

Abstract
Coiled coil is a ubiquitous structural motif in proteins, with two to seven alpha helices coiled together like the strands of a rope, and coiled coil folding and assembly is not completely understood. A GCN4 leucine zipper mutant with four mutations of K3A, D7A, Y17W, and H18N has been designed, and the crystal structure has been determined at 1.6 Å resolution. The peptide monomer shows a helix trunk with short curved N- and C-termini. In the crystal, two monomers cross in 35° and form an X-shaped dimer, and each X-shaped dimer is welded into the next one through sticky hydrophobic ends, thus forming an extended two-stranded, parallel, super long coiled coil rather than a discrete, two-helix coiled coil of the wild-type GCN4 leucine zipper. Leucine residues appear at every seventh position in the super long coiled coil, suggesting that it is an extended super leucine zipper. Compared to the wild-type leucine zipper, the N-terminus of the mutant has a dramatic conformational change and the C-terminus has one more residue Glu 32 determined. The mutant X-shaped dimer has a large crossing angle of 35° instead of 18° in the wild-type dimer. The results show a novel assembly mode and oligomeric state of coiled coil, and demonstrate that mutations may affect folding and assembly of the overall coiled coil. Analysis of the formation mechanism of the super long coiled coil may help understand and design self-assembling protein fibers.
Keywords: leucine zipper, coiled coil, folding and assembly, self assembly, protein design, protofibril, supramolecular structure
Introduction
The yeast general control non-derepressable 4 (GCN4, 281 amino acids) is a transcriptional activator for amino acid biosynthesis in response to amino acid starvation.1 GCN4 dimerizes through the leucine zipper region,2 and the dimer clamps into the major groove of a DNA double helix through two basic regions.3 A leucine zipper exhibits an alpha helix conformation and contains a series of leucine residues at every seventh position, and such positioning will lead to leucine residues to line on one face of the helix, as each turn in an alpha helix contains 3.6 residues. The two helices in a leucine zipper are coiled together through two stripes of leucine residues on their faces. A leucine zipper is a kind of coiled coil. A coiled coil is a ubiquitous structural motif in proteins,4 and consists of two to seven alpha helices that are coiled together like the strands of a rope through hydrophobic forces. Coiled coils contain a repetitive seven residue pattern called a heptad repeat (abcdefg)n, in which the a and d residues are hydrophobic.5 As there are 3.6 residues in each turn of an alpha helix, the a and d residues are located on the same face of an alpha helix. A leucine zipper has leucine residues at the d position in a heptad repeat. Coiled coils are ideal candidates for protein folding and design studies, as they represent the simplest tertiary structure. Moreover, leucine zipper peptides have been employed to self-assemble into biomaterials, which have potential applications in many fields ranging from nanobiotechnology to tissue engineering.6–8
The structure of the wild-type GCN4 leucine zipper has been previously determined, and the leucine zipper is a two-stranded, parallel coiled coil.9 A series of leucine zipper mutants, which result from mutations of the residues on the interface between the two helices of the leucine zipper, have been designed to understand the effects of these mutations on coiled coil folding and assembly, such as the oligomeric state and the helix orientation of the coiled coil.10–15 Besides the leucine zipper, other zippers such as an alanine zipper,16 a tryptophan zipper,17 and a phenylalanine zipper18 also have been designed. GCN4 leucine zipper mutants in previous studies have been designed based on mutations of the a, d, e, g residues on or around the interface between the two helices of the wide-type leucine zipper. And few have been constructed by mutation of non-interfacial residues, which are hydrophilic and are on the other face of the helix opposite to the interface between the two helices. To address the effects of mutation of non-interfacial residues on coiled coil folding and assembly, such as the oligomeric state, the crossing angle, and the parallel or antiparallel configuration of the coiled coil, we constructed a leucine zipper mutant with mutations of four non-interfacial hydrophilic residues, K3A, D7A, Y17W, and H18N. The leucine zipper mutant corresponds to the yeast GCN4 C-terminus (249–281) and the residues are renumbered from 1 to 33. The four mutations correspond to positions b, f, b, c in a heptad repeat, respectively, and the first three mutations from hydrophilic residues to hydrophobic ones make the non-interfacial face of the helix more hydrophobic. The results show a novel longitudinal association of leucine zipper, and provide a structural basis to view the longitudinal association of self-assembling protein fibers.
