucosal ulceration, pointing to the involvement of lymphatic vessels in the early stages of human IBD.39 There are two possible reasons for lymphatic vessel enlargement in inflamed tissue. Lymphatic endothelial cells might, due to the presence of activating inflammatory signals, start to proliferate, resulting in enlarged vessel diameters.9 On the other hand, lymphatics might be enlarged because they are dilated within the edematous tissue and have lost their draining function. This could be caused by increased blood vessel permeability or by an obstruction further down the collecting lymphatic vessels that is created by cell, bacteria, and protein debris, which are abundant in chronically inflamed tissues.17,38
In the normal colon, lymphatic vessels are located longitudinally, in a narrow area beneath the muscularis mucosae, with rare branches that reach the most basal aspect of the crypts and with perpendicular branches that reach deep into submucosa.26 As an additional trait, we analyzed the invasion of lymphatic vessels into the lamina propria between the epithelial crypts. The observation of lymphatics in the mucosa of inflamed colon clearly shows active lymphangiogenesis, because under normal physiological conditions, no lymphatics are found at that site.
Angiogenesis has been reported to have an important function in the pathogenesis of IBD.6 Surprisingly, we found that lymphatic vessel changes observed in mice with colitis were more dramatic than the blood vessel changes observed; area covered by blood vessel endothelium did not change significantly in colitis. These findings indicate that the numerous earlier studies that investigated the CD31+ vasculature in IBD overlooked the fact that lymphatic vessels also express CD31.40 Thus, some of the earlier reported blood vascular changes might indeed represent alterations of the lymphatic vasculature.41,42
The scoring of the lymphatic inflammatory changes observed in Il10−/− mice proved to be a very sensitive method of analysis; it enabled us to map a large-effect QTL that spanned 43.6 Mbp on Chr 3. Using SNP markers and a larger number of F2 mice, we were able to narrow the CI to only 22 Mbp on Chr 3.
Nonetheless, the identification of genes that determine IBD susceptibility remained a challenging task that required a systems genetics approach. High-throughput gene expression analysis is a suitable complementary approach to QTL analysis;43 we performed low-density array qPCR analysis, which enabled us to simultaneously quantify the expression of 87 Chr 3 QTL interval genes and to identify Vcam1 as the most plausible candidate; its expression was significantly higher in colons of C3Bir-Il10−/− compared with B6-Il10−/− mice.
Recently, a sequence-based variation map of 8.27 million SNPs in inbred mouse strains became available, leading to the generation of a mouse haplotype map.32 Laboratory mice have been inbred for the relatively short time of 100 years, so an estimated 97% of polymorphisms are ancestral, providing a 3% chance that a causative QTL lies inside an identical-by-descent block.20,43 Using the haplotype block predictor tool, we were thus able to exclude a large part of the Chr 3 QTL CI, reducing the number of genes in this region from 220 to 89.
The in silico search for polymorphic SNPs that might influence protein abundance, function, or stability led to the identification of four functional SNPs associated with Vcam1. As the polymorphisms reported for the C3H/HeJ strain might not necessarily be present in the C3Bir substrain, we investigated by sequencing the most promising candidate SNP in the Vcam1 gene and confirmed its effect on the secondary protein structure by a prediction tool.
