

Abstract
Background
The C10 family of cysteine proteases includes enzymes that contribute to the virulence of bacterial pathogens, such as SpeB in Streptococcus pyogenes. The presence of homologues of cysteine protease genes in human commensal organisms has not been examined. Bacteroides fragilis is a member of the dominant Bacteroidetes phylum of the human intestinal microbiota, and is a significant opportunistic pathogen.
Results
Four homologues of the streptococcal virulence factor SpeB were identified in the B. fragilis genome. These four protease genes, two were directly contiguous to open reading frames predicted to encode staphostatin-like inhibitors, with which the protease genes were co-transcribed. Two of these protease genes are unique to B. fragilis 638R and are associated with two large genomic insertions. Gene annotation indicated that one of these insertions was a conjugative Tn-like element and the other was a prophage-like element, which was shown to be capable of excision. Homologues of the B. fragilis C10 protease genes were present in a panel of clinical isolates, and in DNA extracted from normal human faecal microbiota.
Conclusions
This study suggests a mechanism for the evolution and dissemination of an important class of protease in major members of the normal human microbiota.
Background
Bacteroides fragilis is a Gram-negative member of the normal human gut microbiota. The Bacteroidetes constitutes one of the major bacterial phyla in the healthy human gut [1]. However, B. fragilis is also an important opportunistic pathogen, and it is the most frequently isolated anaerobic bacterium in clinical specimens, including abdominal abscesses and bloodstream infections [2]. Indeed, while B. fragilis accounts for only 4 to 13% of the normal human fecal microbiota, it is responsible for 63 to 80% of Bacteroides infections [3]. Only a few virulence factors have been described for B. fragilis, with the best characterized being the polysaccharide (PS) capsule [4] and a secreted metalloprotease, fragilysin [5]. The capsule, which displays antigenic variation, promotes the formation of abscesses [4], and the reduction of pro-inflammatory responses to B. fragilis [4,6]. The metalloprotease fragilysin, which has been linked to diarrheal disease [5], has activity against the zonula junctions between cells, and could disrupt tissue integrity [7]. B. fragilis also encodes homologues of C10 proteases [8]. These are members of the CA clan of papain-like proteases. Other C10 proteases include the important virulence factors Streptococcal pyrogenic exotoxin B (SpeB) from Streptococcus pyogenes and Interpain A from Prevotella intermedia. SpeB cleaves a variety of host protein, including immunoglobulin, fibronectin and vitronectin; it also activates IL-1β and releases kinin from kininogen [9]. Interestingly, both SpeB and Interpain A target and inactivate complement factor C3 [10,11]. One further characterized C10 protease is the Periodontain from the oral pathogen Porphyromonas gingivalis, which cleaves α1-proteinase inhibitor promoting degradation of connective tissue components [12].
For both SpeB and another well characterized family of cysteine proteases (C47 family) expressed in staphylococci (Staphopain), the protease genes are found juxtaposed to genes encoding specific protease inhibitors, Spi [13] (a propeptide analogue) and Staphostatin [14] (a lipocalin-like entity), respectively.
The genomes of Bacteroides spp., including B. fragilis, may include plasmids [15], and typically include multiple prophage remnants, pathogenicity islands and both conjugative and non-conjugative transposons (CTn and Tn respectively) [16]. This would facilitate acquisition and dissemination of virulence markers. Indeed, the fragilysin is encoded on a pathogenicity island which has been shown to be mobile [17].
This study centers on the identification and characterization of genes encoding homologues of SpeB, their genetic linkage with putative inhibitors, and the association of these homologous genes with mobile genetic elements.
Results
The B. fragilis genome harbours four paralogous C10 protease genes
A phylogenetic study was undertaken to determine the relatedness of C10 proteases in other members of the Bacteroidetes phylum (Fig. 1). This identified eight-four C10 protease candidates, ranging in size from 269 to 1656 amino acids, in organisms that occupy both human and environmental niches. The larger of these proteins (>600 amino acid residues, average length 803 residues) group together along with SpeB and Interpain A. These larger proteins have additional C-terminal domains, the role of which is yet to be determined [12,18]. The Bfp proteases group with proteins <500 amino acid residues in length (average length 435 residues). Although acceptable bootstrap values were obtained for nodes separating deeper phylogenetic levels, the bootstrap values for the shallower divisions were low. This reflects the unstable phylogeny obtained. However, it is noteworthy that all of the candidate protease sequences had a variation on the two active site motifs indicated in Fig 2.
Figure 1.

Phylogenetic tree of the C10 proteases available on the GenBank and NCBI databases. Cluster analysis was based upon the neighbour-joining method. Numbers at branch-points are percentages of 1000 bootstrap re-samplings that support the topology of the tree. The tree was rooted using C47 family cysteine protease sequences (Staphopains). The locus tag identifiers and the organism name are given. SpeB and the Btp proteases are indicated by a red diamond.
Figure 2.

Amino acid sequence alignment of the Bacteroides fragilis proteases Bfp with the archetype C10 protease SpeB from Streptococcus pyogenes. The alignment was generated with T-coffee [55]. The red back-highlight regions indicate the sequences flanking the critical active site Cys and His residues (vertical black arrowhead).
Of particular interest was the identification of SpeB homologues in B. fragilis. Analysis of the B. fragilis 638R ftp://ftp.sanger.ac.uk/pub/pathogens/bf/, YCH46 [19] and NCTC9343 [7] genome sequences identified genes encoding a paralogous family of C10 cysteine proteases named Bfp1 (BF638R0104, 45390), Bfp2 (BF638R1641, 56666), Bfp3 (BF638R3679, 47323), Bfp4 (BF638R0223, 48433) for B. fragilis protease, encoded by genes bfp1-4 respectively. The locus identifiers for the unpublished 638R genome, followed by the predicted molecular mass of the preproprotein in Daltons are given in parenthesis. bfp1 and bfp2 were present in all three strains whereas bfp3 and bfp4 were present only in B. fragilis 638R (Table 1).
Table 1.
Occurrence of bfp genes in clinical isolates and in the human gut microbiota.
| Strain | bfp1 | bfp2 | bfp3 | bfp4 | Bfgi2 | attB |
|---|---|---|---|---|---|---|
| 638R | + | + | + | + | + | + |
| YCH46a | + | + | - | - | - | + |
| NCTC9343b | + | + | - | - | - | + |
| NCTC9344 | + | + | + | - | + | + |
| NCTC10581 | + | + | - | - | - | + |
| NCTC10584 | - | + | - | - | - | + |
| NCTC11295 | - | + | - | - | - | + |
| NCTC11625 | + | + | - | - | - | + |
| TMD1 | + | + | + | + | + | + |
| TMD2 | + | + | + | + | + | + |
| TMD3 | + | + | + | + | + | + |
a. Based on analysis of genome sequence only, locus identifier BF0154 for bfp1, and BF1628 bfp2. All other strains confirmed by PCR.
b. Locus identifier BF0116 for bfp1 and BF1640 for bfp2.
TMD1-TMD3: total microbiota DNA, from faeces of 3 healthy adult subjects.
Similarity between the predicted Bfp protein sequences and zymogen SpeB ranges from 33-41.2%, with similarity between the paralogues themselves higher (36.7-46.1%) (Table 2). These low values are not surprising, as it has been established that the overall sequence identity and similarity between the CA clan of Papain-like proteases is low [20]. However, the core of the the protease domains of the C10 proteases SpeB (1DKI) and Interpain (3BBA) [18] are similar in structure (root mean squared deviation of 1.220 Å based on 197 Cα positions), even with only 32.5% sequence identity. Critically, the active site residues (Cys165 and His313, SpeB zymogen numbering [21]) are highly conserved (Fig. 2). It is probable that the bfp genes encode active proteases, and thus, may contribute to the pathogenesis of Bacteroides infections in a manner analogous to the role of SpeB in streptococcal pathogenesis [22].
