
Table 1. Data-collection and refinement statistics for DHP B.
Values in parentheses are for the highest resolution shell.
| DHP B | DHP AB complex† | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P212121 |
| Unit-cell parameters | ||
| a (Å) | 60.65 | 60.17 |
| b (Å) | 67.42 | 67.39 |
| c (Å) | 67.48 | 67.65 |
| α = β = γ (°) | 90 | 90, |
| Temperature (K) | 100 | 100 |
| Wavelength (Å) | 1.54 | 0.913 |
| Resolution (Å) | 30.17–1.58 (1.62–1.58) | 40.00–1.52 (1.56–1.52) |
| Rmerge‡ | 9.7 (54.7) | 7.9 (62.8) |
| I/σ(I) | 21.9 (2.5) | 19.3 (2.7) |
| Completeness (%) | 99.7 (99.9) | 97.0 (98.9) |
| Redundancy | 4.8 (4.8) | 4.9 (4.7) |
| Refinement | ||
| No. of reflections | 36546 (2665) | 39585 (2934) |
| Rwork/Rfree§ (%) | 17.0/21.4 | 17.6/21.9 |
| Average B factor (Å2) | ||
| All atoms | 13.82 | 10.13 |
| Protein | 12.52 | 9.07 |
| Water | 23.84 | 20.63 |
| No. of atoms | ||
| Protein | 3002 | 3139 |
| Water | 373 | 287 |
| R.m.s.d.¶ from ideal | ||
| Bond lengths (Å) | 0.024 | 0.011 |
| Bond angles (°) | 2.197 | 1.365 |
| Ramachandran plot†† (%) | ||
| Most favored | 94.0 | 93.6 |
| Allowed | 6.0 | 6.4 |
†
Protein crystallized using a 1:1 ratio of DHP A and DHP B.
‡
R
merge = 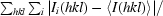
 , where I
i(hkl) is the ith measurement of I(hkl) and 〈I(hkl)〉 is the weighted mean of all measurements of I(hkl).
, where I
i(hkl) is the ith measurement of I(hkl) and 〈I(hkl)〉 is the weighted mean of all measurements of I(hkl).
§
R
work = 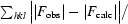
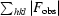 , where F
obs are the observed and F
calc are the calculated structure factors; R
free is the R factor for a subset (5%) of reflections selected before and not included in the refinement.
, where F
obs are the observed and F
calc are the calculated structure factors; R
free is the R factor for a subset (5%) of reflections selected before and not included in the refinement.
¶
Root-mean-square deviation.
††
Calculated using PROCHECK (Laskowski et al., 1993 ▶).