This is the first report of differential mRNA and protein expression levels of VCAM1 in the C3H versus the C57BL/6J mouse strain. Although with the chosen dosage and timing of the blocking antibody treatment only distal colon inflammation was significantly diminished, the ability of a VCAM1 neutralizing antibody to reduce the lymphatic vessel changes associated with colitis in C3Bir-Il10−/− mice supports the concept that VCAM1 has a function in IBD pathogenesis, along with results from genome-wide association studies of human IBD44 and other models.45,46 VCAM1 expression by blood vessel endothelium is required for leukocyte entry into sites of inflammation.7,47 Therefore, reagents designed to block VCAM1 function in vivo could reduce the number of immune effector cells that localize to the colon and diminish IBD severity. Importantly, however, VCAM1 is also upregulated on the lymphatic endothelium during inflammation, which was recently shown to occur in inflamed human small intestine and a mouse model of skin contact hypersensitivity,13,48 and seems to be required for the process of leukocyte transmigration across lymphatic endothelium to the draining lymph nodes.13,49 There are several possibilities how VCAM1 may contribute to the lymphatic vessel phenotype in IBD. Elevated expression of VCAM1 in susceptible individuals could allow for a higher rate of leukocyte infiltration into the tissue, resulting in higher levels of lymphangiogenic factors. In chronic inflammatory states, such as IBD, the persistence of mucosal antigens supports a switch from the innate to the adaptive immune response and a subsequent change in the inflammatory infiltrate. A crucial step allowing for this switch is the transport of antigen-presenting cells from inflamed tissue, via lymphatic vessels, to the draining lymph nodes in which lymphocyte priming occurs. Thus, it is likely that because of the role of VCAM1 in regulating dendritic cell egress from the tissue,13 its elevated expression on lymphatic vessels supports a higher rate of lymphocyte priming in the draining lymph nodes. Increased levels of circulating VCAM1 were shown in Crohn's disease patients and VCAM1 upregulation was reported in bowel EC isolated from IBD patients.50,51 Further support for the role of VCAM1 in IBD pathogenesis comes from studies showing the efficacy of natalizumab, a humanized antibody against the a4 integrin subunit, in treatment of patients with IBD;52 the integrins α4b1 and α4b7 are expressed by leukocytes and are VCAM1 ligands.
We show that a mouse systems genetics approach represents a powerful method for studying complex diseases; we used it to identify VCAM1 as a modifier gene for IBD. Our findings also reveal the importance of lymphatic vessel changes for the pathogenesis of IBD. The molecular evidence we present indicates that reagents designed to target adhesion molecules that mediate leukocyte adhesion to the activated lymphatic vasculature might be effective in treating patients with IBD.
Materials and methods
Mice and tissue preparation
The generation of the C3Bir.129P2-Il10tm1Cgn /J (JR#3968; referred to as C3Bir-Il10−/−) and B6.129P2-Il10tm1Cgn /J (JR#4333; referred to as B6-Il10−/−) congenic mouse stocks that are homozygous for a targeted mutation of the Il10 gene, and of their reciprocal F1 hybrids, has been described previously.23 F1 offspring were previously intercrossed to generate a total of 402 F2 mice at The Jackson Laboratory in a conventional SPF vivarium containing requisite microflora for colitis development. At 6 weeks of age, F2 mice were killed and tissue was collected as described.28 Colons were fixed in Fekete's acid–alcohol–formalin fixative and embedded in paraffin.28 B6-Il10−/− and C3Bir-Il10−/− parental mice were killed at ages 4, 6, and 8 weeks. All mouse experiments performed at the Jackson Laboratory were in accordance with an Institutional Animal Care and Use Committee approved protocol.
Immunostains
Routine hematoxylin–eosin stains were performed on paraffin sections of 402 F2 mice and parental strain mice. In addition, serial sections were stained for the lymphatic marker LYVE1. Slides were deparaffinized and antigen was unmasked by boiling in 0.01 M citrate buffer. Rabbit anti-mouse LYVE1 antibody (kindly provided by Dr N Gale, Regeneron Pharmaceuticals, Tarrytown, NY, USA) was applied, followed by the immunohistochemical reaction as described.16 Colon cryosections were fixed at −20 °C in aceton and at 4 °C in 80% methanol. Primary rabbit anti-mouse LYVE1 antibody (Angiobio, Del Mar, CA, USA) and rat anti-mouse CD31 (BD Biosciences, Franklin Lakes, NJ, USA) were applied at 4 °C overnight and the immunohistochemical reaction or immunofluorescence staining with Alexa Fluor-488 or Alexa Fluor-594-conjugated secondary antibodies (Invitrogen, Carlsbad, CA, USA) followed. To acquire images, an Axioskop2 microscope equipped with Plan-APOCHROMAT 10 × /0.45 objective and AxioCam MRc digital camera was used (Zeiss, Jena, Germany) in conjunction with AxioVision Rel. 4.7.1 software.
