

Abstract
Detecting clonal T-cell receptor (TCR)-γ gene rearrangements (GRs) is an important adjunct test for diagnosing T-cell lymphoma. We compared a recently described assay (BIOMED-2 protocol), which targets multiple variable (V) gene segments in two polymerase chain reaction (PCR) reactions (multi-V), with a frequently referenced assay that targets a single V gene segment in four separate PCR reactions (mono-V). A total of 144 consecutive clinical DNA samples were prospectively tested for T-cell clonality by PCR using laboratory-developed mono-V and commercial multi-V primer sets for TCR-γ GR. The combination of TCR-β, mono-V TCR-γ and multi-V TCR-γ detected more clonal cases (68/144, 47%) than any individual PCR assay. We detected clonal TCR-β GR in 47/68 (69%) cases. Using either mono-V or multi-V TCR-γ primers, the sensitivities for detecting clonality were 52/68 (76%) or 51/68 (75%). Using both mono-V and multi-V TCR-γ primers improved the sensitivity for detecting clonality, 60/68 (88%). Combining either mono-V or multi-V TCR-γ primers with TCR-β primers also improved the sensitivity, 64/68 (94%). Significantly, TCR-γ V11 GRs could only be detected using the mono-V-PCR primers. We conclude that using more than one T-cell PCR assay can enhance the overall sensitivity for detecting T-cell clonality.
The ability to distinguish benign from malignant T-cell proliferations relies on a combination of morphological, immunological and molecular tests. Often a clonal T-cell receptor (TCR) gene rearrangement (GR) is the only definitive marker for neoplasia. While Southern blot analysis is the gold standard for detecting antigen receptor GRs, it is not suitable for small biopsies or formalin-fixed paraffin-embedded (FFPE) tissues. Therefore, most laboratories depend on the polymerase chain reaction (PCR) for detecting TCR GRs. Because of the complexity of the TCR-β locus,1 routine testing has focused on the TCR-γ locus,2,3,4,5,6,7,8,9,10,11,12 which contains fewer variable (V) gene segments.1 Even though TCR-γ comprises only 11 functional V regions, several different PCR strategies are in use.13,14
We rigorously compared the recently described BIOMED-2 TCR-γ assay, which targets two different TCR-γ V gene segments (V1-8/V10 and V9/V11) in two separate reactions (multi-V strategy; Figure 1A)15 against a previously described TCR-γ laboratory-developed assay, which has been cited more than 100 times (ISI Web of knowledge citation index; Thompson Reuters, Philadelphia, PA), which targets a single TCR-γ V gene segment (V1-8, V9, V10, V11) in each of four individual PCR reactions (mono-V strategy; Figure 1A).16 In a sense, both TCR-γ PCR strategies can be considered variants of multiplex strategies because they combine one or two V primers with multiple J primers in each PCR reaction:15,16 the multi-V strategy adds the JP1/JP2 and J1/J2 primers15 and the mono-V strategy adds the JP1/JP2, J1/J2 and JP primers16 (Figure 1B). It has been suggested that because of possible competition between amplicons for reagents and interactions between primers, multiplex PCR may be more susceptible to reduced sensitivity.17,18 Therefore, we compared the mono-V versus the multi-V PCR strategies for detecting TCR-γ gene rearrangements in a large number of clinical DNA samples.
Figure 1.
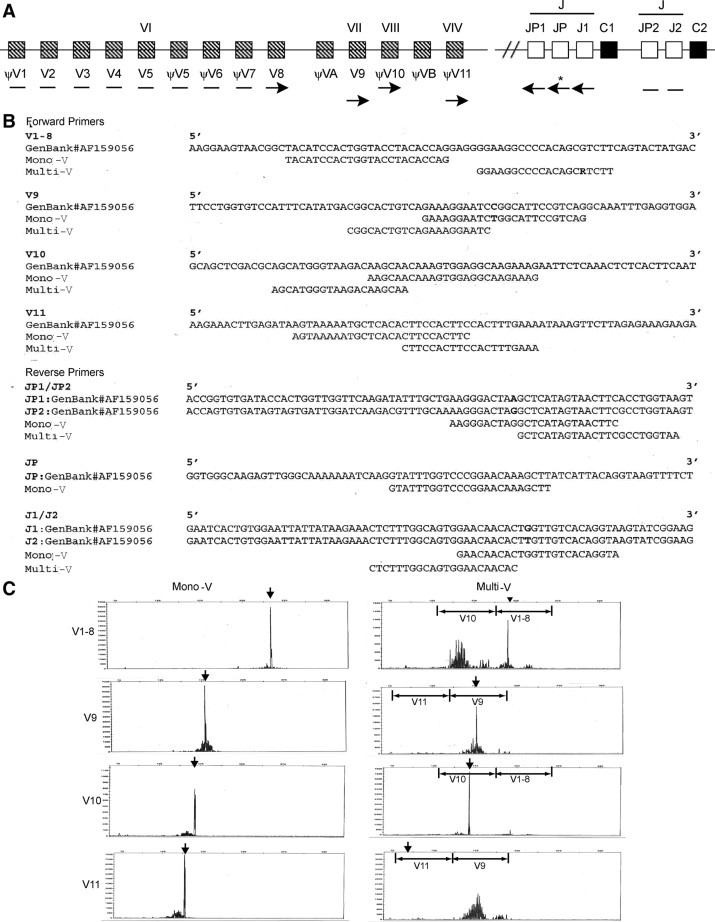
A: Schematic representation of the TCR-γ genomic DNA locus. Approximate positions of the primers for both mono-V and multi-V PCR strategies are marked by arrows. The bars represent consensus primer-binding sequences. * ← JP primer is only used in the mono-V assay. B:TCR-γ variable gene segments V1-8, V9, V10, V11 (forward primers) and J segments (reverse primers): Reference DNA sequences from GenBank (top), mono-V TCR-γ primers (middle), multi-V TCR-γ primers (bottom). All DNA sequences are in 5′ to 3′ orientation. C: Detection of TCR-γ PCR products by capillary gel electrophoresis. Mono-V (left column) and multi-V (right column) PCR assays amplified representative DNA samples from the same patient.
Materials and Methods
Patient Samples
We prospectively tested a total of 144 consecutive patient DNA samples over a period of 7 months in 2008 for TCR-β and TCR-γ GRs. The samples included FFPE biopsies from skin (N = 101) and other tissues (N = 15), including lymph nodes (N = 7), spleen (N = 2), brain (N = 1), lung (N = 1), nasal sinus (N = 1), stomach (N = 1), tongue (N = 1), and uvula (N = 1); and fresh samples from bone marrow (N = 19) and peripheral blood (N = 9). Also, 62 samples were tested simultaneously for immunoglobulin heavy chain (IGH) GRs.
PCR Primers
For the mono-V detection of TCR-γ GR, we used four different PCR reactions that included custom-synthesized fluorescently labeled primers (Sigma-Genosys, The Woodlands, TX) for TCR-γ variable regions V1-8, V9, V10, and V11. The primers were as described previously, but without the GC clamps.16 Each tube in the mono-V strategy contained three primers targeting J segments JP1/JP2, J1/J2 and JP. For the detection of multi-V TCR-γ we used commercial master mixes containing fluorescently-labeled primers based on published BIOMED-2 sequences (InVivoScribe Technologies, San Diego, CA).15 For the multi-V TCR-γ assays, two separate PCR reactions were set up according to the BIOMED-2 protocol using TCR-γ V1-8 and V10 primers in tube A and, TCR-γ V9 and V11 primers in tube B (Figure 1).15 Each tube in the multi-V strategy used two primers targeting J segments JP1/JP2 and J1/J2. It should be noted that since JP1 and JP2 are highly homologous, both the mono-V and the multi-V approaches used a single primer for this target (Figure 1B).16,15 Similarly, because J1 and J2 are highly homologous, only one primer is used to target these J segments (Figure 1B).16,15
The mono-V assay uses each primer at 6.25 pmol/25 μl PCR reaction. The original BIOMED-2 protocol had used each primer at 10 pmol/50 μl PCR reaction.15 However, the proprietary primer concentration in the commercial master-mix is not available.
For the detection of TCR-β and IGH, we used commercial master mixes containing fluorescently-labeled multiplex primers based on published BIOMED-2 sequences (InVivoScribe Technologies).15 For the multiplex detection of TCR-β GRs, three multiplex PCR reactions were set up in tubes A (23 Vβ + 6 Jβ1 + 3 Jβ2 primers), B (23 Vβ + 4 Jβ2 primers) and C (2 Dβ and 13 Jβ primers). For the multiplex detection of IGH, two multiplex PCR reactions were set up in tube A (FR2 primers: 7 VH primers + 1 consensus JH primer) and tube B (FR3 primers: 7 VH primers + 1 consensus JH primer).
