
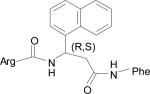
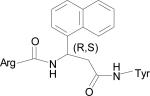
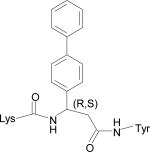
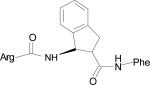
Table 1.
Low energy docked structures of HERP5, 6, 7 and their analogs.
| Code no. | Structure | Docking energy kcal/mol |
|---|---|---|
| HERP5 |
|
–13.87 |
| HERP51 |
|
–14.16 |
| HERP5c3 |
|
–12.59 |
| HERP5a |
|
–12.40 |
| HERP5c |
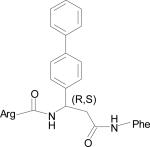
|
–12.06 |
| HERP6 |
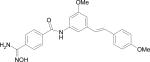
|
–10.54 |
| HERP7 |
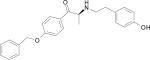
|
–10.65 |
| HERP7h |
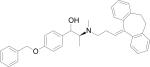
|
–11.23 |
| HERP7f |
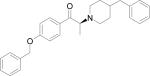
|
–10.97 |
| Arg-Tyr | Arg-Tyr | -3.91 |
Docking energy has an error of 2 kcal/mol in autodock 3.0. The chirality of amino acids was L. For β-amino acid both R & S configurations were considered. The energy difference in the docking calculations was less than 3 kcal/mol between resulting diastereoisomers of HERP5 and its analogs.