Phenotype scoring system
Colon sections were scored for five quantitative traits of inflammatory lymphatic vessel changes (detailed explanation in Supplementary Table S1). The quantification of the lymphatic vasculature was performed as described previously10,14,16,53,54 with slight modifications. LYVE1-immunostained sections of the entire colon (longitudinal sections of the rolled colon) were scanned by light microscopy at low magnification (× 5), and the areas of tissue with the highest density of LYVE1-positive structures with lumina (lymphatic vessels) were selected (`hot spots'). Three hot-spot images of each mouse colon were taken at × 10 magnification and morphometric analysis was performed using the IP-LAB software (Scanalytics, Fairfax, VA, USA). The region of interest inside the hot-spot image was defined as the submucosal colon wall over a length of 500 μm. The submucosal wall area was defined as the area that encompasses the lamina propria in the basis of the epithelial crypts, the muscularis mucosae, submucosa, and muscle layers. The percentage of region of interest covered by lymphatic vessels was computed and the average size of lymphatic vessels was determined. In addition, the invasion of lymphatic vessels into the lamina propria was analyzed. Total score was obtained by summing up the individual trait scores (detailed explanation in Supplementary Table S1). The extent of colon mucosal inflammation in F2 mice was determined with the histopathological colitis scoring system as described previously.28,29 Briefly, scoring of epithelial hyperplasia, ulceration, percent of damaged area, and severity of inflammation in the proximal, mid, and distal colon was performed. Correlation coefficients were calculated for 16 colitis traits and for 5 lymphatic vessel traits and their scores in 389 F2 mice. The MATLAB statistics toolbox (The MathWorks, Natick MA, USA) was used for this analysis (corrcoef function).
Genotyping
DNA was isolated from tail clips. Multiplex genotyping (ABI 370, Applied Biosystems, Foster City, CA, USA) was performed for 110 microsatellite markers, providing a coverage of at least 20 cM on each autosome and the X chromosome (Supplementary Table S2).28 Eighteen SNP markers on Chr 3 were genotyped with Amplifluor technology (Chemicon International, Temecula CA, USA) by The Jackson Laboratory genotyping service. The linkage maps and marker positions reported are based on online Mouse Genome Informatics (MGI 4.2) resource (http://www.informatics.jax.org) (Supplementary Tables S2 and S3).
QTL mapping (genome-wide scan and fine mapping)
Genome-wide scans were carried out by interval mapping.55 The analysis was performed considering a normal distribution of all F2 traits, except for the ingrowth of lymphatic vessels into lamina propria, which was treated as a binary trait. LOD scores were computed at the marker locations across the genome for the five quantitative lymphatic traits. QTLs whose LOD scores exceeded the genome-wide 5% significance levels were further analyzed. The significance threshold was determined by performing a permutation test56 with 512 permutation replicates. Interval estimates of QTL location were obtained through 95% approximate Bayesian CI.57 Pairwise interactions among QTLs were assessed using two-dimensional genome-wide scans.57,58 LOD scores for pairwise scans were completed at 5 cM resolution. Significance thresholds were obtained by performing 1600 simulations on F2 mice genotype and phenotype data and computed at LOD scores 8.30, 6.50, 5.80, 6.98, 3.84 for full, conditional interaction, interaction, additive, and conditional additive models. All QTL analyses were performed with the R/qtl software package, version 1.05–2,59 with R version 2.4.1.60
Fine mapping of the candidate interval was performed by including additional 159 F2 mice and by genotyping all F2 mice (n = 402) for 18 additional SNP markers located within the Chr 3 candidate interval (Figure 2a). LOD scores were computed at 1 cM interval across the genome. SNP marker positions were converted to a pseudo-centiMorgan scale by dividing the physical position in megabase pairs by 2 and adjusting for the known microsatellite marker physical positions (UniSTS annotation of NCBI Mouse Build 37.1). Physical positions of SNP markers were retrieved from NCBI (dbSNP 128) (Supplementary Table S3).