Table 2.
Similarity/identity matrix for Bfp proteases and SpeBa.
| C10 Protease | SpeB | Bfp1 | Bfp2 | Bfp3 | Bfp4 |
|---|---|---|---|---|---|
| SpeB | 19.2 | 22.6 | 16.7 | 21.9 | |
| Bfp1 | 38.1 | 21 | 23.9 | 19.7 | |
| Bfp2 | 33.0 | 36.7 | 20.2 | 22.5 | |
| Bfp3 | 41.2 | 41.7 | 37.7 | 28.5 | |
| Bfp4 | 38.2 | 42.1 | 41.0 | 46.1 |
a Numbers in italics are percentage similarity, numbers in bold type are percentage identities.
Bacterial cysteine protease genes have been found coupled to genes encoding specific inhibitors, therefore, the regions both up and downstream of the four bfp genes were analyzed for candidate inhibitors. Three open reading frames encoding small proteins (116-138 amino acids) within 35 base pairs of the proteases were identified. These were named bfi1A (BF638R0103), bfi1B (BF638R0105) and bfi4 (BF638R0222) (for Bacteroides fragilisinhibitor). The encoded proteins showed no significant identity to the propeptides of any known protease, nor to Spi. Surprisingly, they had identity to the C47 cysteine proteases inhibitors, the Staphostatins, ranging from 15.0-23.4% identity and 32.6-45.7% similarity (Table 3). This is in line with identity between Staphostatin A and Staphostatin B with 20.4% identity and 45.0% similarity. Despite low levels of sequence identity, analysis of the predicted secondary structure and the conservation and alignment of a critical glycine residue in these sequences (indicated in Fig. 3) when compared to Staphostatins, suggested that these bfi genes encode specific protease inhibitors.
Table 3.
Similarity/identity matrix for Bfi putative inhibitors, Staphostatins and Spia.
| Spi | ScpA | SspB | Bfi1A | Bfi1B | Bfi4 | |
|---|---|---|---|---|---|---|
| Spi | 16.4 | 11.9 | 11.1 | 17.2 | 14.3 | |
| ScpBb | 41.7 | 20.4 | 20.2 | 19.4 | 23.4 | |
| SspCb | 31.2 | 45.0 | 20.2 | 18.6 | 15.0 | |
| Bfi1A | 26.7 | 38.8 | 45.7 | 20.3 | 20.4 | |
| Bfi1B | 35.7 | 39.7 | 40.5 | 41.3 | 20.1 | |
| Bfi4 | 31.2 | 39.1 | 32.6 | 38.4 | 39.9 |
a Numbers in italics are percentage similarity, numbers in bold type are percentage identities.
b ScpB and SspC are Staphostatin A and Staphostatin B respectively.
Figure 3.

Structure and sequence based alignments of Staphostatins with putative inhibitors from Bacteroides fragilis. Panel A is a sequence alignment generated with T-coffee. Superimposed on this are secondary structure predictions for all 5 proteins, generated with GorIV [46]. Residues with secondary structure assigned as coil, β-strand, and α-helix are back-highlighted in yellow, red and blue respectively. The glycine residue conserved in Staphostatins is marked with a vertical black arrowhead. Panel B is a sequence alignment of Staphostatin A (1OH1A [56]) and Staphostatin B (1NYCB [14]). The sequence based alignment was generated with T-coffee. This alignment is coloured, as for panel A, according to secondary structure determined from the crystal structures of the two inhibitors. For clarity the spacing is preserved from panel A. These alignments suggest that GorIV is over-predicting helical content in the staphostatins.
To determine the likely cellular location of Bfp and Bfi proteins, the respective sequences were analyzed using LipPred [23], LipoP [24], SignalP [25] and PSORTb [26]. These analyses suggested that Bfi1A has a typical Sec pathway leader sequence and is likely to be exported to the periplasm. Bfi1B, Bfi4, Bfp1, Bfp2 and Bfp4 have predicted lipoprotein signal sequences and are likely to be tethered to the outer membrane [24,27]. Whilst Bfp3 has a lipoprotein leader sequence it is not clear which membrane it is likely to associate with. It should be noted that maturation of C10 zymogens would release the active protease from the anchoring acyl-lipid into the extracellular milieu.
B. fragilis C10 proteases genes, bfp1 and bfp4, are co-transcribed with those for predicted Staphostatin-like inhibitors
For both the streptococcal and staphylococcal systems, the proteases and adjacently encoded inhibitors are co-transcribed [13,28]. To determine if this transcriptional coupling of protease and inhibitor genes was also present in B. fragilis, RNA was isolated from broth grown 638R cells, and analysed by reverse transcriptase PCR, using a series of specific primers for the protease and inhibitor genes (Table 4). Amplicons were detected for all C10 protease structural genes suggesting that all the proteases were transcribed in vitro (Fig. 4, Lanes 2, 6, 7 and 8 for bfp1, bfp2, bfp3 and bfp4 respectively). Amplification of a 1.9 Kb product (Fig. 4, Lane 5) using primers Bfi1A_F and Bfi1B_R supports the hypothesis that bfp1 is co-transcribed on a single mRNA with bfi1A and bfi1B. In addition, amplification of a 1.65 Kb product with primers Bfp4_F and Bfi4_R suggests that bfp4 is transcriptionally coupled to bfi4 (Fig. 4, Lane 9).
Table 4.
Oligonucleotide primers used in this study.
| Primer | Sequence | Commenta |
|---|---|---|
| Bfp1_F | CAGCAGCATATGGACGAAGAAATCATTATTTTGATTAAT | E, L |
| Bfp1_R | CAGCAGGGATCCTTACCACAAAATTTCAGTTCCC | E, L |
| Bfp2_F | CAGCAGCATATGACAAGAAGAGTTGATTCTGCCAG | E |
| Bfp2_R | CAGCAGGGATCCTTATTTATTAGGTGACACTTTAAT | E |
| Bfp3_F | CAGCAGGGATCCAGAAGATAATGTAATTGCTTCTTT | E |
| Bfp3_R | CAGCCAGGAATTCTCATCGGTGTATATTGGTTATC | E |
| Bfp4_F | CAGCAGGGATCCGAAGACAATTTAGAATCTTTAA | E, L |
| Bfp4_R | CAGCAGGGATCCTCATCGCGATATAATAGAATATTC | E |
| Bfi1A_F | CAGCAGGAATTCGAGGATGTAATGGCTATTATG | E, L |
| Bfi1A_R | CAGCAGGGATCCTTACCTTCCAATATAAATGTC | E |
| Bfi1B_F | CAGCAGGGATCCACACCAACCAGATACTCCACC | E |
| Bfi1B_R | CAGCAGGAATTCTTACTCTTTTTTTTCGGCTGTG | E, L |
| Bfi4_F | CAGCAGGAATTCAGGGATGGAGATTGGGATTC | E |
| Bfi4_R | CAGCAGGGATCCTTAATTATCCTTTCCCTTTTGTTT | E, L |
| Bfgi2_Int_F | CCTGATATTAGCTTCTCTATCTTTTTTGCC | I |
| Bfgi2_Int_R | CAGCAGGGATTCCGAAGATAATGTAATTGCTTC | I |
| Bfgi2_attB_F | CCGGGAATGTTTCGTCAGGAATTGATGGTG | I |
| Bfgi2_attB_R | GGTTTATTGATTGTTATTTGTCGGCAAAG | I |
a Primer used in E = Expression studies, L = Linkage studies, I = Integration/Excision studies
Figure 4.