DNA Extraction
For FFPE tissues, the number of 6 μ thick sections that were cut depended on the tissue type and on the cross-sectional area. In general, we trimmed 10 sections from lymph nodes and up to 30 sections from skin biopsies. The sections were immediately placed in 1 ml Citrisolv (Fisher Scientific, Pittsburgh, PA), a xylene substitute, for 5 minutes followed by a 5-minute centrifugation at 13,000 rpm (Biofuge 13, Heraeus Instruments, Hanau, Germany). The pellet was washed twice with ethanol and vacuum-dried. The dried pellet was resuspended in 180 μl of Buffer ATL (QIAmp DNA minikit; Qiagen, Valencia, CA). The tissue was digested by incubation with 20 μl of proteinase K at 56°C for 1 to 48 hours until the tissue was completely lysed. DNA was purified from all samples using the QIAamp DNA minikit (Qiagen). The DNA was quantified using a Nanodrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). The amount of DNA (ng) added to the PCR reaction varied by source: Skin (N = 100, 140 ± 114, mean ± SD), lymph nodes (306 ± 63), peripheral blood (143 ± 65), bone marrow (305 ± 105), other tissue (418 ± 227). We amplified β-globin for all samples as a quality control check of the DNA.
PCR Assay
All PCR reactions were performed on a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA). For DNA amplification, we used the procedure recommended by the manufacturer. A HotStarTaq master mix kit (Qiagen) was used for initiating the mono-V PCR. TCR-γ clonality master mix (InVivoScribe Technologies) and AmpliTaq Gold DNA polymerase (Applied Biosystems) were used for initiating the multi-V PCR. All PCR assays were set up in 25 μl reaction volume except as noted. The PCR protocol for the mono-V primer sets was denaturing at 95°C for 15 minutes followed by 40 cycles of denaturing at 95°C for 30 seconds, annealing at 59°C for 30 seconds, extension at 72°C for 45 seconds, and a final extension at 72°C for 5 minutes; the PCR protocol for the multi-V primer sets was denaturing at 95°C for 7 minutes followed by 35 cycles of denaturing at 95°C for 45 seconds, annealing at 60°C for 45 seconds, extension at 72°C for 90 seconds, and a final extension at 72°C for 10 minutes. The PCR protocol for the TCR-β assay was denaturing at 95°C for 7 minutes followed by 35 cycles of denaturing at 95°C for 45 seconds, annealing at 60°C for 45 seconds, extension at 72°C for 90 seconds, and a final extension at 72°C for 10 minutes. The PCR protocol for the IGH-FR2 assay was denaturing at 95°C for 10 minutes followed by 38 cycles of denaturing at 95°C for 30 seconds, annealing at 62°C for 30 seconds, extension at 72°C for 30 seconds, and a final extension at 72°C for 10 minutes. The PCR protocol for the IGH-FR3 assay was denaturing at 95°C for 10 minutes followed by 35 cycles of denaturing at 94°C for 30 seconds, annealing at 55°C for 30 seconds, extension at 72°C for 60 seconds, and a final extension at 72°C for 10 minutes.
We detected the PCR products by automated capillary gel electrophoresis using a 3130XL Genetic Analyzer (Applied Biosystems). Product sizes were calibrated with Gene Scan-500 LIZ size standards (Applied Biosystems).
PCR Control DNAs
DNA from reactive tonsil was used as a polyclonal control. DNA from Jurkat T-cell or Ramos B-cell lines (American Type Culture Collection, Manassas, VA) was diluted 1:10 in reactive tonsil DNA as a sensitivity control for clonal TCR-β, TCR-γ, or IGH GRs. In addition, DNA provided by InVivoScribe Technologies was used as a positive control for amplifying TCR-γ V11. All control DNAs were used at 250 ng per 25 μl PCR reaction except the commercial clonal and polyclonal control DNAs (InVivoScribe Technologies), which were used at 500 ng per 25 μl reaction.
Morphological Evaluation
Hematoxylin and eosin stained histological sections were reviewed from each FFPE tissue to ensure the presence of adequate diagnostic material and to correlate the morphology with the molecular test results.
Data Analysis
Cases showing up to three sharp peaks more than three times higher than the background using capillary gel electrophoresis with at least one (TCR-β or mono-V TCR-γ or multi-V TCR-γ) PCR assay were interpreted as clonal. Cases with more than three sharp peaks more than three times above the background were interpreted as oligoclonal. Cases with no sharp peaks more than three times above the background were interpreted as polyclonal. The results of TCR-β, TCR-γ, and IGH GRs were tabulated in an Excel spreadsheet (Microsoft, Seattle, WA). Statistical analysis was performed using McNemar's test on SAS 9.1.3 software (SAS Institute Inc., Cary, NC).
Results
Comparison of Mono-V Versus Multi-V PCR Strategies for Detecting TCR-γ Gene Rearrangements
We analyzed the results of simultaneously using both a mono-V and a multi-V PCR assay for TCR-γ GRs in 144 tissue, bone marrow and peripheral blood samples. The DNA sequences targeted by the PCR primers for both mono-V and multi-V strategies are similar, but not identical, for TCR-γ gene segments V1-8, V9, V10, V11, JP1/P2 and J1/2 (Figure 1A). A separate primer for JP is uniquely used in the mono-V assay. The actual DNA sequence alignment for the primers revealed either only a 4-bp gap for V1-8 or significant overlap for the other V gene segments (Figure 1B). We could detect GRs involving V1-8, V9, V10, and V11 using the mono-V assay, but only V1-8, V9, and V10 using the multi-V assay. Surprisingly, the multi-V assay did not detect any TCR-γ V11 GRs (Figure 1C).
Sensitivity of Detecting Clonal TCR Gene Rearrangements
To calculate the percent clonal TCR GRs detected, we used a denominator of 68 cases with either a clonal GR involving TCR-β, or -γ, or both (Table 1). For TCR-γ GRs, we compared mono-V16 versus multi-V PCR primers.15 Although the mono-V PCR assay showed a slightly better sensitivity for detecting clonal cases (52/68, 76%) compared with the multi-V PCR assay (51/68, 75%), this difference was not statistically significant. However, combining results from the mono-V plus multi-V assays, significantly improved the sensitivity of detecting clonal cases (60/68, 88%) compared with the multi-V PCR assay alone (51/68, 75%; P = 0.0078) or compared with the mono-V PCR assay alone (52/68, 76%; P = 0.0078). The combination of TCR-β results with either mono-V or multi-V TCR-γ results detected equal number of clonal cases (64/68, 94%), which was slightly higher but not statistically different from the number of clonal cases detected by combining results from the mono-V and the multi-V TCR-γ assay (60/68, 88%, P = 0.77).
Table 1.
Sensitivity of Various PCR Primer Sets for Detecting Clonality Among 68 Patient DNA Samples with Clonal TCR GRs
| TCR-β | TCR-γ (mono-V) | TCR-γ (multi-V) | Any clonal TCR GR detected N (%) |
|---|---|---|---|
| + | 47 (69) | ||
| + | 51 (75) | ||
| + | 52 (76) | ||
| + | + | 60 (88) | |
| + | + | 64 (94) | |
| + | + | 64 (94) | |
| + | + | + | 68 (100) |
Mono-V and multi-V TCR-γ PCR assays agreed in 127/144 (88%) cases, including 84/144 (58%) clonal GRs and 43/144 (30%) polyclonal GRs. Among 17/144 (12%) discrepancies, T-cell clonality was detected in nine patient DNA samples with the mono-V and in 8 with the multi-V TCR-γ primers. Among DNA samples that were clonal using the mono-V TCR-γ assay, 5/9 were also clonal for TCR-β and 1/9 was clonal for IGH. The discordant cases showed a wide range of presentations, emphasizing the need to interpret the GR studies in the context of clinical and histological features.
Sensitivity of Detecting Particular TCR-γ Variable Gene Segment Rearrangements
We asked whether the primer sets differ in detecting TCR-γ clonality based on the usage of particular TCR-γ variable gene segments (Table 2). The frequency of V segment utilization by the mono-V PCR assay was calculated using as the denominator the number of clonal samples detected by either the mono-V assay (N = 52, column 1) or the total clonal TCR cases (N = 68, column 3). Similarly, the frequency of V segment utilization by the multi-V PCR assay was calculated using as the denominator the number of clonal samples detected using either the multi-V assay (N = 51, column 2) or the total clonal cases (N = 68, column 4). Both mono-V and multi-V PCR assays detected similar proportions of V1-8, V9 and V10 GRs (Figure 1C and Table 2). In contrast, TCR-γ GRs involving V11 were detected exclusively by the mono-V PCR primer set (Figure 1C and Table 2; P = 0.0077). Because of some biallelic rearrangements, the frequency of V segments does not add up to 100% (Table 2).
Table 2.
Sensitivity of Detecting Clonal TCR-γ GRs by V Segment
| TCR-γ variable gene segment | N = 52* mono-V TCR-γ primers N (%) | N = 51† multi-V TCR-γ primers N (%) | N = 68‡ mono-V TCR-γ primers N (%) | N = 68‡ multi-V TCR-γ primers N (%) |
|---|---|---|---|---|
| V1-8 | 31 (60) | 29 (57) | 31 (46) | 29 (43) |
| V9 | 18 (35) | 20 (39) | 18 (26) | 20 (29) |
| V10 | 17 (33) | 20 (39) | 17 (25) | 20 (29) |
| V11 | 9 (17) | 0 (0) | 9 (13) | 0 (0) |
Total clonal TCR-γ cases using mono-V primers, N = 52.
Total clonal TCR-γ cases using multi-V primers, N = 51.
Total clonal TCR cases (mono-V TCR-γ + multi-V TCR-γ + TCR-β), N = 68.