Results
Crystal packing
The crystal belongs to space group P3221, with cell dimensions a = b = 31.15 Å, c = 57.32 Å. There is one leucine zipper peptide (Met 2 – Glu 32) in an asymmetric unit. The leucine zipper peptide exhibits a conformation of helix and fits well into the electron density. Six layers of leucine zipper helices pack along the Z axis of the cell, with each layer about 9.5 Å high (Fig. 1). The helices lie down in the layers, with the axes of the helices in parallel to the a, b plane of the cell. In the same layer, the helices are in parallel to each other, although the helices are not aligned with their N- or C-terminal ends. In different layers, the helices are in different orientations. Two helices from adjacent layers cross in 35° and form an X-shaped dimer. The dimer axis is perpendicular to a 32 screw axis, with two Leu 13 residues at the middle of the dimer crossing the 32 screw axis. In the layers, each X-shaped leucine zipper approaches the next one in a head-to-tail way, thus forming a super long coiled coil. The head and tail between dimers are welded together through hydrophobic residues leucine 5, 26, and valine 9, 30.
Figure 1.
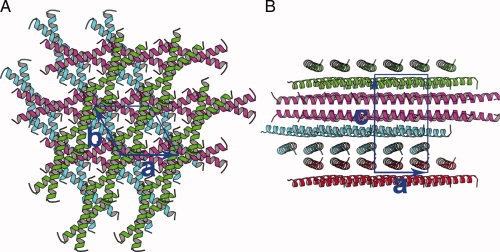
Crystal packing. A, view down to the a, b plane. B, view down to the a, c plane.
Overall structure
The peptide monomer exhibits a long straight helix trunk connected with two short curved tails of the N- and C-termini (Fig. 2). The monomer is in dimensions of 45 Å long and 15 Å wide. The overall structure of the monomer consists of three parts, the N-terminal coil (Met 2-Ala 3), the middle right-handed alpha helix (Gln 4-Ala 30), and the C-terminal coil (Gly 31-Glu 32). The N-terminal residues Met 2 and Ala 3 show a curved conformation different from that of the middle helical part. The starting or the N-cap residue of the helix is Asn 4, which is consistent with the observation that Asn, Asp, Ser, Thr, and Gly preferably appear at the N-cap position of a helix.19 The middle part of the monomer is an alpha helix of 40 Å long. The ending or the C-cap residue of the helix is Val 30. Flexible Gly 31 stops the helix and starts the C-terminal coil, which is consistent with the observation that glycine is common at the C-cap or the following position.19 The C-terminal residues Gly 31 and Glu 32 turn at Gly 31, fall back over the same helix, and form a cap for the helix. Twenty-three water molecules were built into the model and were associated with the monomer.
Figure 2.
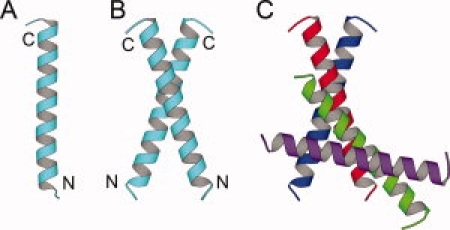
Monomer, dimer, and the interface between the dimers. A, monomer. B, dimer. C, the interdimer interface between the green and red helices.
Dimer formation and interaction
There are two major interfaces between monomers in the crystal. One is the intradimer interface and the other is the interdimer interface (Fig. 2). By the intradimer interface, two helices pack in a parallel way, cross in 35°, and form an X-shaped dimer with dimensions of 45 Å long and 35 Å wide. A “parallel” coiled coil indicates that the N-termini of the helices in the coiled coil point to a similar direction, so that the helices have a similar orientation. As the two helices cross in a large angle of 35°, the interactions are concentrated on the middle parts of the two helices. The core dimer interactions are between Leu 12-Leu 12, Asn 16-Asn 16, and Leu 19-Leu 19. Residues Leu 12, Leu 13, Lys 15, Asn 16, Leu 19, and Glu 20 between the two helices are involved in both hydrophobic and hydrophilic interactions: Leu 12-Leu 12, Leu 12-Leu 13, Leu 13-Leu 12, Lys 15-Glu 20, Asn 16-Asn 16, and Leu 19-Leu 19, and Glu 20-Lys 15. The residues before the hyphen sign are from one helix, whereas the residues after the hyphen sign are from the other helix. By the interdimer interface, two helices pack in an antiparallel way and cross in 25° (Fig. 2C). As the two helices cross in a smaller angle, the interactions are spread over residues on the whole helix surface. The interface consists of residues Glu 10, Leu 13, Ser 14, Trp 17, Asn 21, and two symmetry-related water molecules (HOH 7). The interactions are both hydrophilic and hydrophobic between the side chains from the two helices: Glu 10-Asn 21, Leu 13-Trp 17, Ser 14-Ser 14, Trp 17-Trp 17, Trp 17-Leu 13, Asn 21-Glu 10. Two symmetry-related water molecules (HOH 7) are also involved in linking two pairs of Ser 14-Asn 18 between the two helices.