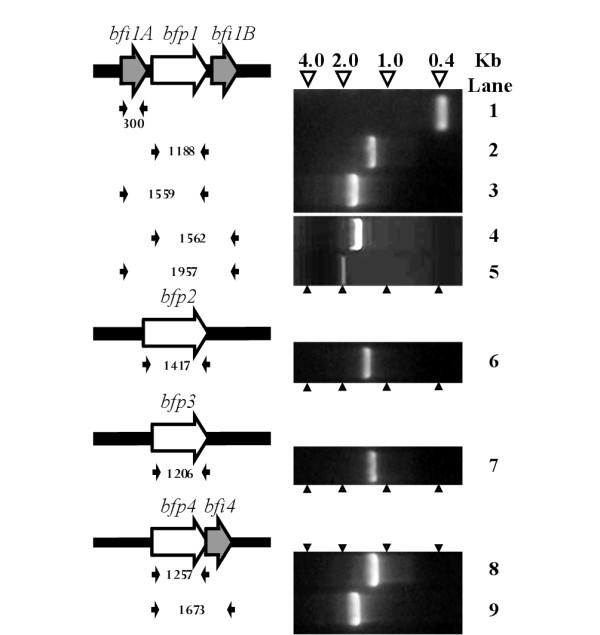
Analysis of expression and transcriptional coupling of bfp genes in Bacteroides fragilis. Horizontal open arrows represent the protease (white) and putative inhibitor (grey) genes. Small filled black arrows represent the positions of the oligonucleotide primers used in the reverse-transcription PCR analysis, the size of the expected amplicon is given in bp between the appropriate sets of pimers. The resulting PCR fragments are presented in the right-hand panels, above which the size markers are indicated.
bfp3 and bfp4 are located on genome insertions
As mentioned above, two of the protease genes (bfp3 and bfp4) were identified only in strain 638R enabling a comparison with the two other sequenced strains of B. fragilis. Using the Artemis comparison tool [29], alignment of the B. fragilis NCTC9343 and B. fragilis 638R genome sequences identified two large insertions in strain 638R associated with the chromosomal locations of bfp3 and bfp4. In B. fragilis 638R, bfp4 was found on a 55.9 Kb insertion, called Bfgi2 in this study. Annotation of this insertion revealed an architecture similar to the CTnERL-type conjugative transposons (CTn) [30] (Fig. 5, panel A and Table 5). Although the expected integrase, excisionase and transfer regions were present in Bfgi1, mobility of this insertion could not be established for broth grown cultures treated with mitomycin C, tetracycline, or UV treatment (data not shown). These treatments are commonly used to initiate excision of CTn elements [31,32]. Bfgi1 showed homology to a region in Porphyromonas gingivalis ATCC 33277 which has previously been characterized as a CTn [33]. However, this region of ATCC 33277 did not encode a C10 protease.
Figure 5.

Insertions in the genome of Bacteroides fragilis 638R carry C10 protease homologues. Genome alignment of B. fragilis strains 638R and NCTC9343 was generated using the Artemis Comparison Tool. The co-ordinates for the insertions are from the unpublished 638R genome. Genes in the insertions are represented by horizontal open coloured arrows and are described below (see also Tables 5 and 6). The G+C content of the insertions is plotted in the lowest section of each panel. The grey horizontal line in each case represents the average G+C content for the genome. For both panels the C10 proteases are represented by horizontal red arrows and the pale blue arrows are genes that are not directly related to the skeleton of the particular mobile genetic element. Panel A. The insertion Bfgi1 has the features of a CTn. The putative integrase and excisionase genes (Int and Ex respectively), ABC transporters (ABC), mobilization genes (Mob), and transfer genes (Tra) are represented by royal blue, dark green, grey and yellow arrows respectively. Panel B. The insertion Bfgi2 has the architecture of a Siphoviridae bacteriophage. The lysis cassette, tail region, head regions, packaging (Pkg) and the replication and modification genes (Rep/Mod) are represented by teal, mid-grey, moss green, royal blue and peach arrows respectively.
Table 5.
Annotation of genes in the B. fragilis 638R Bfgi1 insertion.
| ORF | Protein Length |
Putative function | % Id/Sima | Organismb | Accession no.c |
|---|---|---|---|---|---|
| 1 | 411 | Integrase protein | 59/74 (411) | B. fragilis YCH46 | AAS83518.1 |
| 2 | 119 | Hypothetical protein | 42/64 (114) | B. thetaiotaomicron | AA077037.1 |
| 3 | 162 | Ctn042 | 37/59 (112) | B. fragilis YCH46 | AAS83514.1 |
| 4 | 1828 | DNA Methylase (BmhA) | 57/71 (1339) | B. fragilis YCH46 | AAS83508.1 |
| 5 | 143 | Hypothetical protein | 41/56 (121) | B. thetaiotaomicron | AA077432.1 |
| 6 | 709 | Excisionase | 57/72 (704) | B. fragilis YCH46 | AAS83511.1 |
| 7 | 464 | Hypothetical protein | 41/57 (482) | B. thetaiotaomicron | AA075210.1 |
| 8 | 260 | TetR/AcrR family | 32/58 (204) | B. thetaiotaomicron | AA075614.1 |
| 9 | 161 | Hypothetical protein | 48/71 (108) | P. gingivalis W83 | AA075614.1 |
| 10 | 780 | Putative TonB OM Receptor | 63/78 (780) | B. fragilis YCH46 | BAD47377.1 |
| 11 | 412 | Hypothetical protein | 56/73 (398) | B. fragilis YCH46 | CAH06331.1 |
| 12 | 187 | Putative Ni-Co-Cd resistance protein | 29/42 (110) | Syntrophus aciditrophicus SB | ABC78121.1 |
| 13 | 604 | ABC Transporter | 41/61 (570) | B. thetaiotaomicron | AA075616.1 |
| 14 | 593 | ABC Transporter | 43/63 (591) | B. thetaiotaomicron | AA075615.1 |
| 15 | 172 | RteC | 56/76 (80) | B. thetaiotaomicron | AAA22922.1 |
| 16 | 129 | Peptidase S51 | 44/59 (100) | Listeria monocytogenes | AAT03167.1 |
| 17 | 114 | Hypothetical protein | 69/79 (73) | P. gingivalis W83 | AAQ66123.1 |
| 18 | 138 | Hypothetical protein | 34/53 (135) | B. thetaiotaomicron | AA077558.1 |
| 19 | 431 | C10 protease | 26/43 (454) | B. thetaiotaomicron | AA077558.1 |
| 20 | 112 | Hypothetical protein | 27/72 (80) | Polaribacter irgensii | A4BZ61 |
| 21 | 512 | ECF type σ-factor | 31/50 (502) | B. thetaiotaomicron | AA077884.1 |
| 22 | 148 | Hypothetical protein | 43/58 (46) | Campylobacter upsaliensis | EAL52724.1 |
| 23 | 671 | MobC | 51/91 (660) | B. fragilis YCH46 | AAS83500.1 |
| 24 | 408 | MobB | 53/71 (348) | B. fragilis YCH46 | AAS83499.1 |
| 25 | 137 | MobA | 46/66 (136) | B. fragilis YCH46 | AAS83498.1 |
| 26 | 260 | TraA | 53/71 (246) | B. fragilis YCH46 | AAG17826.1 |
| 27 | 142 | TraB | 34/51 (133) | B. fragilis YCH46 | BAD48110.1 |
| 28 | 135 | TraC | 34/55 (63) | B. fragilis YCH46 | AAS83495.1 |
| 29 | 271 | TraA | 37/53 (251) | B. fragilis YCH46 | BAD49765.1 |
| 30 | 196 | TraD | 26/37 (182) | B. thetaiotaomicron | AA077408.1 |
| 31 | 123 | TraE | 73/79 (78) | B. fragilis YCH46 | BAD48110.1 |
| 32 | 126 | TraF | 56/66 (87) | B. fragilis YCH46 | AAS83492.1 |