Clinical and Molecular Features of Patients with TCR-γ V11 Gene Segment Rearrangements
The mono-V PCR assay detected a clonal TCR-γ V11 peak that was three times above the background in nine patient DNA samples associated with a clear-cut malignancy or a premalignant condition (Table 3). Besides TCR-γ V11, an additional clonal antigen receptor GR was detected in 8/9 (89%) of these DNA samples: TCR-γ (N = 6), TCR-β (N = 5), IGH (N = 1). In only 1/9 (11%) DNA samples did we find a clonal TCR-γ V11 GR without rearrangement of any other antigen receptor (patient 7, Table 3). However, this patient had myelodysplasia with T/NK-cell large granular lymphocytosis (LGL) in the peripheral blood. We considered this to be a true clonal TCR GR, because it is well known that patients with a myelodysplastic syndrome can have an associated clonal T/NK-cell LGL.19
Table 3.
Clinical and Molecular Features of Patients with Clonal TCR-γ V11 Gene Rearrangements
|
TCR-β |
TCR-γ primer sets |
IGH |
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mono-V |
Multi-V |
FR2 | FR3 | |||||||||||||||||
| A | B | C | V1-8 | V9 | V10 | V11 |
V1-8 |
V9 |
V10 |
V11 |
||||||||||
| TA 60°C |
TA 55°C |
TA 60°C |
||||||||||||||||||
| Patient no. | Age/ sex | DNA per reaction (ng) | Final diagnosis | JP1/JP2 | JP | J1/J2 | No Added MgCl2* | +1 mM MgCl2† | ||||||||||||
| 1 | 49/M | 33 | CTCL | + | + | + | + | +‡ | − | ND | − | ND | + | +‡ | − | − | ND | ND | − | − |
| 2 | 84/F | 220 | CTCL | + | − | + | − | − | − | + | − | − | − | − | − | − | + | − | NT | NT |
| 3 | 59/F | 245 | CTCL | − | − | + | + | − | − | + | − | − | + | − | − | − | + | + | NT | NT |
| 4 | 72/F | 175 | CTCL | − | − | − | + | − | − | ND | ND | ND | + | − | − | − | ND | ND | NT | NT |
| 5 | 55/F | 190 | CTCL | − | + | +‡ | + | − | − | + | − | − | + | − | − | − | + | +‡ | NT | NT |
| 6 | 80/F | 305 | T/NKL | − | − | − | − | − | − | + | − | + | + | + | − | − | + | + | NT | NT |
| 7 | 72/M | 153 | MDS-LGL | − | − | − | − | − | − | + | − | + | − | − | − | − | − | − | − | − |
| 8 | 27/F | 88 | LYP | − | − | − | − | − | + | ND | ND | ND | − | − | + | − | ND | ND | − | − |
| 9 | 86/M | 53 | B-NHL | − | + | − | − | − | − | ND | − | ND | − | − | − | − | − | ND | + | + |
+, Clonal gene rearrangement; −, polyclonal gene rearrangement; B-NHL, B-cell non-Hodgkin lymphoma; CTCL, cutaneous T-cell lymphoma; LYP, lymphomatoid papulosis; MDS-LGL, myelodysplastic syndrome T/NK-cell large granular lymphocytosis; ND, not done because of insufficient DNA; NT, not tested; T/NK L, T/natural killer cell lymphoma. Anatomic sites: skin (patients 1, 2, 3, 4, 8, 9); peripheral blood (patients 5, 7); nasal sinus (patient 6). Fixation: formalin-fixed paraffin-embedded tissue (patients 1, 2, 3, 4, 6, 8, 9); fresh or frozen (patients 5, 7).
PCR master mix used as provided by the manufacturer (Mg2+ concentration not disclosed by the manufacturer).
Addition of 1 μl 25 mmol/L MgCl2 per 25 μl PCR reaction to the commercial multi-V master mix.
Two peaks three times above the background.
J Segment Usage in TCR-γ V11 Gene Rearrangements
A significant difference between the mono-V and the multi-V PCR primer sets for amplifying TCR-γ GR is the inclusion of J1/J2, JP1/JP2 and JP in the former, but only J1/J2 and JP1/JP2 in the latter. Therefore, we tested the ability of individual J segment primers to amplify the target DNA sequence for V11 GRs. Some J primers could not be tested due to lack of adequate remaining patient DNA. Using the mono-V forward V11 primer, the TCR-γ V11-J amplicons could be reproduced in all cases with sufficient patient DNA: JP1/JP2 (5/5, 100%); JP (0/7, 0%); J1/J2 (2/5, 40%) (Table 3). Although the multi-V primer sets lacked JP, none of the TCR-γ V11 GRs depended on this J segment for successful amplification, as shown using the mono-V primers (Table 3).
Comparison of TCR-γ V11 Gene Segment Amplification in Control DNA by Mono-V Versus Multi-V PCR Primers
Next, we compared the ability of the V11 primers from the mono-V and the multi-V strategies to amplify TCR-γ GRs. We analyzed polyclonal and clonal control DNAs that were either extracted in our laboratory (250 ng per 25 μl PCR reaction) from reactive tonsil or Jurkat T-cell line or obtained from a commercial source (500 ng per 25 μl PCR reaction) (InVivoScribe Technologies). Primers for both V11 (mono-V; Figure 2, A and C) and V9 (multi-V; Figure 2, B and D), but not V11 (multi-V; Figure 2, B and D), could amplify TCR-γ target DNA sequences from commercial polyclonal control DNA and from reactive tonsil DNA. V11 primers for both mono-V (Figure 2E) and for multi-V (Figure 2F) could amplify commercial clonal control DNA. In contrast, although the mono-V primer for V11 (Figure 2, G and J) could amplify both 100% Jurkat T-cell line DNA and 10% Jurkat diluted in reactive tonsil DNA, the multi-V primer for V11 (Figure 2, H and K) apparently could amplify neither DNA test sample. By increasing the scale sensitivity 17.5-fold, we noted that a minute amount of 100% Jurkat T-cell line DNA appeared to be amplified (Figure 2I). Since tube B of the multi-V assay contained both V9 and V11 primers, we suspected that either the V9 primer slightly cross-reacted with the V11 target DNA sequence or that the reaction conditions for the V11 primer were suboptimal.
Figure 2.
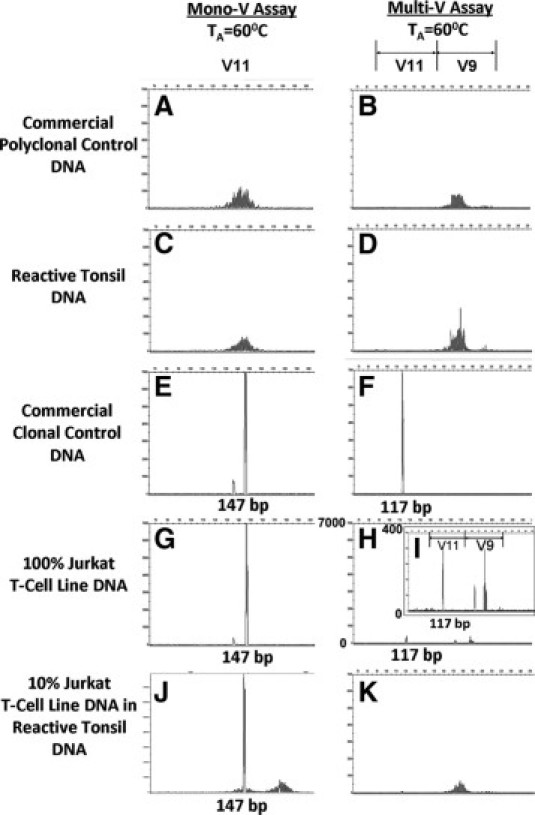
Comparison of TCR-γ V11 gene segment amplification by mono-V versus multi-V PCR assays in control DNA. All PCR assays were set in 25 μl reaction volume. The commercial polyclonal (A and B) and clonal (E and F) control DNAs obtained from In vivoScribe were used at 500 ng per reaction. The reactive tonsil DNA (C and D), Jurkat T-cell line DNA (G–I), and 10% Jurkat T-cell line DNA diluted in reactive tonsil DNA (J and K) were used at 250 ng per reaction. TA, annealing temperature of the PCR reaction. I shows the same PCR products as H but with a 17.5-fold increase in scale sensitivity.
Thermodynamic Features of TCR-γ Primers
Because there was the possibility that the V11 multi-V primer could amplify Jurkat T-cell line DNA, albeit at very low efficiency, we analyzed the thermodynamic features of all mono-V and multi-V primers using the salt-adjusted melting temperature (Tm) equation of Marmur-Schildkraut-Doty, Tm = 81.5 + 16.6 × (log10[Na+] + [K+]) + 0.41 × (% G + C) – 675/n.20 In this equation, n is the number of bases in the oligonucleotide and [Na+] and [K+] are the final concentrations of the sodium and potassium ions in the reaction (Table 4).
Table 4.