Super leucine zipper
The X-shaped dimer can polymerize longitudinally to form a super long coiled coil (Fig. 3). The distances between Leu 5-Leu 5, Val 9-Val 9, Leu 12-Leu 12, Asn 16-Asn 16, and Leu 19-Leu 19, Val 23-Val 23, Leu 26-Leu 26, Val 30-Val 30 of the a, d residues of the X-shaped dimer are 7.3, 5.2, 3.3, 2.9, 3.7, 4.4, 4.1, 8.3 Å, respectively. While the close contacts are between the middle parts of the two helices, the distances between Leu 5-Leu 5, Val 9-Val 9, and Val 30-Val 30 at the ends of the two helices are large. Two helical N-termini of each X-shaped dimer approach two helical C-termini of the next X-shaped dimer, and cross into each other using their v-shaped openings, thus forming a super long coiled coil. The interactions are hydrophobic in the welding region where the two N-termini and two C-termini cross into each other (Figs. 3, 4). Two parts of Leu 26-Val 30 from one dimer fill in the space between two parts of Leu 5-Val 9 of the next dimer, so that two Leu 26 and two Leu 5 residues as well as two Val 30 and two Val 9 residues are aligned, respectively (Fig. 4). Side chains of the two Lys 8 residues move largely to expose two parts of Leu 5-Val 9 in one dimer in order that the two parts of Leu 5-Val 9 cross into two parts of Leu 26-Val 30 of another dimer. The contacts among the two Leu 26 residues and two Leu 5 residues or the two Val 30 residues and two Val 9 residues are in the range of van der Waals' force. The interfacial contacts in the super long coiled coil are valine and leucine residues alternating at the a, d positions, except that occasionally asparagine 16 appears at the a positions (Fig. 3). That the leucine residues appear at every seventh position in the super long coiled coil suggests that this is an extended super leucine zipper.
Figure 3.
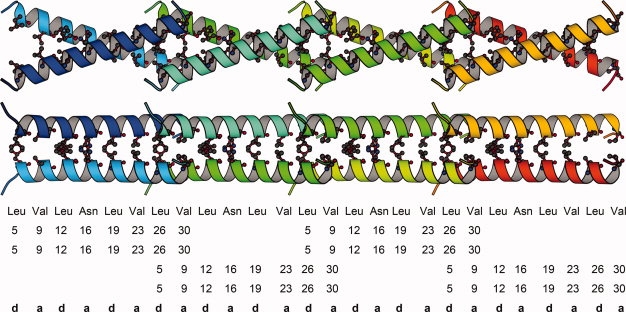
Top and side views of the super leucine zipper. Only four dimer repeats are shown. The interfacial residues are represented in ball-and-stick mode, with residue type, residue number, and corresponding positions in a heptad repeat (abcdefg)n shown below.
Figure 4.
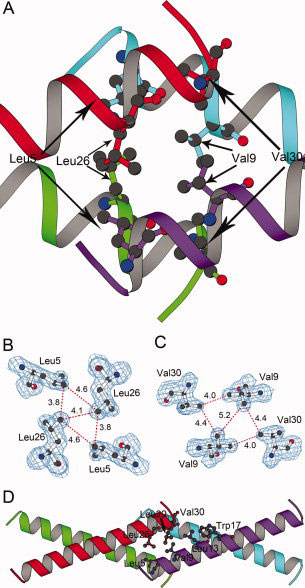
The welding region of the super leucine zipper. A, The welding region mainly consists of four leucine residues and four valine residues. The four helices are presented in the same view as in Figure 4D, but the helix width is narrowed to view the leucine and valine residues in ball-and-stick mode. B, Four leucine residues and 2Fo-Fc map contoured at 1σ level. The distances between the atoms are in the unit of Ångström. C, Four valine residues and 2Fo-Fc map contoured at 1σ level. The distances between the atoms are in the unit of Ångström. D, The red helix attaches to the purple helix from a side, forming a parallel-attaching dimer, which interacts with another parallel-attaching dimer consisting of the green and cyan helices. Hydrophobic residues Val 30, Leu 29, Leu 26 from the red helix and hydrophobic residues Trp 17, Leu 13, Val 9, Leu 5 from the purple helix are shown in ball-and-stick mode, and attract each other through hydrophobic interactions.