Quantitative gene expression analysis by TaqMan low-density array
For gene expression studies, B6-Il10−/− and C3Bir-Il10−/− mice (originating from The Jackson Laboratory) were maintained in a strict hygienic barrier system in which they did not develop any signs of IBD (Hannover Medical School facility). A piece of proximal colon was harvested from five 10-week-old mice of each genotype, washed in cold PBS and immersed in RNAlater solution (Ambion, Austin, TX, USA). Tissue was homogenized using a TissueLyser (Qiagen, Hilden, Germany). RNA was isolated using the RNeasy Mini kit (Qiagen). RNA quality was assessed with a Bioanalyzer (Agilent, Santa Clara, CA, USA) and total RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). A custom low-density array in 384-well microfluidic card format was designed to contain available TaqMan assays for 87 genes located within the Chr 3 QTL interval (107–128 Mbp), and five endogenous control genes, three endothelial cell marker, and one inflammatory marker gene (Supplementary Table S4). cDNA (600 ng per sample) was mixed with TaqMan master mix and PCR was performed using a 7900HT Fast Real-Time PCR System (Applied Biosystems). The results were analyzed using Real-Time StatMiner software (Integromics, Madrid, Spain). For differential expression analysis, a LIMMA modified t-test was used to calculate ΔΔCt quantities, which were used for fold-change computation. Values from two (Actb and Pgk1) of five endogenous control genes were used for the normalization, after finding the best combination using the built-in Normfinder option.61 The false discovery rate was controlled by correcting the P-values with the Benjamini–Hochberg method.
Bioinformatics tools and SNP analysis
The haplotype block viewer tool (http://mouse.perlegen.com/mouse/) was used to align the haplotype blocks of C57BL/6J and C3H/HeJ mouse strains and determine the blocks in which these strains differed. As previously reported,32 the pairwise scores were determined using a Hidden Markov Model trained to detect low-SNP-density and high-SNP-density regions. Haplotype block regions were detected when the matching Hidden Markov Model changes state from low- to high density or vice versa.32 The Chr 3 QTL region, as defined by the 95% CI, was investigated for all of the known polymorphisms between the C57BL/6J and C3H/HeJ mouse strains. For this purpose, we used the Mouse Phenome Database SNP feature (http://www.jax.org/phenome/SNP) and Sanger Mouse Genomes Project SNP tool (http://www.sanger.ac.uk/cgi-bin/modelorgs/mousegenomes/snps.pl) with all genes and polymorphisms mapped to NCBI Build 37 and dbSNP 128. DNA extracted from two C3Bir-Il10−/− mice and two B6-Il10−/− mice was used for amplification and subsequent sequencing of a 416 bp PCR product using the primers Vcam1-9-F `5′-AGT-CTC-GAT-GGTCCT-CAG-TG-3′ and Vcam1-9-R `5′-GGT-TCT-GGGAAG-TCT-GAA-TG-3′. PCR was carried out using REDExtract-N-AMP-PCR ReadyMix (Sigma-Aldrich, Munich, Germany). PCR products were gel purified using the Nucleo Spin Extract II Kit (Machery&Nagel, Düren, Germany) and sequenced by SequenceLaboratories Göttingen GmbH (Göttingen, Germany). Sequences were translated (http://www.expasy.ch/tools/dna.html) and secondary structures predicted by GOR4 (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page = npsa_gor4.html).