Thermodynamic Properties of TCR-γ Primers
| Primer set | Gene segment | Primer sequence | Tm (°C) | GC (%) | PCR product size range (bp)* |
|---|---|---|---|---|---|
| Mono-V | V1-8 | 5′-TACATCCACTGGTACCTACACCAG-3′ | 59.6 | 50 | 240–270 |
| V9 | 5′-GAAAGGAATCTGGCATTCCGTCAG-3′ | 59.6 | 50 | 160–200 | |
| V10 | 5′-AAGCAACAAAGTGGAGGCAAGAAAG-3′ | 57.9 | 44 | 145–175 | |
| V11 | 5′-AGTAAAAATGCTCACACTTCCACTTC-3′ | 56.4 | 38.5 | 130–160 | |
| Range | 56.4–59.6 | 38.5–50 | |||
| JP1/JP2 | 5′-GAAGTTACTATGAGCTTAGTCCCTT-3′ | 56.3 | 40 | ||
| J1/J2 | 5′-TACCTGTGACAACAAGTGTTGTTC-3′ | 56.2 | 41.7 | ||
| JP | 5′-AAGCTTTGTTCCGGGACCAAATAC-3′ | 57.9 | 45.8 | ||
| Range | 56.2–57.9 | 40–45.8 | |||
| Multi-V | V1-8 | 5′-GGAAGGCCCCACAGCRTCTT-3′ | 62.5 | 64.3 | 200–255 |
| V9 | 5′-CGGCACTGTCAGAAAGGAATC-3′ | 57.6 | 52.4 | 155–225 | |
| V10 | 5′-AGCATGGGTAAGACAAGCAA-3′ | 53.4 | 45 | 145–200 | |
| V11 | 5′-CTTCCACTTCCACTTTGAAA-3′ | 51.3 | 40 | 90–110† | |
| Range | 51.3–62.5 | 40–64.3 | |||
| JP1/JP2 | 5′-TTACCAGGCGAAGTTACTATGAGC-3′ | 57.4 | 45.8 | ||
| J1/J2 | 5′-GTGTTGTTCCACTGCCAAAGAG-3′ | 57.7 | 50 | ||
| Range | 57.4–57.7 | 45.8–50 |
Mono-V and multi-V size ranges based on measuring PCR products that could be amplified from DNA extracted from 10 reactive tonsils.
DNA from only 1/10 reactive tonsils could be amplified using the multi-V V11 primer set.
There were striking thermodynamic differences between mono-V and multi-V V11 primers. The multi-V V11 primer had a significantly lower Tm (51.3°C) compared with the other V9 and J primers included in commercial tube B (Tm range: 57.4°C−57.7°C). We hypothesized that this Tm discrepancy probably prevented the multi-V V11 primers from successfully annealing to the DNA target at the manufacturer's recommended annealing temperature (TA) of 60°C, which is relatively high compared with the calculated Tm for the multi-V V11 primer of only 51.3°C (Table 4). We also noted that the multi-V V11 primer had the lowest GC% of all primers contained in tube B of the multi-V BIOMED-2 PCR assay and the mono-V V11 primer had the lowest GC percent of all primers contained in the mono-V PCR assay (Table 4).
Effect of Multi-V Combination on Individual TCR-γ V9 and V11 Primers
To examine whether the poor sensitivity of the multi-V TCR-γ V11 BIOMED-2 primer resulted from combining more than one V segment primer in the same reaction, we tested the ability of individual BIOMED-2 V9, V11, and combined BIOMED-2 V9 plus V11 multi-V primers to amplify TCR-γ GRs in control DNA material (Figure 3). All PCR reactions contained 1 μg final amount of DNA, a mixture of J1/2 and JP1/P2 primers and HotStarTaq master mix kit (Qiagen) in 50 μl reaction volume. As expected, the V9 primer by itself (Figure 3, left column) amplified reactive tonsil DNA alone (Figure 3A) and amplified reactive tonsil DNA when mixed with Jurkat T-cell line DNA (Figure 3G). Importantly, the V11 primer by itself (Figure 3, middle column) could amplify a clonal GR from 100% Jurkat T-cell line DNA (Figure 3E), but not from 10% Jurkat T-cell line DNA diluted in reactive tonsil DNA (Figure 3H). Similarly, the combination of V9 plus V11 primers in the same PCR reaction (Figure 3, right column) could amplify a clonal V11 GR from 100% Jurkat T-cell line DNA (Figure 3F), but not from 10% Jurkat T-cell line DNA diluted in reactive tonsil DNA (Figure 3I). The multi-V TCR-γ V11 primer completely failed to amplify any polyclonal GRs from reactive tonsil (Figure 3, top and bottom rows), when used alone (Figure 3, B and H) or in combination with the V9 primer (Figure 3, C and I).
Figure 3.
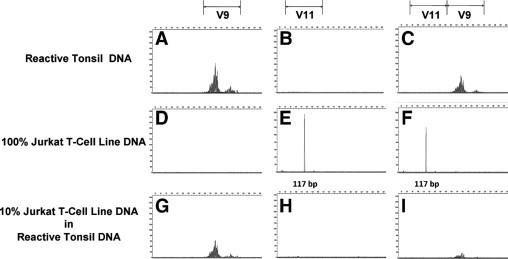
Effect of combining individual multi-V TCR-γ V9 and V11 BIOMED-2 primers. We synthesized multi-V TCR-γ V9 and V11 primers based on their published DNA sequences.15 We added the primers individually (A, D, G and B, E, H) and in combination with each other (C, F, I) to amplify TCR-γ GRs from control DNA. Reactive tonsil DNA (A–C), 100% Jurkat T-cell line DNA (D–F), and 10% Jurkat T-cell line DNA diluted in reactive tonsil DNA (G–I) were used at 1 μg in 50 μl reaction volume. All PCR reactions included a mixture of J1/2 and JP1/P2 primers.
Optimization of the V9 + V11 Multi-V TCR-γ PCR Assay
Since it appeared that the manufacturer's recommended TA of 60°C for the multi-V PCR reaction was too high for the calculated Tm of 51.3°C for the multi-V V11 primer, we tested whether lowering the TA could improve the sensitivity of detecting clonal TCR-γ V11 GRs by the multi-V PCR assay. At TA equal to 60°C (Figure 4, left column), the multi-V PCR assay failed to amplify the V11 gene segment from reactive tonsil DNA (Figure 4A), 100% Jurkat T-cell line DNA (Figure 4D), 10% Jurkat T-cell line DNA diluted in reactive tonsil DNA (Figure 4G), and a representative clinical DNA sample (Figure 4J; patient 3, Table 3). As expected, by lowering the TA from 60°C to 55°C (middle column) or to 50°C (right column), the V11 gene segment could be amplified from the same reactive tonsil DNA (Figure 4, B and C), 100% Jurkat T-cell line DNA (Figure 4, E and F) and 10% Jurkat T-cell line DNA diluted in reactive tonsil DNA (Figure 4, H and I). By lowering the TA, we were able to amplify clonal TCR-γ V11 GRs from 4/6 (67%) available clinical DNA samples (Table 3, patients 2, 3, 5, 6; data not shown except patient 3, Figure 4, K and L; same patient as in Figure 4J) that had shown a clonal TCR-γ V11 GRs by the mono-V assay. For a patient with concurrent V9 GR at TA of 60°C (patient 6, Table 3), the clonal V9 GR could not be detected at TA of 55°C and 50°C (data not shown). Thus, while lowering the TA from 60°C to 55°C appears to improve the sensitivity for V11, it seems to have the opposite effect on the sensitivity for V9 in this sample.
Figure 4.
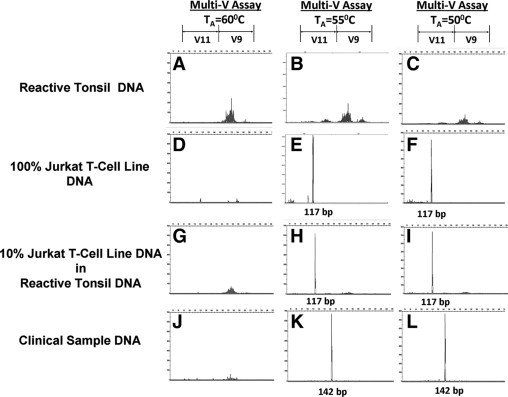
Effect of annealing temperature (TA) on TCR-γ V11 gene segment amplification using the V9 + V11 multi-V primer set. All PCR assays were set in 25 μl reaction volume. Reactive tonsil DNA (A–C), Jurkat T-cell line DNA (D–F), 10% Jurkat T-cell line DNA diluted in reactive tonsil DNA (G–I), and a representative clinical DNA sample (J–L; patient 3, Table 3) were used at 250 ng per PCR reaction. PCR amplification was performed at TA of 60°C (A, D, G, J), 55°C (B, E, H, K), or 50°C (C, F, I, L).