The interactions described above that two N-termini and two C-termini cross into each other can be viewed in another way. Rather than the red and green helices in different layers are assembled first to form an X-shaped dimer, the red and purple helices in the same layer may be assembled first to form a parallel-attaching dimer (Fig. 4D). The C-terminus of the red helix attaches to the N-terminus of the purple helix in the same layer, and hydrophobic interactions are between Val 30, Leu 29, Leu 26 from the red helix and Leu 13, Val 9, Leu 5 from the purple helix. Hydrophobic residues Val 30, Leu 29, Leu 26 from the red helix are attracted by hydrophobic residues Trp 17, Leu 13, Val 9, Leu 5, which form an oblique hydrophobic ridge on the purple helix. Similar attraction and attachment also exists in the green and cyan helices in an adjacent layer. Between the two layers, a 180° rotation operation exists, so that one attaching region consisting of Val 30, Leu 29, Leu 26, Leu 13, Val 9, Leu 5 from one layer have close contacts with another attaching region consisting of Val 30, Leu 29, Leu 26, Leu 13, Val 9, Leu 5 from an adjacent layer. A crossing angle of 35° is between the two parallel-attaching dimers from the two layers. As a result, the two N-termini and two C-termini are clustered together through hydrophobic interactions, which form the welding region of the super leucine zipper.
Comparison with the wild-type GCN4 leucine zipper
Compared to the wild-type GCN4 leucine zipper (Protein Data Bank [PDB] code 2ZTA),9 the leucine zipper mutant of this study has four mutations of A3K, A7D, W17Y, and N18H. The leucine zipper mutant shows a straight helix with two curved tails, whereas the wild-type leucine zipper is a smoothly bent helix. The helix of the mutant starts at Gln 4 and ends at Val 30, and both the N-terminal Met 2-Ala 3 and the C-terminal Gly 31-Glu 32 are in a curved conformation. The helix of the wild-type leucine zipper starts at Arg 1 and ends at Gly 31, and the C-terminal Glu 32-Arg 33 are not determined while Glu 32 of the leucine zipper mutant is determined. In the crystal, the leucine zipper mutant forms a two-stranded, parallel, super long coiled coil, whereas the wild-type leucine zipper is a discrete, two-stranded, parallel two-helix coiled coil. The X-shaped mutant dimer has a crossing angle of 35° instead of 18° in the wild-type dimer. The root mean square differences (rmsds) between the Cα atoms of the mutant and the wild-type leucine zipper are shown in Table I. Removing residue Met 2 from the structural comparisons results in a more than 0.5 Å rmsd drop, indicating that compared to the wild-type leucine zipper, the N-terminal Met 2 of the mutant has a large conformational change. The N-terminus (Met 2-Ala 3) of the mutant is in a curved conformation instead of the helical conformation of the wild-type leucine zipper. The conformational change at the N-terminus of the mutant may be due to the flexibility of the N-terminus and crystal packing forces between helices.
Table I.
RMSDs Between the Cα Atoms of the Mutant and the Wild-Type GCN4 Leucine Zippera
| Residue range | Valueb | Valuec |
|---|---|---|
| 2–31 | 1.36 | 1.57 |
| 3–31 | 0.78 | 1.03 |
| 4–31 | 0.77 | 1.03 |
| 4–30 | 0.75 | 0.84 |
| 4–29 | 0.68 | 0.78 |
| 8–27 | 0.43 | 0.52 |
The mutant consists of only one chain, Chain A, whereas the wild-type GCN4 leucine zipper refers to the structure of PDB code 2ZTA and consists of two chains, Chains A and B. The unit for the values is Ångström (Å).
RMSDs between Chain A of the mutant and Chain A of the wild-type.
RMSDs between Chain A of the mutant and Chain B of the wild-type.