FACS analysis
For FACS analysis, C3H/HeJ and C57BL/6J wild-type mice originating from The Jackson Laboratory were maintained in the Rodent Center HCI (ETH Zurich, Switzerland) under conventional, non-SPF conditions. Age- and sex-matched mice were used to obtain colon tissue. Colons were opened longitudinally, washed in cold PBS, and the mucus was gently scraped off in 1 mM DTT for 3 min. Then, the colon was cut into small pieces and incubated at 37 °C in 8 mg ml−1 collagenase IV (Invitrogen), 0.5 mg ml−1 DnaseI (Roche, Rotkreuz, Switzerland), 5 mM CaCl2 in PBS for 30 min while rotating. Tissue suspensions were passed through a 70 μm cell strainer (BD Biosciences) while flushing with 2% fetal bovine serum in PBS. Cell suspensions were centrifuged at 500 g for 10 min at RT and resuspended in PBS containing 2% fetal bovine serum and 1 mM EDTA. The following antibodies were used: allophycocyaninconjugated rat anti-mouse CD31; peridinin-chlorophyll-protein-conjugated rat anti-mouse CD45 (BD Biosciences); fluorescein isothiocyanate-conjugated rat anti-mouse VCAM1 (eBiosciences, San Diego, CA, USA); hamster anti-mouse podoplanin (clone 8.1.1; Developmental Studies Hybridoma Bank, Iowa City, IA, USA); and anti-hamster phycoerythrin (Invitrogen) and isotype control antibodies (BD Biosciences). FACS was performed using a BD FACSCanto and the FACSDiva software (BD Biosciences). Data were analyzed with FlowJo software (Treestar, Ashland, TN, USA). Four independent experiments were performed. Animal experiments were approved by the Kantonales Veterinäramt Zurich.
In vivo blockade of VCAM1 in the C3Bir-Il10−/− model of IBD
C3Bir-Il10−/− mice were housed in a conventional SPF facility containing colitis-requisite microflora at The Jackson Laboratory-West (Sacramento, CA, USA). Six week-old mice received intraperitoneal injections of 100 μg of the blocking rat anti-mouse VCAM1 antibody 6C7.147 or control rat IgG1 (BioLegend, San Diego, CA, USA), every second day for 2 weeks. Each group consisted of four males and four females, grouped according to similar body weights. At day 15, mice were killed and the colon was removed, rolled, and cryopreserved in optimal cutting temperature compound (Sakura Finetek, Zoeterwoude, NE, USA). Cryosections (8 μm) were immunostained for LYVE1 (Angiobio) and counterstained with hematoxylin. The colon sections were scored with the scoring system for lymphatic vessel changes described above, and also with the previously described colon mucosal inflammation histopathological scoring system.28,29
Supplementary Material
Acknowledgements
We thank C Halin for scientific advice and G Churchill for inputs on QTL analysis. We thank J Scholl and L Nguyen for technical assistance and B Snijder for assistance with data analysis. This work was supported by National Institutes of Health grant CA69184, Swiss National Science Fund grant 3100A0-108207, Austrian Science Foundation grant S9408-B11, Cancer League Zurich, Oncosuisse and The European Commission grant LSHC-CT-2005-518178 (MD), National Institutes of Health grant DK44240, and Broad Biomedical Research Program (EHL and JPS).
Footnotes
Conflict of interest The authors declare no conflict of interest.
Supplementary Information accompanies the paper on Genes and Immunity website (http://www.nature.com/gene)
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 4.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (New York, NY) 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franke A, Balschun T, Karlsen TH, Sventoraityte J, Nikolaus S, Mayr G, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–1323. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 6.Deban L, Correale C, Vetrano S, Malesci A, Danese S. Multiple pathogenic roles of microvasculature in inflammatory bowel disease: a Jack of all trades. Am J Pathol. 2008;172:1457–1466. doi: 10.2353/ajpath.2008.070593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 8.Cueni LN, Detmar M. The lymphatic system in health and disease. Lymphat Res Biol. 2008;6:109–122. doi: 10.1089/lrb.2008.