| 33 | 828 | TraG | 72/83 (829) | B. fragilis YCH46 | BAD466872.1 |
| 34 | 209 | TraI | 65/80 (209) | B. fragilis YCH46 | BAD46870.1 |
| 35 | 366 | TraJ | 70/86 (303) | B. fragilis YCH46 | AAS83488.1 |
| 36 | 207 | TraK | 75/84 (207) | B. fragilis YCH46 | AAS83487.1 |
| 37 | 110 | TraL | 37/58 (72) | B. fragilis YCH46 | BAD48102.1 |
| 38 | 454 | TraM | 49/64 (439) | B. fragilis YCH46 | BAD46866.1 |
| 39 | 310 | TraN | 70/84 (300) | B. fragilis YCH46 | AAG17839.1 |
| 40 | 194 | TraO | 55/72 (177) | B. fragilis YCH46 | BAD46864.1 |
| 41 | 292 | TraP | 52/67 (292) | B. fragilis YCH46 | BAD46863.1 |
| 42 | 153 | TraQ | 60/76 (139) | B. fragilis YCH46 | BAD48097.1 |
| 43 | 171 | Lysozyme | 53/73 (147) | B. fragilis YCH46 | BAD46861.1 |
| 44 | 116 | DNA Binding protein | 75/80 (103) | P. gingivalis W83 | AAQ66295.1 |
| 45 | 530 | Hemerythrin | 41/62 (508) | Alkaliphilus metalliredigens | EA081668.1 |
| 46 | 426 | Ctn003 | 41/57 (441) | B. fragilis YCH46 | BAD46856.1 |
| 47 | 176 | Anti-restriction protein | 52/71 (175) | B. fragilis YCH46 | BAD48093.1 |
| 48 | 138 | Ctn002 | 48/62 (115) | B. fragilis YCH46 | BAD46855.1 |
| 49 | 200 | Hypothetical protein | 74/77 (31) | B. fragilis YCH46 | BAD48092.1 |
a Percentage identity/similarity, the number in parenthesis is the number of amino acids used in the calculations.
b The organism encoding the B. fragilis 638R gene homologue.
cAccession number of the highest scoring BLAST hit with an annotated function.
The bfp3 gene was located on a 39 Kb insertion, called Bfgi2 in this study. Analysis of this region predicted functional modules, e.g. DNA metabolism, DNA packaging, prophage head, tail and lysis proteins, consistent with a bacteriophage genomic structure similar to the Siphoviridae family of bacteriophages (Fig. 5, panel B and Table 6). These phage are known to infect bacteria that reside in the gut, and are the most frequently identified phage infecting B. fragilis [34]. Similarly to other Siphoviridae, Bfgi2 inserts into the 3' end of the tRNAArg gene [31]. The attB site overlaps the tRNAArg gene, however integration of Bfgi2 regenerates a functional tRNAArg gene. Bfgi2 had homology only with a region of a genome for an unidentified Bacteroides sp. (Bacteroides sp. 3_2_5), which included a homologue of bfp3.
Table 6.
Annotation of genes in the B. fragilis 638R Bfgi2 insertion.
| ORF | Protein Length |
Putative function | % Id/Sima | Organism (Bacteriophage)b | Accession no.c |
|---|---|---|---|---|---|
| 1 | 446 | Integrase | 47/63 (436) | Bacteroides uniformis | AAF74437.1 |
| 2 | 751 | Polysialic acid transport protein, KpsD | 72/84 (676) | B. fragilis YCH46 | BAD48680.1 |
| 3 | 163 | Hypothetical protein | 37/49 (156) | B. fragilis YCH46 | BAD49193.1 |
| 4 | 172 | N-acetylmuramyl-L-alanine amidase | 60/75 (150) | B. thetaiotaomicron | AA077433.1 |
| 5 | 151 | Holin | 25/54 (99) | B. subtillus (phi-105) | NP_690778.1 |
| 6 | 1215 | Phage related protein, tail component | 26/49 (173) | Actinobacillus pleuropneumonia | ZP_00134779.1 |
| 7 | 697 | Hypothetical protein | 21/40 (300) | Flavobacterium (11b) | YP_112519.1 |
| 8 | 1034 | Tail tape measure protein | 31/50 (119) | Burkholderia cepacia (BcepNazgul) | NP_918983.1 |
| 9 | 195 | Hypothetical protein | 32/54 (150) | B. fragilis YCH46 | BAD49201.1 |
| 10 | 126 | Hypothetical protein | 29/52 (86) | B. fragilis YCH46 | BAD49202.1 |
| 11 | 425 | Phage major capsid | 32/50 (252) | Vibrio phage VP882 | AAS38503.2 |
| 12 | 204 | Prohead protease | 42/59 (157) | Lactobacillus casei (A2) | CAD43895.1 |
| 13 | 450 | Phage portal protein | 34/52 (365) | Pseudomonas (D3) | AAD38955.1 |
| 14 | 543 | Terminase (Large subunit) | 38/58 (493) | Streptococcus agalactiae (λSa04) | ABA45667.1 |
| 15 | 145 | Terminase (Small subunit) | 26/43 (122) | Lactococcus lactis (Bil309) | NP_076733.1 |
| 16 | 139 | Hypothetical protein | 28/59 (171) | Clostridium difficile 630 | CAJ67750.1 |
| 17 | 104 | HNH Endonuclease | 41/59 (74) | Geobacillus (GBSVI) | ABC61271.1 |
| 18 | 142 | Hypothetical protein | 98/100 (136) | B. fragilis YCH46 | BAD49213.1 |
| 19 | 104 | Hypothetical protein | 97/100 (93) | B. fragilis YCH46 | BAD49214.1 |
| 20 | 320 | Hypothetical protein | 99/100 (294) | B. fragilis YCH46 | BAD49215.1 |
| 21 | 113 | Hypothetical protein | 99/99 (109) | B. fragilis YCH46 | BAD49216.1 |
| 22 | 428 | Ctn003 | 39/53 (420) | B. fragilis YCH46 | AAS83476.1 |
| 23 | 175 | Ctn002 | 35/48 (134) | B. fragilis YCH46 | AA583475.1 |
| 24 25 |
253 137 |
Putative DNA Methylase | 100/100 (253) | Lactococcus lactis (Tuc2009) | NP_108695.1 |
| 26 | 124 | Hypothetical protein | 88/88 (116) | B. fragilis YCH46 | BAD49220.1 |
| 27 | 150 | NinG recombination protein | 98/98 (125) |
A. actinomycetemcomitans (AaPhi23) bacteriophage bb bacteriophage |
NP_852744.1 |
| 28 | 126 | Hypothetical protein | 93/94 (116) | B. fragilis YCH46 | YP_099756.1 |
| 29 | 149 | DNA Topoisomerase I | 32/51 (82) | Pediococcus pentosaceus ATCC25745 | YP_80446.1 |
| 30 | 106 | Excisionase | 42/61 (52) | Colwellia psychrerythraea 34H | YP_268668.1 |
| 31 | 198 | Hypothetical protein | 66/74 (110) | B. fragilis YCH46 | BAD49224.1 |
| 32 | 137 | Peptidase S24 | 29/50 (81) | Flavobacterium johnsoniae | EASS8507.1 |
| 33 | 121 | Hypothetical protein | 35/52 (120) | Pelobacter carbinolicus | YP_358455.1 |
| 34 | 431 | C10 protease | 28/45 (375) | B. thetaiotaomicron | NP_811364.1 |
a Percentage identity/similarity, the number in parenthesis is the number of amino acids used in the calculations.
b The organism, with associated bacteriophage in parenthesis where applicable.
cAccession number of the highest scoring BLAST hit with an annotated function.