Although the MgCl2 concentration was not specified in the commercial kit, the BIOMED-2 protocol had recommended 1.5 mmol/L MgCl2 for the TCR-γ PCR assays.15 When the MgCl2 was increased by adding 1 μl of 25 mmol/L MgCl2 stock solution to 25 μl of the multi-V PCR reaction, the multi-V TCR-γ primers could successfully amplify TCR-γ GRs involving the V11 gene segment even at the recommended TA of 60°C. Assuming that the manufacturer uses the same 1.5 mmol/L MgCl2 per reaction as recommended by the original BIOMED-2 protocol, the revised final MgCl2 concentration would be 2.5 mmol/L per PCR reaction.15 This was true even for 10% Jurkat T-cell line DNA diluted in reactive tonsil DNA and for 3/5 (60%) patient DNA samples (patient 3, 5, 6; Table 3), which had been previously proven to have a clonal TCR-γ V11 GRs by the mono-V assay. Thus, for the detection of clonal TCR-γ V11 GRs, the same 3/4 clinical DNA samples (patient 3, 5, 6; Table 3) that had benefited from lowering the TA from 60°C to 55°C or 50°C also benefited from raising the Mg2+ concentration from 1.5 mmol/L to 2.5 mmol/L. Overall, we believe that suboptimal TA and Mg2+ concentration help explain why the multi-V PCR assay failed to amplify TCR-γ V11 GRs in clinical DNA samples (patients 2, 3, 5, 6; Table 3).
Substitution of Mono-V V11 for Multi-V V11 Primer
Next, we tested the ability of an alternative primer to amplify TCR-γ V11 GRs by replacing the multi-V V11 primer in tube B of the BIOMED-2 PCR assay with the mono-V V11 primer from the laboratory-developed assay, which has a Tm of 56.4°C that is more similar to other primers in tube B, at the recommended TA of 60°C. Indeed, when mono-V V11 primer was mixed with multi-V V9 and J primers, and HotStarTaq master mix (Qiagen), we could successfully amplify polyclonal TCR-γ V11 GRs from reactive tonsil DNA, and a clonal TCR-γ V11 GR from 10% Jurkat T-cell line DNA diluted in reactive tonsil DNA as well as from 2/3 (66%) clinical DNA samples (patients 5, 6; Table 3), which had been previously proven to have a clonal TCR-γ V11 GRs by the mono-V assay (data not shown). Thus, the multi-V strategy itself is not the problem, but the specific DNA sequence of the V11 primer is suboptimal under these PCR conditions.
Testing PCR Reagents for Defects
Finally, to eliminate the possibility of defective reagents, we re-tested six available clinical DNA samples with clonal V11 GRs, which had been previously successfully amplified using mono-V TCR-γ primers, using a new lot of multi-V reagents at TA of 60°C. In only 1/6 (17%) of the clinical DNA samples could we detect a clonal TCR-γ V11 peak. However, these peaks were much smaller than those obtained at TA of 55°C (data not shown). Furthermore, we were unable to amplify TCR-γ V11 from either polyclonal tonsil DNA or 10% Jurkat T-cell line DNA diluted in reactive tonsil DNA using the new reagents. Therefore, defective multi-V PCR reagents were not responsible for the failure to detect either monoclonal or polyclonal TCR-γ V11 GRs.
Effect of Amount of DNA on Detecting TCR-γ V11 GRs by Multi-V Assay
We extracted different total amounts of DNA (ng) per sample depending on the tissue source: skin (3288 ± 4218, N = 100; mean ± SD, N), lymph nodes (8060 ± 3052, N = 7), peripheral blood (3220 ± 1537, N = 7), bone marrow (9928 ± 5261, N = 18), other tissues (12,997 ± 9,085, N = 8). Therefore, we could not use 500 to 2000 ng of DNA for all PCR reactions, as recommended by the manufacturer (InVivoScribe Technologies). The total amount of DNA (ng) that we included in each 25 μl PCR reaction varied according to availability: skin (140 ± 114, N = 100, mean ± SD, N), lymph nodes (306 ± 63, N = 7), peripheral blood (143 ± 65, N = 7), bone marrow (305 ± 105, N = 18), other tissues (418 ± 227, N = 8). The information on total amount of DNA extracted and total amount of DNA included per PCR reaction could not be retrieved for 4/144 samples.
To investigate whether the amount of DNA used in the PCR reaction could have affected the sensitivity of detecting TCR-γ V11 GRs by the multi-V assay, we tested Jurkat T-cell line DNA in a concentration range recommended by the manufacturer. We amplified 0.5, 1, and 2 μg total DNA per 50 μl PCR reaction using purified Jurkat T-cell line DNA, reactive tonsil DNA, and 50% Jurkat T-cell line DNA diluted in reactive tonsil DNA (data not shown). The V9 plus V11 multi-V primer set amplified polyclonal V9 GRs from both 1 μg of reactive tonsil DNA and amplified a monoclonal V11 GR from 1 μg of Jurkat T-cell line DNA. But using the multi-V assay, we could not amplify a clonal TCR-γ V11 from a 1:1 mixture of Jurkat T-cell line DNA (0.5 μg) and reactive tonsil DNA (0.5 μg). We obtained identical results using 0.5 μg and 2 μg total DNA per PCR reaction.
Discussion
A PCR assay for TCR-γ GRs is a convenient way to detect T-cell clonality because most αβ-positive T-cell lymphomas retain a nonproductive TCR-γ GR, which can be used as a clonal marker. Since TCR-γ is a relatively simple genetic locus, it can be analyzed by PCR using relatively few primers.15 Several monoplex TCR-γ PCR assays have been developed with varying efficacies.5,6,7,8,12,16,21 Recently, a standardized BIOMED-2 multiplex PCR protocol has been proposed for the detection of clonal antigen receptor GRs.15 The 2008 College of American Pathologists molecular oncology proficiency testing surveys A and B indicated that, for detecting clonality, more laboratories used “laboratory-developed” PCR assays (52/94, 55% and 45/96, 47%) than the commercial PCR assay based on the BIOMED-2 protocol (19/94, 20% and 25/96, 26%) (College of American Pathologists Participant Summary of the 2008 Molecular Oncology Proficiency Testing Program, Northfield, IL). The BIOMED-2 protocol was based on the use of fresh/frozen tissue in a limited number of T-cell malignancies. In the original study, only a total of 18 DNA samples with Southern blots that confirmed T-cell clonality were tested;15 among these, just four clonal cases were detected in FFPE tissues and 14 clonal cases were detected in matching fresh/frozen tissues. Later validations of this PCR strategy also mainly relied on fresh/frozen tissues.3,22,23
Since it is known that simultaneous amplification of multiple targets can reduce the sensitivity of the PCR assay for one or more targets,17 we compared the BIOMED-2 multiplex primers that target two TCR-γ V gene segments per reaction (multi-V) with an approach that targets a single V segment per reaction (mono-V) in a real-life clinical setting that necessitates the use of predominantly FFPE tissue. It is important to note that the mono-V primers are similar, but not identical, to the multi-V primers; which, in theory, could lead to differences in the sensitivities between the mono-V and multi-V approaches.
We demonstrated a statistically significant improvement in sensitivity for detecting T-cell clonality by combining mono-V and multi-V TCR-γ PCR assays, or by combining TCR-β results with either mono-V or multi-V TCR-γ results. The value of targeting multiple TCR-γ V gene segments for detecting T-cell clonality has been well-established.2,6 In a multicenter study involving 24 laboratories, PCR assays that targeted multiple V gene segments showed significantly better sensitivity compared with those targeting a single V gene segment.2 Similarly, in a study that examined the utilization of V segments in clonal TCR-γ GRs, the maximum number of clonal cases was detected only when multiple V segments were targeted.6 In addition, the complementary nature of BIOMED-2 TCR-β and TCR-γ PCR assays in detecting clonal TCR GRs has been proven in several multicenter studies using freshly collected or frozen samples of mature T-cell neoplasms.3,23,24 Our results, using predominantly FFPE tissue, confirm these findings. Furthermore, we believe that independently confirming either positive or negative multi-V TCR-γ assay results with another set of mono-V TCR-γ primers can add to the clinical robustness of the TCR-γ PCR test. Also, we have tested a greater number of FFPE tissue samples than other investigators.5,6,7,8,9,10,11,12
In a meta-analysis of TCR-γ V segment utilization, we found that the frequencies of V1-8, V9 and V10 gene usage in the current study were similar to those reported by others (Table 5).4,5,6,7,8,9,10,11,12 However, using the mono-V PCR assay, we detected a slightly higher percentage of cases (9/52, 17%) with V11 GRs compared with others (9/64; 14%, study no. 10, Table 5). In contrast, the multi-V PCR assay completely failed to detect any clonal TCR-γ V11 GRs in our hands. It is important to note that percentages are not calculated the same way in each of the studies listed in Table 5. Only one study (no. 1, Table 5) analyzed all TCR-γ gene rearrangements that were detected and, therefore, their percentages added up to 100%.11 The other studies, including ours, have reported the percentage of cases with a clonal V segment. Therefore, due to biallelic antigen receptor gene rearrangements, the total percentage of TCR-γ GRs exceeds 100%. An estimate of the expected V11 usage, therefore, differs depending on the method of analysis. The reported frequencies of V segment utilization are also influenced by whether a complete primer set was used for multiple TCR-γ V segments and whether the V segment GRs were reported individually. Overall, the frequencies of clonal rearrangements involving TCR-γ V1-8, V9, and V10 in our study were comparable with previous reports. Also, we report a comparable proportion of cases with one and two clonal GRs per DNA sample (Table 5). In our study, we detected two cases with three clonal GRs. Interestingly, cases proven to be malignant lymphoma by Southern blot analysis have shown up to three or even four TCR-γ GRs by PCR.3,9 The determination of which of these multiple PCR peaks represents the actual clonal rearrangement can be difficult without performing further testing. Possible explanations for this phenomenon include biallelic GRs, presence of a second or a minor unrelated clone, genomic instability, and aneuploidy.9
Table 5.