Discussion
In the crystals, the wild-type GCN4 leucine zipper is a discrete dimer, whereas the mutant forms an extended super long coiled coil composed of sticky-ended dimers. Change of the oligomeric state may be due to either mutations of four non-interfacial residues or change of the crystallization condition or both. The following describes the reason of the formation of the super long coiled coil due to mutations. There are two faces on a GCN4 leucine zipper helix. One face consists of hydrophobic residues Leu 5, Val 9, Leu 12, Leu 19, Val 23, Leu 26, Val 30, and is referred to the leucine face. The other face opposite to the leucine face consists of polar residues, and is referred to the non-leucine face, on which Tyr 17 is located. As hydrophobic residues are lined on the leucine face, two GCN4 leucine zipper helices can dimerize through the leucine faces so that hydrophobic residues are buried, leaving polar residues outside to interact with polar solvent environment. Mutation of Tyr 17 to Trp 17 introduces a large hydrophobic residue on the non-leucine face. Two helices are not sufficient to bury both the hydrophobic residues on the leucine faces and two Trp 17 residues on the non-leucine faces, so more helices need to participate in. While the leucine zipper mutant can form a conventional X-shaped dimer in a larger crossing angle of 35° through hydrophobic residues on the leucine faces, each of the two Trp 17 residues on the leucine face at the middle of the X-shaped dimer may induce another helix attaching from a side (Fig. 4D). Trp 17, Leu 13, Val 9, and Leu 5 at four residues' interval separation on the helix surface form an oblique hydrophobic ridge from the non-leucine face to the leucine face, and attract hydrophobic residues Val 30, Leu 29, and Leu 26 from the attaching helix. Two new attaching helices also form an X-shaped dimer, which further attract other two helices to attach from a side. By such an attaching mechanism, the X-shaped dimer elongates and a super leucine zipper has formed. In addition, each of the two exposed Trp residues in an X-shaped dimer attracts one Trp 17 residue from another X-shaped dimer in a different layer through the interdimer interface, and the incoming Trp 17 mediates the attaching hydrophobic interactions between Trp 17 and Leu 29 of the parallel-attaching dimer.
While a specific description on the formation of the super leucine zipper has been given above, a general principle of folding and assembly of coiled coil is discussed below. Compared to the alanine zipper that forms a three-stranded coiled coil,16 the tryptophan zipper17 and phenylalanine zipper18 form five-stranded coiled coils. As the hydrophobic residues are too large, the tryptophan and phenylalanine zippers would have some hydrophobic residues exposed to polar solvent environment should they form three-stranded coiled coils. In other words, three helices are not sufficient to bury those bulky hydrophobic residues, so that more helices need to participate in and therefore the high oligomeric state five-stranded coiled coils result. While the replacement of small hydrophobic residues by large hydrophobic residues results in the formation of a high oligomeric state coiled coil, mutations of hydrophilic residues to hydrophobic ones result in more hydrophobic surface area on the helix, and also result in the formation of a high oligomeric state coiled coil. For example, mutations of eight hydrophilic residues to hydrophobic alanine residues result in a seven-helix coiled coil.15 These data seem to suggest a principle of folding and assembly of coiled coil that the more hydrophobic the helix, the higher oligomeric number the coiled coil. In the case of the leucine zipper mutant, the first three mutations of K3A, D7A, Y17W, and H18N result in more hydrophobic pimples or more hydrophobic surface area on the helix. Unlike the wild-type GCN4 leucine zipper that forms a discrete dimer, the leucine zipper mutant forms a super leucine zipper, which may be viewed as a hybrid of the two-stranded and four-stranded coiled coils. The four-stranded part refers to the welding region of the super leucine zipper, which contains two N-termini and two C-termini crossing into each other. The increasing hydrophobicity on the helix surface may lead to the two-stranded to four-stranded transition and the formation of the four-stranded part of the coiled coil, which is consistent with the principle that the more hydrophobic the helix, the higher oligomeric number the coiled coil. The oligomeric number of the coiled coil may be roughly predicted based on its direct proportion to the hydrophobicity of the helix, and the hydrophobicity of the helix can be calculated based on the percentage of the hydrophobic area over the whole helix surface area.