The regions flanking the C10 loci in a range of Bacteroidetes (B. thetaiotaomicron (AE015928), B. uniformis (AAYH00000000), B. ovatus (AAXF00000000), B. intestinalis (ABJL00000000), Parabacteroides distasonis (CP000140), Porphyromonas gingivalis (AP009380, AE015924) and Prevotella intermedia (ID: 246198) were examined for the presence of markers for mobile genetic elements (e.g. the Tra functional module, or phage structural modules for instance tail, and capsid). The GenBank accession code or JCVI taxon numbers are given in parenthesis. A cassette of Tra genes (A through O, locus tags PG1473-1486) was found 35.3 Kb away from prtT in Porphyromonas gingivalis strain W83 (locus tag 1427) and again in strain ATCC 33277 Tra I to Q were found (locus tags PGN_592 to PGN_599) 40.5 Kb away from PrtT (PGN_0561) in that strain. However, no complete CTn or phage could be found adjacent to these or any other C10 protease gene.
The Bfgi2 element harbouring the bfp3 gene is capable of excision
The putative att sequence for the integration of Bfgi2 was identified by analysis of the sequence at the boundaries of the inserted DNA in strain 638R compared with NCTC9343. A short 16 bp direct repeat sequence was identified flanking the Bfgi2 insertion (Fig. 6, panel A). PCR primers Bfgi2_attB_F and Bfgi2_attB_R (Table 4) were used in a PCR reaction to detect the excision of the Bfgi2 prophage from mitomycin C treated B. fragilis 638R cells. The resulting 595 bp PCR product is consistent with excision of Bfgi2 from the B. fragilis 638R genome (Fig. 6, panel B, Lane 2), and reconstruction of an intact tRNAArg gene (Fig. 6, panel C). Sequencing of this PCR product indicated the presence of a single copy of the 16 bp repeat region, the proposed attB site for Bfgi2 (Fig. 6, panel C).
Figure 6.
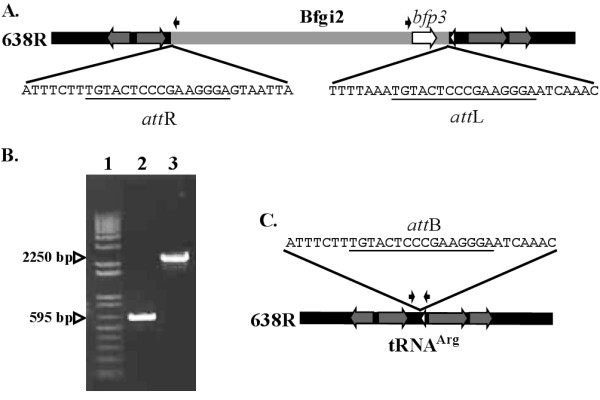
The prophage carrying bfp3 is capable of excision. Panel A. The Bfgi2 prophage (grey bar) is flanked by the B. fragilis 638R genome (black bar). The bfp3 gene (open white arrow), tRNAArg (white arrowhead) and genes flanking Bfgi2 (mid-grey) are shown. The attR and attL sequences (underlined) are shown in the expanded sequence. The locations of primers used in these studies are shown by small black arrows (see Table 4). Panel B. Agarose gel electrophoretic analysis of PCR reactions to test for excision of the prophage (Lane 2) and for the circular intermediate of the 'phage (Lane 3). Lane 1 contains DNA size markers. Panel C. Schematic representation of the 638R genome, after excision of the Bfgi2 element. Colour scheme is as for panel A. The regenerated attB site (underlined) is shown in the expanded sequence.
The mitomycin C-treated cells were also analysed for the presence of the Bfgi2 circular intermediate. The primers Bfgi2_Int_F and Bfgi2_Int_R (Table 4) were designed directed outwards across the proposed attL and attR sites. Using these primers, amplification of product should only occur if a circularized form of Bfgi2 is present in the cell. The size (2.25 Kb) sequence of the resulting PCR product confirmed the presence of the circular intermediate (Fig. 6 panel B, Lane 3). Attempts to show plaque formation using NCTC9343 as an indicator strain did not produce any visible plaques. This could be due to the phenomenon of limited host range for the bacteriophage. However, given that Bfgi2 circular intermediate was detected it is tempting to speculate that it is, or is a derivative of an active phage and such phage could be transmitted to a non-lysogenized strain of B. fragilis, bringing with it a copy of a C10 protease.
C10 protease genes are present in clinical isolates of B. fragilis and in the healthy human faecal microbiota
In addition to the 3 genome strains, a panel of 5 clinical isolates of B. fragilis from several human infection sites (Table 7) were tested by allele-specific PCR for the C10 protease genes they harbour. The results indicated that this panel of strains have a complement of bfp genes more similar to NCTC9343 than to 638R (Table 1). The distribution of bfp genes in the clinical isolates is not identical, and none of the 5 isolates carried all four bfp genes. The bfp1-4 genes were detected in 3, 5, 1 and 0 clinical isolates respectively. The bfp4 gene was not be detected in any of these clinical strains, while bfp1 was not detected in two strains (NCTC 10584 and NCTC 11295). In contrast, bfp2 was encoded by all strains. In B. fragilis strain YCH46, there is a CTnERL-type conjugative transposon 353 bp distance from the bfi1A-bfp1-bfi1B gene cluster. However, this conjugative transposon is not present in either of the other two sequenced B. fragilis genomes, 638R and NCTC 9343. The bfp3 gene was only detected in one clinical isolate (NCTC 9344), with a concomitant detection of the Bfgi2 insertion. In all cases a 595 bp fragment was successfully amplified using the primer pair Bfgi2_attB_F and Bfgi2_attB_R (not shown), indicating the presence of a free integration site for Bfgi2 in all strains. It should be noted that for NCTC 9344 and 638R, there was a lower product yield and although not quantitative this is likely due to the integration of Bfgi2 in a sub-population of the cells.
Table 7.
Bacterial strains used in this study
| B. fragilis strain | Source of isolate | Reference |
|---|---|---|
| 638R | Clinical isolate, human | [57] |
| YCH46a | Bacteraemia, human | [19] |
| NCTC9343 | Appendix abscess, human | [58] |
| NCTC9344 | Septic operation wound, human | [59] |
| NCTC10581 | Empyema fluid, human | [60] |
| NCTC10584 | Pus, human | [58] |
| NCTC11295 | Pus from fistula, human | [61] |
| NCTC11625 | Post-operative wound infection, human | [62] |
a. Analysis of genome sequence only.
Presence of bfp genes in the healthy human intestinal microbiota was investigated by PCR analysis performed on total DNA extracted from faeces from three adult subjects. The amplification of the appropriately sized DNA fragments indicated that all 4 bfp genes characterized in this study were present in all three subjects whose samples were tested (Table 1). Interestingly, this analysis also indicated the presence of an integrated Bfgi2 prophage in these faecal samples, as well as free attB sites.