Meta-Analysis of Reported Frequencies of TCR-γ Variable Gene Segment Utilization and Number of Clonal GRs per Case
|
TCR-γ variable gene segment utilization in clonal cases‡ |
Number of V segments rearranged per case |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Study no. | Clonality assay | Primer set | Tissue* | Diagnosis† | DNA amount (ng) | V1-8 | V9 | V10 | V11 | 1 | 2 | 3 | ≥ 4 | Year | Reference no. |
| 1 | SB | NS | LN | PTCL | 10,000 | 23/32 (72) | 8/32 (25) | 0/32 (0) | 1/32 (3) | NS | NS | NS | NS | 1994 | 11 |
| 2 | Mono-V | LD | Skin | CTCL | 2000 | 51/61 (84) | 26/61 (43) | NT | NT | 45/61 (74) | 16/61 (26) | NS | NS | 1994 | 12 |
| 3 | Multi-V | LD | Blood | ALL | 100–200 | 14/30 (47) | 2/30 (7) | 2/30 (7) | 1/30 (3) | 19/30 (63) | 11/30 (37) | 0/30 (0) | 0/30 (0) | 1996 | 4 |
| 4 | Mono-V | LD | Skin | CTCL | 80–120 | 20/24 (83) | 1/4 (25) | 2/3 (67) | 0/1 (0) | NS | NS | NS | NS | 1999 | 8 |
| 5 | Mono-V | LD | Skin | CTCL | 2000 | 39/49 (80) | 17/49 (35) | 1/49 (2) | 1/49 (2) | 40/49 (82) | 9/49 (18) | 0/49 (0) | 0/49 (0) | 2001 | 7 |
| 6* | Mono-V | LD | NS | PTCL | 500 | 55/98 (56) | 22/98 (23) | 17/98 (17) | 4/98 (4) | 25/60 (42) | 32/60 (53) | 3/60 (5) | 0/60 | 2003 | 6 |
| 7 | Multi-V | BM | LN | AITL | 500–2000 | 66/93 (71) | 44/93 (47) | 55/93 (59) | 8/93 (9) | 29/90 (31) | 42/90 (45) | 16/90 (17) | 3/90 (3) | 2006 | 10 |
| 8 | Multi-V | LD | Blood | LGL | NS | 37/49 (76) | 9/49 (18) | 15/49 (31) | 3/49 (6) | 31/49 (63) | 18/49 (37) | 0/49 (0) | 0/49 (0) | 2007 | 5 |
| 9 | Multi-V | BM | NS | PTCL | 100 | 126/166 (76) | NS | NS | NS | 74/166 (45) | 76/166 (46) | 18/166 (11) | NS | 2007 | 3 |
| 10 | Multi-V | BM | LN | ALCL | 500–2000 | 41/64 (64) | 30/64 (47) | 27/64 (42) | 9/64 (14) | 33/64 (52) | 21/64 (33) | 8/64 (13) | 2/64 (3) | 2008 | 9 |
| 11 | Mono-V | LD | Skin | CTCL | 20–855 | 31/52 (60) | 18/52 (35) | 17/52 (33) | 9/52 (17) | 31/52 (60) | 19/52 (37) | 2/52 (4) | 0/52 (0) | 2009 | This study |
| 12 | Multi-V | BM | Skin | CTCL | 20–855 | 29/51 (57) | 20/51 (39) | 20/51 (39) | 0/51 (0) | 34/51 (67) | 16/51 (31) | 1/51 (2) | 0/51 (0) | 2009 | This study |
Numerator/denominator (%) = Number of cases with clonal gene rearrangement/total number of clonal cases (percent).
AITL, angioimmunoblastic T-cell lymphoma; ALCL, anaplastic large cell lymphoma; ALL, acute lymphoblastic leukemia; BM, BIOMED-215; CTCL, cutaneous T-cell lymphoma; LD, laboratory developed (home brew); LGL, large granular lymphocytosis; LN, lymph node; mono-V, mono-V PCR; multi-V, multi-V PCR; NS, not specified in the publication; NT, not tested; PTCL, peripheral T-cell lymphoma; SB, Southern blot.
†Most frequent diagnosis in the study group.
Most frequent tissue type.
Meta-analysis: reanalysis of published studies for TCR-γ variable gene segment usage among total clonal cases; frequency of clonal V segment gene usage as percent of clonal cases, except study 6 where frequency of V segment usage is presented as percent of all gene rearrangements. The sum of percentages is greater than 100% because more than one TCR-γ variable gene segment can be rearranged simultaneously in a single DNA sample.
Because the multi-V TCR-γ V11 PCR assay completely failed to amplify any of the patient DNA samples with proven TCR-γ V11 GRs, we reviewed the design of the multi-V primer sets.15 Since we could amplify the manufacturer-supplied clonal control DNA using tube B, which contains the multi-V V11 primer, correct synthesis of fluorescently labeled primers was confirmed. For the multi-V protocol, it was reported by van Dongen et al15 that initial attempts at multiplexing all V gene segments in a single reaction tube were not successful because of preferential amplification of smaller size PCR products and because some V1-8 and V10/11 gene rearrangements could not be detected. This approach was subsequently replaced by a strategy that combined the most commonly rearranged V segments (V1-8 and V9) with the least commonly rearranged V segments (V10 and V11) in two separate tubes (tube A: V1-8 plus V10; tube B: V9 plus V11). This also permitted specific identification of rearranged V segments by PCR amplicon size.15 Well-known potential sources of decreased sensitivity in multiplex PCR assays include formation of primer-dimers, suboptimal primer-to-template ratio, preferential amplification of one target over the other, and competition for reagents between the primers.18 The actual primer concentration in the commercial multi-V master mix is not available to compare primer concentrations as a potential source of difference in the sensitivity. It is unlikely that the interactions between the V9 and V11 multi-V primers affected the sensitivity of V11 primer, as PCR reactions containing V11 alone or in combination with multi-V V9 primer could successfully amplify clonal GRs from 100% Jurkat T-cell line DNA. However, the multi-V V11 primer lost the ability to amplify clonal GRs from Jurkat T-cell line DNA when it was diluted in reactive tonsil DNA. This suggests that either the presence of additional targets or the dilution of DNA had a detrimental effect on the sensitivity of the multi-V TCR-γ V11 primer. Significantly, the multi-V V11 primer failed to amplify any polyclonal GRs from the control DNA when used alone or in combination with the V9 primer, suggesting overall suboptimal efficiency in amplifying V11 targets using the manufacturer's recommended PCR reaction conditions.
We identified a relatively low Tm of 51.3°C for the multi-V TCR-γ V11 primer as one of the contributing factors for the failure to amplify TCR-γ V11 GRs under the manufacturer's recommended TA equal to 60°C. While a TA of 56–60°C is suitable for the majority of the primers in the mono-V TCR-γ PCR reactions, in general, multiplex assays can benefit from lowering the TA by 4–6°C to allow amplification of all multiplex DNA targets.18 In addition, by increasing the Mg2+ concentration (1 mmol/L MgCl2 added to the commercial master-mix with undisclosed Mg2+ concentration), we could amplify 4/6 clinical DNA samples that had been previously proven to have a sharp TCR-γ V11 peak using the mono-V assay, but were not detected by the original multi-V assay. These findings are consistent with the need for optimizing the Mg2+ concentration, which has been emphasized in a study involving B- and T-cell clonality analysis of FFPE tissues.25 When many DNA targets are amplified simultaneously, those that are either smaller or more efficiently amplified can adversely affect the others by competing for reagents.18,26 The annealing of the multi-V V11 primer could also have been adversely influenced by the AAA sequence at the 3′ end.27 In general, it has been recommended that primer design avoid A and especially T at the 3′ end.27
Since 2/6 retested cases still did not show a clonal TCR-γ GR under optimized PCR conditions, it is possible that other factors such as the amount of DNA used per PCR reaction may have limited the sensitivity of the multi-V assay. Due to the small size of cutaneous biopsies, in many cases it was not possible to obtain large amounts of DNA. Others, including the original BIOMED-2 study, have shown that the use of a smaller amount of DNA (80–300 ng per reaction) from cutaneous and gastric biopsies can be adequate to detect T-cell clonality.8,28,29 Since the initial validation of BIOMED-2 primers was performed in both fresh/frozen and paraffin-embedded tissue using 100 ng of DNA,15 we used 184 ± 146 ng of DNA (mean ± SD; range: 10–920 ng) per 25 μl reaction based on the size of tissue compared with 500 to 2000 ng per 50 μl reaction that is recommended by the manufacturer (InVivoScribe Technologies). It is possible that this lower amount of DNA is optimal for mono-V but not for multi-V PCR amplification of TCR-γ V11. This could also explain why the BIOMED-2 protocol could detect TCR-γ V11 GRs using predominantly lymph nodes, which permitted the extraction of larger amounts of DNA (studies 8, 9; Table 5).9,10 However, the lower amount of DNA per reaction did not seem to affect the amplification of V1-8, V9 and V10 by the multi-V assay. Using a greater amount of DNA per PCR reaction, as recommended by the manufacturer, we still were unable to amplify a clonal TCR-γ V11 GR from Jurkat T-cell line DNA diluted to 10% or even to 50% in reactive tonsil DNA. Interestingly, a clonal TCR-γ V11 GR could be amplified when the same amount of Jurkat T-cell line DNA that was contained in the dilution was used without any tonsil DNA. This suggests the possibility that a sample containing a mixture of V9 and V11 target sequences could lower the sensitivity of detecting clonal TCR-γ V11 GRs with the multi-V PCR assay.