In crystals, some coiled coils exist in discrete assemblies,9,14–17 whereas other coiled coils form elongated assemblies through coiled coils stacking end to end.10–14,18 Unlike all other elongated assemblies, which contain coiled coils stacking end to end or in which the coiled coils are blunt ended, the super leucine zipper of this study contains the welding regions formed by helices offset by ∼1 heptad or the super leucine zipper is assembled with sticky-ended coiled coils. The sticky-ended leucine zippers have been employed to assemble into protein fibers, which may have potential application as novel scaffolds in tissue engineering.6 In a self-assembling fiber system,8,20 two leucine zipper helices with sticky ends assemble into a long fiber. The fiber can be up to 70 nm thick and involves hundreds of two-stranded coiled coil fibrils bundled together. The questions are what drives the thickening process of the fiber and whether the thickening can be controlled. In the crystal of this study, there are no much interactions between parallel super leucine zippers, and disordered water molecules are filled between parallel super leucine zippers. If the super leucine zipper bundles up to form a matured fiber, the interactions between the super leucine zippers should be hydrophilic because of polar residues on the helix surface and water molecules in between. The thickening may be limited if exposed polar residues on the surface of the super leucine zipper have a same kind of charge. In contrast, the thickening may be boosted if exposed polar residues on two opposite faces of the fiber have opposite charges. In another nanorope assembly system,21 the self-assembling leucine zipper peptide has an insertion of two alanine residues at the middle of the helix, and as a result of the insertion the N- and C-terminal hydrophobic ridges are on the two opposite faces of the helix. In the proposed nanorope assembly model,21 the C-terminal hydrophobic ridge of the first helix is attached to the N-terminal hydrophobic ridge of the second helix, whereas the C-terminal hydrophobic ridge of the second helix is attached to the N-terminal hydrophobic ridge of the third helix. The overall nanorope assembly elongates in a staggered mode, in which the helices are offset by half length of the helix or ∼2 heptad. The similar staggered assembling mode has been observed in the super leucine zipper of this study, in which the two opposite hydrophobic ridges are the oblique N-terminal ridge of Leu 5, Val 9, Leu 13, and Trp 17 and the short C-terminal ridge of Leu 26, Leu 29, and Val 30. The leucine zipper helices elongate in one layer and form a nanorope-like assembly (Fig. 3), and two nanorope-like assemblies related by a 180° rotation form the super leucine zipper. The interactions between the two nanorope-like assemblies are due to the hydrophobic residues on the leucine faces, which are also responsible for the dimer formation of the wild-type GCN4 leucine zipper.
Material and Methods
Synthesis and crystallization
The leucine zipper peptide was synthesized by Biosynthesis Inc. with a sequence of RMAQLEAKVEELLSKNWNLENEVARLKKLVGER. Using the hanging drop vapor diffusion method, crystallization conditions of number 24 from the Hampton Crystal Screen kit produced crystals at room temperature. The peptide solution contained 8 mg/mL leucine zipper peptide, 0.1 M NaAc, pH 4.5, and the reservoir solution contained 0.8 mL of 0.2 M CaCl2, 0.1 M NaAc, pH 4.5, 20% (v/v) isopropanol. The hanging drops were set up by mixing 1 μL protein solution with 1 μL reservoir solution. The crystals were flash-cooled directly in liquid N2 for data collection.
Data collection and processing and molecular replacement
Diffraction data were collected to 1.6 Å resolution at the X25 beamline of Brookhaven National Laboratory, and were processed with HKL2000.22 The crystal belonged to the primitive hexagonal Bravais lattice, with cell dimensions a = b = 31.15 Å and c = 57.32 Å. Because the systematic absences were not clear, the space group identification by HKL2000 did not determine the space group of the crystal. The structure factor amplitudes were obtained by Truncate23 and Cad of the CCP4 suite.24 Using a standard helix of 28 polyalanine residues as a search model and all data between 19.64 and 1.6 Å, molecular replacement solutions were searched with all the possible space groups. MolRep25 from the CCP4 package produced a correct solution with space group P32. MolRep truncated the data to 19.64–1.94 Å, used an integration radius of 17.13 Å, and found two rotation and translation solutions (35.98°, 76.52°, 88.91°, 0.046, 0.122, 0.000) and (108.87°, −166.45°, 26.67°, 0.831, 0.317, 0.480) for space group P32. The R-factor between 19.64 and 1.94 Å resolution was 49.9%, with two 28-polyalanine helices in an asymmetric unit.