Discussion
This study has established the presence of homologues of the streptococcal virulence factor SpeB in a significant gut microorganism, B. fragilis. The amplification of bfp1-4 specific sequences from mRNA samples supports the idea that these protease genes are expressed in vivo and in two cases the protease genes (bfp1 and bfp4) are coupled to genes encoding proteins resembling Staphostatins-like inhibitors. A role in protection of the bacterial cells from ectopic protease has been mooted for these inhibitors [35]. From sequence analysis, the Bacteroides inhibitors are likely to localize to the periplasm and cell membranes, which could be an additional mechanism to protect the bacterial cell from proteolytic damage, similar to roles suggested for Spi and the Staphostatins.
The presence of two Bfp protease genes on mobile genetic elements parallels some of the paradigms for the acquisition of virulence determinants by other microorganisms. For example the Panton-Valentine Leukocidin of Staphylococcus aureus [36], SpeC of S. pyogenes [37], diphtheria toxin of Corynebacterium diphtheria [38] and cholera toxin of Vibrio cholera [39] as well as the fragilysin of B. fragilis [40] are all encoded by mobile genetic elements. Although the latter case has yet to be conclusively established, the other examples cited, and many others in the literature, illustrate an augmentation of virulence in the recipient organism. Thus, the acquisition of additional copies of a protease with homology to SpeB by lateral gene transfer may increase the ability of B. fragilis to cause disease. However, establishing the mechanism of transfer of these protease genes and the role of the encoded proteases in B. fragilis opportunistic infections will require further studies.
Conclusion
The phylum Bacteroidetes constitutes a major proportion of the healthy human intestinal microbiota. Variations in the Bacteroidetes proportion are linked to disease, and selected species are significant causes of human infectious disease. Alterations in the composition or function of the Bacteroidetes component of the intestinal microbiota might plausibly be involved in diseases involving immune dysregulation, including Inflammatory Bowel Disease, or Irritable Bowel Syndrome. Bacterial proteases are particularly relevant in this context, because they might be involved in the perturbed regulation of host matrix metalloproteases, which is a feature of IBD [41]. Thus the linkage of C10 proteases genes to mobile genetic elements in B. fragilis, and the demonstrated presence of these coding sequences in the healthy adult gut microbiota, is potentially significant. Experiments to investigate the expression and function of these genes in vivo are in progress.
Methods
Bacterial strains and culture conditions
Bacteroides fragilis strains used in this study are presented in Table 7. All strains were purchased from the United Kingdom National Culture Collection (UKNCC) except 638R which was a kind gift from Dr Sheila Patrick, Queen's University, Belfast. Both B. fragilis strains and B. thetaiotaomicron VPI-5482 [42] were grown in an anaerobic chamber at 37°C. Cultures were grown without shaking in Brain Heart Infusion (BHI) broth supplemented with 50 μg/ml hemin and 0.5 μg/ml menadione. Media for plating was made from Brain Heart Infusion agar supplemented with 5% defibrinated sheep blood, 50 μg/ml hemin and 0.5 μg/ml menadione.
Bioinformatics and sequence analysis
Members of the C10 protease family in B. fragilis were detected by BLAST analysis [43]. Sequences were aligned by CLUSTAL W [44] or T-Coffee [45]. Protein secondary structure was predicted using GorIV [46] and JPred [47]. Protein export signals were identified using the algorithms using LipPred [23], LipoP [48], SignalP [25] and PSORTb [26]. Phylogenetic and molecular evolutionary analyses were conducted using genetic-distance-based neighbour-joining algorithms [49] within MEGA Version 4.0 http://www.megasoftware.net/. Bootstrap analysis for 1000 replicates was performed to estimate the confidence of tree topology [50]. MegaBLAST [51] was used to search all NCBI genomes for Bfgi1 and Bfgi2.
Molecular techniques
Standard techniques were employed for molecular analysis [52]. Bacteroides genomic DNA was prepared as described by [53]. Total microbial DNA was extracted from human faeces, collected under an ethically approved protocol, by a glass beads-Qiagen Stool kit method previously described [54]. PCR reactions were carried using 10-30 ng of genomic DNA from B. fragilis 638R as template and using Phusion Polymerase (New England Biolabs). The primers Bfp3_F and Bfgi2_Int_F (Table 4) were used for detecting the attP sites for Bfgi2.
Bfgi2_attB_F and Bfgi2_attB_R (Table 4) were used for determining the attB attachment sites for Bfgi2 integration. The primers TraQ_F and Int_F were used in testing for the presence of the circular intermediate for Bfgi1. Primers to detect the circular intermediate for both Bfgi1 and Bfgi2 were designed, pointing outwards, flanking the ends of each predicted element. Primers to detect the attB site in Bfgi2 were designed, pointing inwards, flanking the proposed excision point for the Bfgi2 prophage DNA.
Total RNA isolation for Reverse Transcription analysis
B. fragilis 638R and B. thetaiotaomicron VPI-5482 were cultured under anaerobic conditions until early logarithmic phase and the cultures were then immediately centrifuged for 15 minutes at 4000 × g. Total RNA extraction from B. fragilis 638R and B. thetaiotaomicron VPI-5482 was carried out using the FastRNA® Pro Blue Kit according to manufacturer's instructions (Q-Biogene, UK). Total RNA was subjected to DNase treatment using Turbo DNase (Ambion, UK) and stored at -80°C. RNA integrity was analyzed visually using denaturing 1.2% agarose gel electrophoresis and quantified using a NanoDrop (Thermo Fisher Scientific, USA). Reverse transcription PCR for C10 proteases was performed using the Superscript III One-step RT-PCR system (Invitrogen, USA). Primers used in RT-PCR reactions are documented in Table 4. Primers were added to a final concentration of 200 nM and 200 ng of total RNA added. As a control for DNA contamination, RT-PCR minus reactions was set up where the control reaction only received primers after the reverse transcriptase step. Aliquots (20 μl from 25 μl) of all samples were analyzed by standard agarose gel electrophoresis.
Induction of Bfgi1 and Bfgi2 excision from the B. fragilis 638R genome
B. fragilis 638R was grown overnight and then sub-cultured by a 1 in 50 dilution into fresh broth and grown until late log phase. The culture was then exposed to either Mitomycin C (0.2 μg/ml), Tetracycline (0.5 μg/ml) UV light (1 mJ/cm2) then grown for a further 12 hours.
Authors' contributions
RFT performed and designed experiments, and co-wrote the manuscript. TFK designed experiments and interpreted the data. PWOT designed experiments, analyzed data and co-wrote the manuscript. JCC conceived the study, designed the experiments, interpreted the data and co-wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Roibeard F Thornton, Email: roibeard.thornton@gmail.com.
Todd F Kagawa, Email: kagawatf@gmail.com.
Paul W O'Toole, Email: pwotoole@ucc.ie.
Jakki C Cooney, Email: jakki.cooney@ul.ie.
Acknowledgements
The authors gratefully acknowledge financial support from the following sources: University of Limerick PhD studentship to RFT; Science Foundation Ireland grant 08/RFP/BMT1596 to JCC; PWOT is supported by the (Govt. of Ireland) Dept. Agriculture Fisheries and Food FHRI award to the ELDERMET project, and by CSET (Alimentary Pharmabiotic Centre) and PI awards from Science Foundation Ireland. The B. fragilis 638R genome sequence data were provided by the Pathogen Genome Sequencing group at the Wellcome Trust Sanger Institute and can be obtained from ftp://ftp.sanger.ac.uk/pub/pathogens/bf/. Permission of J. Parkhill and S. Patrick to use this data is gratefully acknowledged.