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ, et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest. 2005;115:247–257. doi: 10.1172/JCI22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kunstfeld R, Hirakawa S, Hong YK, Schacht V, Lange-Asschenfeldt B, Velasco P, et al. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood. 2004;104:1048–1057. doi: 10.1182/blood-2003-08-2964. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Q, Lu Y, Proulx ST, Guo R, Yao Z, Schwarz EM, et al. Increased lymphangiogenesis in joints of mice with inflammatory arthritis. Arthritis Res Ther. 2007;9:R118. doi: 10.1186/ar2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerjaschki D, Regele HM, Moosberger I, Nagy-Bojarski K, Watschinger B, Soleiman A, et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol. 2004;15:603–612. doi: 10.1097/01.asn.0000113316.52371.2e. [DOI] [PubMed] [Google Scholar]
- 13.Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. 2006;203:2763–2777. doi: 10.1084/jem.20051759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kajiya K, Hirakawa S, Detmar M. Vascular endothelial growth factor-A mediates ultraviolet B-induced impairment of lymphatic vessel function. Am J Pathol. 2006;169:1496–1503. doi: 10.2353/ajpath.2006.060197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halin C, Tobler NE, Vigl B, Brown LF, Detmar M. VEGF-A produced by chronically inflamed tissue induces lymphangiogenesis in draining lymph nodes. Blood. 2007;110:3158–3167. doi: 10.1182/blood-2007-01-066811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kajiya K, Kunstfeld R, Detmar M, Chung JH. Reduction of lymphatic vessels in photodamaged human skin. J Dermatol Sci. 2007;47:241–243. doi: 10.1016/j.jdermsci.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kovi J, Duong HD, Hoang CT. Ultrastructure of intestinal lymphatics in Crohn's disease. Am J Clin Pathol. 1981;76:385–394. doi: 10.1093/ajcp/76.4.385. [DOI] [PubMed] [Google Scholar]
- 18.Kataru RP, Jung K, Jang C, Yang H, Schwendener RA, Baik JE, et al. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood. 2009;113:5650–5659. doi: 10.1182/blood-2008-09-176776. [DOI] [PubMed] [Google Scholar]
- 19.Kim KE, Koh YJ, Jeon BH, Jang C, Han J, Kataru RP, et al. Role of CD11b+ macrophages in intraperitoneal lipopolysaccharide-induced aberrant lymphangiogenesis and lymphatic function in the diaphragm. Am J Pathol. 2009;175:1733–1745. doi: 10.2353/ajpath.2009.090133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wade CM, Kulbokas EJ, 3rd, Kirby AW, Zody MC, Mullikin JC, Lander ES, et al. The mosaic structure of variation in the laboratory mouse genome. Nature. 2002;420:574–578. doi: 10.1038/nature01252. [DOI] [PubMed] [Google Scholar]
- 21.Peters LL, Robledo RF, Bult CJ, Churchill GA, Paigen BJ, Svenson KL. The mouse as a model for human biology: a resource guide for complex trait analysis. Nat Rev. 2007;8:58–69. doi: 10.1038/nrg2025. [DOI] [PubMed] [Google Scholar]
- 22.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 23.Bristol IJ, Farmer MA, Cong Y, Zheng XX, Strom TB, Elson CO, et al. Heritable susceptibility for colitis in mice induced by IL-10 deficiency. Inflamm Bowel Dis. 2000;6:290–302. doi: 10.1002/ibd.3780060407. [DOI] [PubMed] [Google Scholar]
- 24.Kalima TV. The structure and function of intestinal lymphatics and the influence of impaired lymph flow on the ileum of rats. Scand J Gastroenterol. 1971;10:1–87. [PubMed] [Google Scholar]
- 25.Kalima TV, Saloniemi H, Rahko T. Experimental regional enteritis in pigs. Scand J Gastroenterol. 1976;11:353–362. [PubMed] [Google Scholar]
- 26.Fogt F, Pascha TL, Zhang PJ, Gausas RE, Rahemtulla A, Zimmerman RL. Proliferation of D2-40-expressing intestinal lymphatic vessels in the lamina propria in inflammatory bowel disease. Int J Mol Med. 2004;13:211–214. [PubMed] [Google Scholar]