Model building and structure refinement
After 10% data were selected as an Rfree data set, rigid body refinement in CNS26 changed Rwork/Rfree from 47.35%/47.51% to 47.54%/43.29% between 19.64 and 3 Å. Using the data between 19.64 and 1.9 Å, positional and individual B-factor refinement in CNS lowered Rwork/Rfree from 47.58%/44.24% to 39.60%/38.28%. The data were extended to 19.64–1.6 Å, and Rwork/Rfree were lowered to 39.54%/37.34% after positional and B-factor refinement. 2Fo-Fc and Fo-Fc maps were calculated, and the positions of the two tryptophan-17 residues were located based on the strong positive electron density of their side chains. Side chains of all the residues were built based on the positions of the two Trp 17 residues and other positive electron density consistent with the sequence. After addition of 36 water molecules, Rwork/Rfree were 27.65%/29.28% between 19.64 and 1.6 Å. In space group P32, the two helices in the asymmetric unit seem to suggest that a crystallographic two-fold axis exists between the two helices and is perpendicular to the 32 screw axis, so that the space group may be P3212 or P3221 instead of P32. The structure factor amplitudes in space group P32 were further processed in space group P3212 or P3221. Using one helical chain previously built in space group P32 as a search model, an auto-run of Phaser27 found a molecular replacement solution for space group P3221. The R-factor was 30.93% between 19.64 and 1.6 Å. Positional and B-factor refinements in CNS were applied to the model. Arg 1 and Arg 33 were removed owing to the lack of electron density. Twenty-three water molecules were added. Glu 11, Leu 12, Asn 16, and Asn 21 were built in double conformations. The final Rwork/Rfree were 25.77%/28.72% for the diffraction data between 19.64 and 1.6 Å (Table II). The atomic coordinates and structure factors have been deposited in the Protein Data Bank with the accession code 3I1G. Figures 1–4 were produced with Molscript.28
Table II.
Data Processing and Refinement Statistics
| A. Data processing | |
| Space group | P3221 |
| Unit cell dimensions (Å) | |
| a, b | 31.15 |
| c | 57.32 |
| Resolution (Å) | 19.64–1.6 (1.66–1.6)a |
| Unique reflections | 4104(176) |
| Completeness (%) | 89.7(37.7) |
| Redundancyb | 3.2(1.3) |
| Rsym (%)b | 5.9(24.0) |
| I/σ(I)b | 31.1(2.0) |
| B. Refinement | |
| Rwork (%) | 25.77 |
| Rfree (%) | 28.72 |
| Root mean square (RMS) deviations from ideality | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.9 |
| Dihedrals (°) | 15.7 |
| Impropers (°) | 0.59 |
| Average B-factors (Å2) | |
| Main-chain | 26.25 |
| Side-chain | 26.69 |
| 23 water molecules | 44.15 |
| Wilson plot | 29.40 |
| Ramachandran plot | |
| Residues in most favored regions | 100.0% |
| Residues in additional allowed regions | 0.0% |
| Residues in generously allowed regions | 0.0% |
| Residues in disallowed regions | 0.0% |
Values in parentheses refer to the highest resolution shell.
The values corresponding to these parameters were obtained by data processing using space group P32.
Acknowledgments
This work was supported by the AFSOR grant F49620-03-1-0365 to Professor Joanne I. Yeh. The author thanks Professor Joanne I. Yeh for permitting me to publish this work. The author thanks Antoni Tortajada for crystallizing the leucine zipper mutant peptide.