We further investigated nine clinical DNA samples that were discrepant for the detection of clonal TCR-γ V11 GRs in the mono-V versus multi-V PCR assays. The mono-V TCR-γ V11 GRs were interpreted as clonal in nine cases because, in addition to clinical and morphological features for malignancy, we could identify additional TCR-γ GRs that involved other V segments or GRs that simultaneously involved either TCR-β or IGH in 8/9 (89%) of these individuals. Clonal TCR GRs have been reported in B-cell lymphomas, myelodysplastic syndromes, lymphomatoid papulosis and T/NK-cell disorders.19,29,30,31,32 Our findings show that the JP primer was not used in any clonal TCR-γ V11 GRs. These findings support the exclusion of the JP primer from the multi-V assay of the BIOMED-2 study.15 This also suggests that the multi-V BIOMED-2 PCR assay failed to amplify any clonal TCR-γ V11 GR that could have been amplified in cases with either the JP1/JP2 or J1/J2 primers, which are included in the multi-V reaction mixture.
Pseudoclonality in cutaneous biopsies, which represented a majority of the samples in our study, is well documented in the literature with reported frequencies ranging from 0% to 24%.33,34,35 We think that the mono-V assay could be susceptible to amplifying false-positive pseudoclonality since we detected a frequency of clonal TCR-γ V11 GRs that is higher than that reported by others (studies 1, 3, 5, 6, 7, 8, 10; Table 5). We encountered several challenging clinical cases that support this observation. For example, Table 3 shows that we detected two rearranged J segments in TCR-γ V11 GRs for cases 6 and 7, which is extremely rare. At the same time, cases 7 and 9 in Table 3 did not show any clonal TCR-γ V11 GRs with the multi-V TCR-γ assay, even under the optimized conditions. These results suggest the possibility that the mono-V TCR-γ assay might be susceptible to detecting pseudoclonality. Therefore, it is critical to avoid overcalling potential pseudoclonal results, which could occur because of a low number of lymphocytes in the sample, a pseudolymphoma of the skin, or an oligoclonal T-cell expansion in an inflammatory process.22,33 We believe that the true frequency of clonal TCR-γ V11 GRs is likely to be found somewhere in between the extremes listed in Table 5, 0% to 17%. However, a detailed analysis of J segment utilization and parallel testing under different PCR conditions is not feasible for routine clinical testing. Therefore, such cases showing sharp clonal peaks by mono-V PCR in the presence of suggestive clinical features are likely to be interpreted as clonal, whereas they may be pseudoclonal. It has been suggested that the detection of TCR-γ PCR products by Genescan analysis (capillary gel electrophoresis) alone may be associated with false-positive results.15 However, due to multiple practical advantages, Genescan analysis is among the most widely used detection methods in current laboratory practice.36 (College of American Pathologists Participant Summary of the 2008 Molecular Oncology Proficiency Testing Program, Northfield, IL) A strong caution must be advised in interpreting and acting on the results of the mono-V and the multi-V PCR assays when a clonal GR is detected by only one assay or when a TCR-γ V11 GR is the only clonal peak. This is especially important in the absence of confirmation by a second method such as Southern blot analysis or a TCR-β PCR assay.
The lack of Southern blot correlation is a limitation of our study, which is shared by many other clinical laboratories that offer only PCR-based clonality assays. Therefore, our study more closely reflects the reality of molecular diagnosis in the 21st century that we have, for better or worse, become accustomed to. Yet the question of the standard for clonality in this study and in clinical laboratories is important. The use of Southern blot analysis in routine clinical diagnosis is limited when only a small amount of DNA can be extracted from the sample, such as a punch or shave skin biopsy. Degradation of DNA in paraffin-embedded tissue is another limiting factor.23 In a recent study, PCR was actually superior to Southern blot analysis in detecting T-cell clonality.23 On one hand, TCR-β analysis might be of help, but it is well known that not every case with a clonal TCR-γ GR is accompanied by TCR-β clonality and vice versa.3,23 Morphological information also has its flaws since one uses TCR clonality assays to solve diagnostic dilemmas raised by morphological examination. Therefore, the results of gene rearrangement studies should be interpreted with caution and correlated with all of the available clinical and pathological information before rendering a diagnosis of malignant lymphoma.
References
- 1.van Dongen JJ, Wolvers-Tettero IL. Analysis of immunoglobulin and T cell receptor genes. i: Basic and technical aspects. Clin Chim Acta. 1991;198:1–91. doi: 10.1016/0009-8981(91)90246-9. [DOI] [PubMed] [Google Scholar]
- 2.Arber DA, Braziel RM, Bagg A, Bijwaard KE. Evaluation of T cell receptor testing in lymphoid neoplasms: results of a multicenter study of 29 extracted DNA and paraffin-embedded samples. J Mol Diagn. 2001;3:133–140. doi: 10.1016/S1525-1578(10)60664-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruggemann M, White H, Gaulard P, Garcia-Sanz R, Gameiro P, Oeschger S, Jasani B, Ott M, Delsol G, Orfao A, Tiemann M, Herbst H, Langerak AW, Spaargaren M, Moreau E, Groenen PJ, Sambade C, Foroni L, Carter GI, Hummel M, Bastard C, Davi F, Delfau-Larue MH, Kneba M, van Dongen JJ, Beldjord K, Molina TJ. Powerful strategy for polymerase chain reaction-based clonality assessment in T-cell malignancies report of the biomed-2 concerted action bhm4 ct98–3936. Leukemia. 2007;21:215–221. doi: 10.1038/sj.leu.2404481. [DOI] [PubMed] [Google Scholar]
- 4.Fodinger M, Buchmayer H, Schwarzinger I, Simonitsch I, Winkler K, Jager U, Knobler R, Mannhalter C. Multiplex PCR for rapid detection of T-cell receptor-gamma chain gene rearrangements in patients with lymphoproliferative diseases. Br J Haematol. 1996;94:136–139. doi: 10.1046/j.1365-2141.1996.6372268.x. [DOI] [PubMed] [Google Scholar]
- 5.Gra OA, Sidorova JV, Nikitin EA, Turygin AY, Surzhikov SA, Melikyan AL, Sudarikov AB, Zasedatelev AS, Nasedkina TV. Analysis of T-cell receptor-gamma gene rearrangements using oligonucleotide microchip: a novel approach for the determination of T-cell clonality. J Mol Diagn. 2007;9:249–257. doi: 10.2353/jmoldx.2007.060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawnicki LC, Rubocki RJ, Chan WC, Lytle DM, Greiner TC. The distribution of gene segments in T-cell receptor gamma gene rearrangements demonstrates the need for multiple primer sets. J Mol Diagn. 2003;5:82–87. doi: 10.1016/s1525-1578(10)60456-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li N, Bhawan J. New insights into the applicability of T-cell receptor gamma gene rearrangement analysis in cutaneous T-cell lymphoma. J Cutan Pathol. 2001;28:412–418. doi: 10.1034/j.1600-0560.2001.028008412.x. [DOI] [PubMed] [Google Scholar]
- 8.Signoretti S, Murphy M, Cangi MG, Puddu P, Kadin ME, Loda M. Detection of clonal T-cell receptor gamma gene rearrangements in paraffin-embedded tissue by polymerase chain reaction and nonradioactive single-strand conformational polymorphism analysis. Am J Pathol. 1999;154:67–75. doi: 10.1016/s0002-9440(10)65252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan BT, Seo K, Warnke RA, Arber DA. The frequency of immunoglobulin heavy chain gene and T-cell receptor gamma-chain gene rearrangements and Epstein-Barr virus in alk+ and alk- anaplastic large cell lymphoma and other peripheral T-cell lymphomas. J Mol Diagn. 2008;10:502–512. doi: 10.2353/jmoldx.2008.080054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan BT, Warnke RA, Arber DA. The frequency of b- and T-cell gene rearrangements and Epstein-Barr virus in T-cell lymphomas: a comparison between angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, unspecified with and without associated B-cell proliferations. J Mol Diagn. 2006;8:466–475. doi: 10.2353/jmoldx.2006.060016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Theodorou I, Raphael M, Bigorgne C, Fourcade C, Lahet C, Cochet G, Lefranc MP, Gaulard P, Farcet JP. Recombination pattern of the TCR gamma locus in human peripheral T-cell lymphomas. J Pathol. 1994;174:233–242. doi: 10.1002/path.1711740402. [DOI] [PubMed] [Google Scholar]