References
- Rajilic-Stojanovic M, Smidt H, de Vos WM. Diversity of the human gastrointestinal tract microbiota revisited. Environ Microbiol. 2007;9:2125–2136. doi: 10.1111/j.1462-2920.2007.01369.x. [DOI] [PubMed] [Google Scholar]
- Avila-Campos MJ, Liu C, Song Y, Rowlinson MC, Finegold SM. Determination of bft gene subtypes in Bacteroides fragilis clinical isolates. J Clin Microbiol. 2007;45:1336–1338. doi: 10.1128/JCM.02108-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerdeno-Tarraga AM, Patrick S, Crossman LC, Blakely G, Abratt V, Lennard N, Poxton I, Duerden B, Harris B, Quail MA. Extensive DNA inversions in the Bacteroides fragilis genome control variable gene expression. Science. 2005;307:1463–1465. doi: 10.1126/science.1107008. [DOI] [PubMed] [Google Scholar]
- Tzianabos AO, Onderdonk AB, Smith RS, Kasper DL. Structure-function relationships for polysaccharide-induced intra-abdominal abscesses. Infect Immun. 1994;62:3590–3593. doi: 10.1128/iai.62.8.3590-3593.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obiso RJ Jr, Azghani AO, Wilkins TD. The Bacteroides fragilis toxin fragilysin disrupts the paracellular barrier of epithelial cells. Infect Immun. 1997;65:1431–1439. doi: 10.1128/iai.65.4.1431-1439.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaleznik DF, Kasper DL. The role of anaerobic bacteria in abscess formation. Annu Rev Med. 1982;33:217–229. doi: 10.1146/annurev.me.33.020182.001245. [DOI] [PubMed] [Google Scholar]
- Wu S, Lim KC, Huang J, Saidi RF, Sears CL. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc Natl Acad Sci USA. 1998;95:14979–14984. doi: 10.1073/pnas.95.25.14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potempa J, Pike RN. Bacterial peptidases. Contrib Microbiol. 2005;12:132–180. doi: 10.1159/000081693. full_text. [DOI] [PubMed] [Google Scholar]
- von Pawel-Rammingen U, Bjorck L. IdeS and SpeB: immunoglobulin-degrading cysteine proteinases of Streptococcus pyogenes. Curr Opin Microbiol. 2003;6:50–55. doi: 10.1016/S1369-5274(03)00003-1. [DOI] [PubMed] [Google Scholar]
- Terao Y, Mori Y, Yamaguchi M, Shimizu Y, Ooe K, Hamada S, Kawabata S. Group A streptococcal cysteine protease degrades C3 (C3b) and contributes to evasion of innate immunity. J Biol Chem. 2008;283:6253–6260. doi: 10.1074/jbc.M704821200. [DOI] [PubMed] [Google Scholar]
- Potempa M, Potempa J, Kantyka T, Nguyen KA, Wawrzonek K, Manandhar SP, Popadiak K, Riesbeck K, Eick S, Blom AM. Interpain A, a cysteine proteinase from Prevotella intermedia, inhibits complement by degrading complement factor C3. PLoS Pathog. 2009;5:e1000316. doi: 10.1371/journal.ppat.1000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson D, Potempa J, Kordula T, Travis J. Purification and characterization of a novel cysteine proteinase (periodontain) from Porphyromonas gingivalis. Evidence for a role in the inactivation of human alpha1-proteinase inhibitor. J Biol Chem. 1999;274:12245–12251. doi: 10.1074/jbc.274.18.12245. [DOI] [PubMed] [Google Scholar]
- Kagawa TF, O'Toole PW, Cooney JC. SpeB-Spi: a novel protease-inhibitor pair from Streptococcus pyogenes. Mol Microbiol. 2005;57:650–666. doi: 10.1111/j.1365-2958.2005.04708.x. [DOI] [PubMed] [Google Scholar]
- Rzychon M, Filipek R, Sabat A, Kosowska K, Dubin A, Potempa J, Bochtler M. Staphostatins resemble lipocalins, not cystatins in fold. Protein Sci. 2003;12:2252–2256. doi: 10.1110/ps.03247703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Tribble GD, Bayley DP. Genetic elements of Bacteroides species: a moving story. Plasmid. 1998;40:12–29. doi: 10.1006/plas.1998.1347. [DOI] [PubMed] [Google Scholar]
- Kuwahara T, Yamashita A, Hirakawa H, Nakayama H, Toh H, Okada N, Kuhara S, Hattori M, Hayashi T, Ohnishi Y. Genomic analysis of Bacteroides fragilis reveals extensive DNA inversions regulating cell surface adaptation. Proc Natl Acad Sci USA. 2004;101:14919–14924. doi: 10.1073/pnas.0404172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco AA, Cheng RK, Chung GT, Wu S, Oh HB, Sears CL. Molecular evolution of the pathogenicity island of enterotoxigenic Bacteroides fragilis strains. J Bacteriol. 1999;181:6623–6633. doi: 10.1128/jb.181.21.6623-6633.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallorqui-Fernandez N, Manandhar SP, Mallorqui-Fernandez G, Uson I, Wawrzonek K, Kantyka T, Sola M, Thogersen IB, Enghild JJ, Potempa J, Gomis-Ruth FX. A new autocatalytic activation mechanism for cysteine proteases revealed by Prevotella intermedia interpain A. J Biol Chem. 2008;283:2871–2882. doi: 10.1074/jbc.M708481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara T, Sarker MR, Ugai H, Akimoto S, Shaheduzzaman SM, Nakayama H, Miki T, Ohnishi Y. Physical and genetic map of the Bacteroides fragilis YCH46 chromosome. FEMS Microbiol Lett. 2002;207:193–197. doi: 10.1111/j.1574-6968.2002.tb11050.x. [DOI] [PubMed] [Google Scholar]
- Berti PJ, Storer AC. Alignment/phylogeny of the papain superfamily of cysteine proteases. J Mol Biol. 1995;246:273–283. doi: 10.1006/jmbi.1994.0083. [DOI] [PubMed] [Google Scholar]
- Kagawa TF, Cooney JC, Baker HM, McSweeney S, Liu M, Gubba S, Musser JM, Baker EN. Crystal structure of the zymogen form of the group A Streptococcus virulence factor SpeB: an integrin-binding cysteine protease. Proc Natl Acad Sci USA. 2000;97:2235–2240. doi: 10.1073/pnas.040549997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukomski S, Sreevatsan S, Amberg C, Reichardt W, Woischnik M, Podbielski A, Musser JM. Inactivation of Streptococcus pyogenes extracellular cysteine protease significantly decreases mouse lethality of serotype M3 and M49 strains. J Clin Invest. 1997;99:2574–2580. doi: 10.1172/JCI119445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PD, Toseland CP, Attwood TK, Flower DR. LIPPRED: A web server for accurate prediction of lipoprotein signal sequences and cleavage sites. Bioinformation. 2006;1:176–179. doi: 10.6026/97320630001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juncker AS, Willenbrock H, von Heijne G, Brunak S, Nielsen H, Krogh A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003;12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, Brinkman FS. PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics. 2005;21:617–623. doi: 10.1093/bioinformatics/bti057. [DOI] [PubMed] [Google Scholar]
- Seydel A, Gounon P, Pugsley AP. Testing the '+2 rule' for lipoprotein sorting in the Escherichia coli cell envelope with a new genetic selection. Mol Microbiol. 1999;34:810–821. doi: 10.1046/j.1365-2958.1999.01647.x. [DOI] [PubMed] [Google Scholar]
- Rzychon M, Sabat A, Kosowska K, Potempa J, Dubin A. Staphostatins: an expanding new group of proteinase inhibitors with a unique specificity for the regulation of staphopains, Staphylococcus spp. cysteine proteinases. Mol Microbiol. 2003;49:1051–1066. doi: 10.1046/j.1365-2958.2003.03613.x. [DOI] [PubMed] [Google Scholar]
- Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- Whittle G, Hamburger N, Shoemaker NB, Salyers AA. A bacteroides conjugative transposon, CTnERL, can transfer a portion of itself by conjugation without excising from the chromosome. J Bacteriol. 2006;188:1169–1174. doi: 10.1128/JB.188.3.1169-1174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M, Canchaya C, Bernini V, Altermann E, Barrangou R, McGrath S, Claesson MJ, Li Y, Leahy S, Walker CD. Comparative genomics and transcriptional analysis of prophages identified in the genomes of Lactobacillus gasseri, Lactobacillus salivarius, and Lactobacillus casei. Appl Environ Microbiol. 2006;72:3130–3146. doi: 10.1128/AEM.72.5.3130-3146.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salyers AA, Shoemaker NB, Stevens AM, Li LY. Conjugative transposons: an unusual and diverse set of integrated gene transfer elements. Microbiol Rev. 1995;59:579–590. doi: 10.1128/mr.59.4.579-590.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito M, Hirakawa H, Yamashita A, Ohara N, Shoji M, Yukitake H, Nakayama K, Toh H, Yoshimura F, Kuhara S. Determination of the genome sequence of Porphyromonas gingivalis strain ATCC 33277 and genomic comparison with strain W83 revealed extensive genome rearrangements in P. gingivalis. DNA Res. 2008;15:215–225. doi: 10.1093/dnares/dsn013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queralt N, Jofre J, Araujo R, Muniesa M. Homogeneity of the morphological groups of bacteriophages infecting Bacteroides fragilis strain HSP40 and strain RYC2056. Curr Microbiol. 2003;46:163–168. doi: 10.1007/s00284-002-3813-7. [DOI] [PubMed] [Google Scholar]
- Shaw LN, Golonka E, Szmyd G, Foster SJ, Travis J, Potempa J. Cytoplasmic control of premature activation of a secreted protease zymogen: deletion of staphostatin B (SspC) in Staphylococcus aureus 8325-4 yields a profound pleiotropic phenotype. J Bacteriol. 2005;187:1751–1762. doi: 10.1128/JB.187.5.1751-1762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou D, Kaneko J, Narita S, Kamio Y. Prophage, phiPV83-pro, carrying panton-valentine leukocidin genes, on the Staphylococcus aureus P83 chromosome: comparative analysis of the genome structures of phiPV83-pro, phiPVL, phi11, and other phages. Biosci Biotechnol Biochem. 2000;64:2631–2643. doi: 10.1271/bbb.64.2631. [DOI] [PubMed] [Google Scholar]
- Goshorn SC, Schlievert PM. Bacteriophage association of streptococcal pyrogenic exotoxin type C. J Bacteriol. 1989;171:3068–3073. doi: 10.1128/jb.171.6.3068-3073.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird W, Groman N. Prophage map of converting corynebacteriophage beta. J Virol. 1976;19:208–219. doi: 10.1128/jvi.19.1.208-219.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas V, Miyake J, Balsley H, Roark J, Telles S, Leeds S, Zurita I, Breitbart M, Bartlett D, Azam F, Rohwer F. Widespread occurrence of phage-encoded exotoxin genes in terrestrial and aquatic environments in Southern California. FEMS Microbiol Lett. 2006;261:141–149. doi: 10.1111/j.1574-6968.2006.00345.x. [DOI] [PubMed] [Google Scholar]
- Buckwold SL, Shoemaker NB, Sears CL, Franco AA. Identification and characterization of conjugative transposons CTn86 and CTn9343 in Bacteroides fragilis strains. Appl Environ Microbiol. 2007;73:53–63. doi: 10.1128/AEM.01669-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lampe B, Barthel B, Coupland SE, Riecken EO, Rosewicz S. Differential expression of matrix metalloproteinases and their tissue inhibitors in colon mucosa of patients with inflammatory bowel disease. Gut. 2000;47:63–73. doi: 10.1136/gut.47.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight-matrix choice. Nucl Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Garnier J, Gibrat JF, Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996;266:540–553. doi: 10.1016/s0076-6879(96)66034-0. full_text. [DOI] [PubMed] [Google Scholar]
- Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003;12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, Schaffer AA. Database indexing for production MegaBLAST searches. Bioinformatics. 2008;24:1757–1764. doi: 10.1093/bioinformatics/btn322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fristch EF, Maniatis T. A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 1989. Molecular cloning. [Google Scholar]
- Ausubel FM, Brent R, Kingston R, More D, Seidman J. Current protocols in molecular biology. supplement 27. J Wiley and Sons, New York; 1987. p. 241. [Google Scholar]
- Claesson MJ, O'Sullivan O, Wang Q, Nikkila J, Marchesi JR, Smidt H, de Vos WM, Ross RP, O'Toole PW. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One. 2009;4:e6669. doi: 10.1371/journal.pone.0006669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan O, Suhre K, Abergel C, Higgins DG, Notredame C. 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J Mol Biol. 2004;340:385–395. doi: 10.1016/j.jmb.2004.04.058. [DOI] [PubMed] [Google Scholar]
- Dubin G, Krajewski M, Popowicz G, Stec-Niemczyk J, Bochtler M, Potempa J, Dubin A, Holak TA. A novel class of cysteine protease inhibitors: solution structure of staphostatin A from Staphylococcus aureus. Biochemistry. 2003;42:13449–13456. doi: 10.1021/bi035310j. [DOI] [PubMed] [Google Scholar]
- Privitera G, Dublanchet A, Sebald M. Transfer of multiple antibiotic resistance between subspecies of Bacteroides fragilis. J Infect Dis. 1979;139:97–101. doi: 10.1093/infdis/139.1.97. [DOI] [PubMed] [Google Scholar]
- Elhag KM, Bettelheim KA, Tabaqchali S. Serological studies of Bacteroides fragilis. J Hyg (Lond) 1977;79:233–241. doi: 10.1017/s0022172400053043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala J, Quesada A, Vadillo S, Criado J, Piriz S. Penicillin-binding proteins of Bacteroides fragilis and their role in the resistance to imipenem of clinical isolates. J Med Microbiol. 2005;54:1055–1064. doi: 10.1099/jmm.0.45930-0. [DOI] [PubMed] [Google Scholar]
- Macy JM, Ljungdahl LG, Gottschalk G. Pathway of succinate and propionate formation in Bacteroides fragilis. J Bacteriol. 1978;134:84–91. doi: 10.1128/jb.134.1.84-91.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida FS, Nakano V, Avila-Campos MJ. Occurrence of enterotoxigenic and nonenterotoxigenic Bacteroides fragilis in calves and evaluation of their antimicrobial susceptibility. FEMS Microbiol Lett. 2007;272:15–21. doi: 10.1111/j.1574-6968.2007.00732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudder P, Uemura K, Dolby J, Fukuda MN, Feizi T. Isolation and characterization of an endo-beta-galactosidase from Bacteroides fragilis. Biochem J. 1983;213:485–494. doi: 10.1042/bj2130485. [DOI] [PMC free article] [PubMed] [Google Scholar]