References
- 1.Hinnebusch AG, Fink GR. Positive regulation in the general amino acid control of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1983;80:5374–5378. doi: 10.1073/pnas.80.17.5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Struhl K. Helix-turn-helix, zinc-finger, and leucine-zipper motifs for eukaryotic transcriptional regulatory proteins. Trends Biochem Sci. 1989;14:137–140. doi: 10.1016/0968-0004(89)90145-X. [DOI] [PubMed] [Google Scholar]
- 3.Ellenberger TE, Brandl CJ, Struhl K, Harrison SC. The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted α helixes: crystal structure of the protein-DNA complex. Cell. 1992;71:1223–1237. doi: 10.1016/s0092-8674(05)80070-4. [DOI] [PubMed] [Google Scholar]
- 4.Cohen C, Parry DAD. α-helical coiled coils: more facts and better predictions. Science. 1994;263:488–489. doi: 10.1126/science.8290957. [DOI] [PubMed] [Google Scholar]
- 5.Crick FHC. The packing of α-helices: simple coiled-coils. Acta Crystallgr. 1953:689–697. [Google Scholar]
- 6.Holmes TC. Novel peptide-based biomaterial scaffolds for tissue engineering. Trends Biotechnol. 2002;20:16–21. doi: 10.1016/s0167-7799(01)01840-6. [DOI] [PubMed] [Google Scholar]
- 7.Fairman R, Åkerfeldt KS. Peptides as novel smart materials. Curr Opin Struct Biol. 2005;15:453–463. doi: 10.1016/j.sbi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Woolfson DN, Ryadnov MG. The design of coiled-coil structures and assemblies. In fibrous proteins: Coiled-coils, collagen and elastomers. Curr Opin Chem Biol. 2006;10:559–567. [Google Scholar]
- 9.O'Shea EK, Klemm JD, Kim PS, Alber T. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science. 1991;254:539–544. doi: 10.1126/science.1948029. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez L, Woolfson DN, Alber T. Buried polar residues and structural specificity in the GCN4 leucine zipper. Nat Struct Biol. 1996;3:1011–1018. doi: 10.1038/nsb1296-1011. [DOI] [PubMed] [Google Scholar]
- 11.Harbury PB, Kim PS, Alber T. Crystal structure of an isoleucine-zipper trimer. Nature. 1994;371:80–83. doi: 10.1038/371080a0. [DOI] [PubMed] [Google Scholar]
- 12.Eckert DM, Malashkevich VN, Kim PS. Crystal structure of GCN4-pIQI, a trimeric coiled coil with buried polar residues. J Mol Biol. 1998;284:859–865. doi: 10.1006/jmbi.1998.2214. [DOI] [PubMed] [Google Scholar]
- 13.Akey DL, Malashkevich VN, Kim PS. Buried polar residues in coiled-coil interfaces. Biochemistry. 2001;40:6352–6360. doi: 10.1021/bi002829w. [DOI] [PubMed] [Google Scholar]
- 14.Deng YQ, Liu J, Zheng Q, Eliezer D, Kallenbach NR, Lu M. Antiparallel four-stranded coiled coil specified by a 3-3-1 hydrophobic heptad repeat. Structure. 2006;14:247–255. doi: 10.1016/j.str.2005.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, Zheng Q, Deng YQ, Cheng CS, Kallenbach NR, Lu M. A seven-helix coiled coil. Proc Natl Acad Sci USA. 2006;103:15457–15462. doi: 10.1073/pnas.0604871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, Lu M. An alanine-zipper structure determined by long range intermolecular interactions. J Biol Chem. 2002;277:48708–48713. doi: 10.1074/jbc.M208773200. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Yong W, Deng YQ, Kallenbach NR, Lu M. Atomic structure of a tryptophan-zipper pentamer. Proc Natl Acad Sci USA. 2004;101:16156–16161. doi: 10.1073/pnas.0405319101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu J, Zheng Q, Deng YQ, Kallenbach NR, Lu M. Conformational transition between four and five-stranded phenylalanine zippers determined by a local packing interaction. J Mol Biol. 2006;361:168–179. doi: 10.1016/j.jmb.2006.05.063. [DOI] [PubMed] [Google Scholar]
- 19.Richardson JS, Richardson DC. Amino acid preferences for specific locations at the ends of alpha helices. Science. 1988;240:1648–1652. doi: 10.1126/science.3381086. [DOI] [PubMed] [Google Scholar]
- 20.Pandya MJ, Spooner GM, Sunde M, Thorpe JR, Rodger A, Woolfson DN. Sticky-end assembly of a designed peptide fiber provides insight into protein fibrillogenesis. Biochemistry. 2000;39:8728–8734. doi: 10.1021/bi000246g. [DOI] [PubMed] [Google Scholar]
- 21.Wagner DE, Phillips CL, Ali WM, Nybakken GE, Crawford ED, Schwab AD, Smith WF, Fairman R. Toward the development of peptide nanofilaments and nanoropes as smart materials. Proc Natl Acad Sci USA. 2005;102:12656–12661. doi: 10.1073/pnas.0505871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.French GS, Wilson KS. On the treatment of negative intensity observations. Acta Crystallgr. 1978;34:517–525. [Google Scholar]
- 24.Collaborative Computational Project (CCP4) The CCP4 suite: programs for protein crystallography. Acta Crystallgr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 25.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallgr. 1997;30:1022–1025. [Google Scholar]
- 26.Brünger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallgr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 27.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallgr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallgr. 1991;24:946–950. [Google Scholar]