- 12.Wood GS, Tung RM, Haeffner AC, Crooks CF, Liao S, Orozco R, Veelken H, Kadin ME, Koh H, Heald P, Barnhill RL, Sklar J. Detection of clonal T-cell receptor gamma gene rearrangements in early mycosis fungoides/Sezary syndrome by polymerase chain reaction and denaturing gradient gel electrophoresis (PCR/DGGE) J Invest Dermatol. 1994;103:34–41. doi: 10.1111/1523-1747.ep12389114. [DOI] [PubMed] [Google Scholar]
- 13.Cozzio A, French LE. T-cell clonality assays: how do they compare? J Invest Dermatol. 2008;128:771–773. doi: 10.1038/jid.2008.49. [DOI] [PubMed] [Google Scholar]
- 14.Cairns SM, Taylor JM, Gould PR, Spagnolo DV. Comparative evaluation of pcr-based methods for the assessment of T cell clonality in the diagnosis of T cell lymphoma. Pathology. 2002;34:320–325. doi: 10.1080/003130202760120463. [DOI] [PubMed] [Google Scholar]
- 15.van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, Delabesse E, Davi F, Schuuring E, Garcia-Sanz R, van Krieken JH, Droese J, Gonzalez D, Bastard C, White HE, Spaargaren M, Gonzalez M, Parreira A, Smith JL, Morgan GJ, Kneba M, Macintyre EA. Design and standardization of pcr primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the biomed-2 concerted action bmh4-ct98–3936. Leukemia. 2003;17:2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 16.Greiner TC, Raffeld M, Lutz C, Dick F, Jaffe ES. Analysis of T cell receptor-gamma gene rearrangements by denaturing gradient gel electrophoresis of gc-clamped polymerase chain reaction products: correlation with tumor-specific sequences. Am J Pathol. 1995;146:46–55. [PMC free article] [PubMed] [Google Scholar]
- 17.Markoulatos P, Georgopoulou A, Kotsovassilis C, Karabogia-Karaphillides P, Spyrou N. Detection and typing of hsv-1, hsv-2, and vzv by a multiplex polymerase chain reaction. J Clin Lab Anal. 2000;14:214–219. doi: 10.1002/1098-2825(2000)14:5<214::AID-JCLA3>3.0.CO;2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markoulatos P, Siafakas N, Moncany M. Multiplex polymerase chain reaction: a practical approach. J Clin Lab Anal. 2002;16:47–51. doi: 10.1002/jcla.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Epling-Burnette PK, Painter JS, Rollison DE, Ku E, Vendron D, Widen R, Boulware D, Zou JX, Bai F, List AF. Prevalence and clinical association of clonal T-cell expansions in myelodysplastic syndrome. Leukemia. 2007;21:659–667. doi: 10.1038/sj.leu.2404590. [DOI] [PubMed] [Google Scholar]
- 20.von Ahsen N, Wittwer CT, Schutz E. Oligonucleotide melting temperatures under pcr conditions: nearest-neighbor corrections for mg(2+), deoxynucleotide triphosphate, and dimethyl sulfoxide concentrations with comparison to alternative empirical formulas. Clin Chem. 2001;47:1956–1961. [PubMed] [Google Scholar]
- 21.Shadrach B, Warshawsky I. A comparison of multiplex and monoplex T-cell receptor gamma pcr. Diagn Mol Pathol. 2004;13:127–134. doi: 10.1097/01.pdm.0000126419.92931.a3. [DOI] [PubMed] [Google Scholar]
- 22.Langerak AW, Molina TJ, Lavender FL, Pearson D, Flohr T, Sambade C, Schuuring E, Al Saati T, van Dongen JJ, van Krieken JH. Polymerase chain reaction-based clonality testing in tissue samples with reactive lymphoproliferations: usefulness and pitfalls: a report of the biomed-2 concerted action bmh4-ct98–3936. Leukemia. 2007;21:222–229. doi: 10.1038/sj.leu.2404482. [DOI] [PubMed] [Google Scholar]
- 23.Sandberg Y, van Gastel-Mol EJ, Verhaaf B, Lam KH, van Dongen JJ, Langerak AW. Biomed-2 multiplex immunoglobulin/T-cell receptor polymerase chain reaction protocols can reliably replace southern blot analysis in routine clonality diagnostics. J Mol Diagn. 2005;7:495–503. doi: 10.1016/S1525-1578(10)60580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Krieken JH, Langerak AW, Macintyre EA, Kneba M, Hodges E, Sanz RG, Morgan GJ, Parreira A, Molina TJ, Cabecadas J, Gaulard P, Jasani B, Garcia JF, Ott M, Hannsmann ML, Berger F, Hummel M, Davi F, Bruggemann M, Lavender FL, Schuuring E, Evans PA, White H, Salles G, Groenen PJ, Gameiro P, Pott C, Dongen JJ. Improved reliability of lymphoma diagnostics via pcr-based clonality testing: report of the biomed-2 concerted action bhm4-ct98–3936. Leukemia. 2007;21:201–206. doi: 10.1038/sj.leu.2404467. [DOI] [PubMed] [Google Scholar]
- 25.Bereczki L, Kis G, Bagdi E, Krenacs L. Optimization of pcr amplification for b- and T-cell clonality analysis on formalin-fixed and paraffin-embedded samples. Pathol Oncol Res. 2007;13:209–214. doi: 10.1007/BF02893501. [DOI] [PubMed] [Google Scholar]
- 26.Elnifro EM, Ashshi AM, Cooper RJ, Klapper PE. Multiplex pcr: optimization and application in diagnostic virology. Clin Microbiol Rev. 2000;13:559–570. doi: 10.1128/cmr.13.4.559-570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viljoen GJN, Louis H, Crowther JR, editors. Molecular diagnostic PCR handbook. Vol. 2. Springer; New York: 2005. pp. 20–49. [Google Scholar]
- 28.Hummel M, Oeschger S, Barth TF, Loddenkemper C, Cogliatti SB, Marx A, Wacker HH, Feller AC, Bernd HW, Hansmann ML, Stein H, Moller P. Wotherspoon criteria combined with b cell clonality analysis by advanced polymerase chain reaction technology discriminates covert gastric marginal zone lymphoma from chronic gastritis. Gut. 2006;55:782–787. doi: 10.1136/gut.2005.080523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandberg Y, Heule F, Lam K, Lugtenburg PJ, Wolvers-Tettero IL, van Dongen JJ, Langerak AW. Molecular immunoglobulin/t- cell receptor clonality analysis in cutaneous lymphoproliferations: experience with the biomed-2 standardized polymerase chain reaction protocol. Haematologica. 2003;88:659–670. [PubMed] [Google Scholar]
- 30.Saunthararajah Y, Molldrem JL, Rivera M, Williams A, Stetler-Stevenson M, Sorbara L, Young NS, Barrett JA. Coincident myelodysplastic syndrome and T-cell large granular lymphocytic disease: clinical and pathophysiological features. Br J Haematol. 2001;112:195–200. doi: 10.1046/j.1365-2141.2001.02561.x. [DOI] [PubMed] [Google Scholar]
- 31.Garcia MJ, Martinez-Delgado B, Granizo JJ, Benitez J, Rivas C. IGH, TCR-gamma, and TCR-beta gene rearrangement in 80 B- and T-cell non-Hodgkin's lymphomas: study of the association between proliferation and the so-called “aberrant” patterns. Diagn Mol Pathol. 2001;10:69–77. doi: 10.1097/00019606-200106000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Lamberson C, Hutchison RE, Shrimpton AE. A pcr assay for detecting clonal rearrangement of the TCR-gamma gene. Mol Diagn. 2001;6:117–124. doi: 10.1054/modi.2001.25321. [DOI] [PubMed] [Google Scholar]
- 33.Boer A, Tirumalae R, Bresch M, Falk TM. Pseudoclonality in cutaneous pseudolymphomas: a pitfall in interpretation of rearrangement studies. Br J Dermatol. 2008;159:394–402. doi: 10.1111/j.1365-2133.2008.08670.x. [DOI] [PubMed] [Google Scholar]
- 34.Ponti R, Quaglino P, Novelli M, Fierro MT, Comessatti A, Peroni A, Bonello L, Bernengo MG. T-cell receptor gamma gene rearrangement by multiplex polymerase chain reaction/heteroduplex analysis in patients with cutaneous T-cell lymphoma (mycosis fungoides/Sezary syndrome) and benign inflammatory disease: correlation with clinical, histological and immunophenotypical findings. Br J Dermatol. 2005;153:565–573. doi: 10.1111/j.1365-2133.2005.06649.x. [DOI] [PubMed] [Google Scholar]
- 35.Theriault C, Galoin S, Valmary S, Selves J, Lamant L, Roda D, Rigal-Huguet F, Brousset P, Delsol G, Al Saati T. Pcr analysis of immunoglobulin heavy chain (IGH) and TCR-gamma chain gene rearrangements in the diagnosis of lymphoproliferative disorders: results of a study of 525 cases. Mod Pathol. 2000;13:1269–1279. doi: 10.1038/modpathol.3880232. [DOI] [PubMed] [Google Scholar]
- 36.Greiner TC, Rubocki RJ. Effectiveness of capillary electrophoresis using fluorescent-labeled primers in detecting T-cell receptor gamma gene rearrangements. J Mol Diagn. 2002;4:137–143. doi: 10.1016/s1525-1578(10)60694-0. [DOI] [PMC free article] [PubMed] [Google Scholar]