- 27.Van Kruiningen HJ, Colombel JF. The forgotten role of lymphangitis in Crohn's disease. Gut. 2008;57:1–4. doi: 10.1136/gut.2007.123166. [DOI] [PubMed] [Google Scholar]
- 28.Farmer MA, Sundberg JP, Bristol IJ, Churchill GA, Li R, Elson CO, et al. A major quantitative trait locus on chromosome 3 controls colitis severity in IL-10-deficient mice. Proc Natl Acad Sci USA. 2001;98:13820–13825. doi: 10.1073/pnas.241258698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bleich A, Mahler M, Most C, Leiter EH, Liebler-Tenorio E, Elson CO, et al. Refined histopathologic scoring system improves power to detect colitis QTL in mice. Mamm Genome. 2004;15:865–871. doi: 10.1007/s00335-004-2392-2. [DOI] [PubMed] [Google Scholar]
- 30.Prevo R, Banerji S, Ferguson DJ, Clasper S, Jackson DG. Mouse LYVE-1 is an endocytic receptor for hyaluronan in lymphatic endothelium. J Biol Chem. 2001;276:19420–19430. doi: 10.1074/jbc.M011004200. [DOI] [PubMed] [Google Scholar]
- 31.Rogers MS, Rohan RM, Birsner AE, D'Amato RJ. Genetic loci that control the angiogenic response to basic fibroblast growth factor. FASEB J. 2004;18:1050–1059. doi: 10.1096/fj.03-1241com. [DOI] [PubMed] [Google Scholar]
- 32.Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, et al. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–1053. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- 33.Sundberg JP, Elson CO, Bedigian H, Birkenmeier EH. Spontaneous, heritable colitis in a new substrain of C3H/HeJ mice. Gastroenterology. 1994;107:1726–1735. doi: 10.1016/0016-5085(94)90813-3. [DOI] [PubMed] [Google Scholar]
- 34.Garnier J, Gibrat JF, Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996;266:540–553. doi: 10.1016/s0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- 35.Meresse B, Rutgeerts P, Malchow H, Dubucquoi S, Dessaint JP, Cohard M, et al. Low ileal interleukin 10 concentrations are predictive of endoscopic recurrence in patients with Crohn's disease. Gut. 2002;50:25–28. doi: 10.1136/gut.50.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beckwith J, Cong Y, Sundberg JP, Elson CO, Leiter EH. Cdcs1, a major colitogenic locus in mice, regulates innate and adaptive immune response to enteric bacterial antigens. Gastroenterology. 2005;129:1473–1484. doi: 10.1053/j.gastro.2005.07.057. [DOI] [PubMed] [Google Scholar]
- 37.Bleich A, Buchler G, Beckwith J, Petell LM, Affourtit JP, King BL, et al. Cdcs1 a major colitis susceptibility locus in mice; subcongenic analysis reveals genetic complexity. Inflamm Bowel Dis. doi: 10.1002/ibd.21146. e-pub ahead of print 23 October 2009; PMID: 19856416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heatley RV, Bolton PM, Hughes LE, Owen EW. Mesenteric lymphatic obstruction in Crohn's disease. Digestion. 1980;20:307–313. doi: 10.1159/000198452. [DOI] [PubMed] [Google Scholar]
- 39.Robb-Smith AH. A bird's-eye view of Crohn's disease. Proc R Soc Med. 1971;64:157–161. [PMC free article] [PubMed] [Google Scholar]
- 40.Hirakawa S, Hong YK, Harvey N, Schacht V, Matsuda K, Libermann T, et al. Identification of vascular lineage-specific genes by transcriptional profiling of isolated blood vascular and lymphatic endothelial cells. Am J Pathol. 2003;162:575–586. doi: 10.1016/S0002-9440(10)63851-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Danese S, Sans M, Spencer DM, Beck I, Donate F, Plunkett ML, et al. Angiogenesis blockade as a new therapeutic approach to experimental colitis. Gut. 2007;56:855–862. doi: 10.1136/gut.2006.114314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chidlow JH, Jr, Langston W, Greer JJ, Ostanin D, Abdelbaqi M, Houghton J, et al. Differential angiogenic regulation of experimental colitis. Am J Pathol. 2006;169:2014–2030. doi: 10.2353/ajpath.2006.051021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stylianou IM, Affourtit JP, Shockley KR, Wilpan RY, Abdi FA, Bhardwaj S, et al. Applying gene expression, proteomics and single-nucleotide polymorphism analysis for complex trait gene identification. Genetics. 2008;178:1795–1805. doi: 10.1534/genetics.107.081216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vermeire S, Rutgeerts P, Van Steen K, Joossens S, Claessens G, Pierik M, et al. Genome wide scan in a Flemish inflammatory bowel disease population: support for the IBD4 locus, population heterogeneity, and epistasis. Gut. 2004;53:980–986. doi: 10.1136/gut.2003.034033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soriano A, Salas A, Salas A, Sans M, Gironella M, Elena M, et al. VCAM-1, but not ICAM-1 or MAdCAM-1, immunoblockade ameliorates DSS-induced colitis in mice. Lab Invest. 2000;80:1541–1551. doi: 10.1038/labinvest.3780164. [DOI] [PubMed] [Google Scholar]
- 46.Burns RC, Rivera-Nieves J, Moskaluk CA, Matsumoto S, Cominelli F, Ley K. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn's disease in mice. Gastroenterology. 2001;121:1428–1436. doi: 10.1053/gast.2001.29568. [DOI] [PubMed] [Google Scholar]
- 47.Engelhardt B, Laschinger M, Schulz M, Samulowitz U, Vestweber D, Hoch G. The development of experimental autoimmune encephalomyelitis in the mouse requires alpha4-integrin but not alpha4beta7-integrin. J Clin Invest. 1998;102:2096–2105. doi: 10.1172/JCI4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawa Y, Shibata K, Braithwaite MW, Suzuki M, Yoshida S. Expression of immunoglobulin superfamily members on the lymphatic endothelium of inflamed human small intestine. Microvasc Res. 1999;57:100–106. doi: 10.1006/mvre.1998.2132. [DOI] [PubMed] [Google Scholar]
- 49.Ledgerwood LG, Lal G, Zhang N, Garin A, Esses SJ, Ginhoux F, et al. The sphingosine 1-phosphate receptor 1 causes tissue retention by inhibiting the entry of peripheral tissue T lymphocytes into afferent lymphatics. Nat Immunol. 2008;9:42–53. doi: 10.1038/ni1534. [DOI] [PubMed] [Google Scholar]
- 50.Binion DG, West GA, Ina K, Ziats NP, Emancipator SN, Fiocchi C. Enhanced leukocyte binding by intestinal microvascular endothelial cells in inflammatory bowel disease. Gastroenterology. 1997;112:1895–1907. doi: 10.1053/gast.1997.v112.pm9178682. [DOI] [PubMed] [Google Scholar]
- 51.Jones SC, Banks RE, Haidar A, Gearing AJ, Hemingway IK, Ibbotson SH, et al. Adhesion molecules in inflammatory bowel disease. Gut. 1995;36:724–730. doi: 10.1136/gut.36.5.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stefanelli T, Malesci A, De La Rue SA, Danese S. Anti-adhesion molecule therapies in inflammatory bowel disease: touch and go. Autoimmun Rev. 2008;7:364–369. doi: 10.1016/j.autrev.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 53.Hirakawa S, Kodama S, Kunstfeld R, Kajiya K, Brown LF, Detmar M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med. 2005;201:1089–1099. doi: 10.1084/jem.20041896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kajiya K, Sawane M, Huggenberger R, Detmar M. Activation of the VEGFR-3 pathway by VEGF-C attenuates UVB-induced edema formation and skin inflammation by promoting lymphangiogenesis. J Invest Dermatol. 2009;129:1292–1298. doi: 10.1038/jid.2008.351. [DOI] [PubMed] [Google Scholar]
- 55.Lander ES, Botstein D. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics. 1989;121:185–199. doi: 10.1093/genetics/121.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sen S, Churchill GA. A statistical framework for quantitative trait mapping. Genetics. 2001;159:371–387. doi: 10.1093/genetics/159.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sugiyama F, Churchill GA, Higgins DC, Johns C, Makaritsis KP, Gavras H, et al. Concordance of murine quantitative trait loci for salt-induced hypertension with rat and human loci. Genomics. 2001;71:70–77. doi: 10.1006/geno.2000.6401. [DOI] [PubMed] [Google Scholar]
- 59.Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics (Oxford, England) 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- 60.Ihaka R, Gentelman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314. [Google Scholar]
- 61.Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.