

Abstract
Cancer stem cells (CSCs) are characterized by their self-renewing potential, and by their ability to differentiate and phenocopy the original tumor in orthotopic xenografts. Long term propagation of glioblastoma (GBM) cells in serum containing medium results in loss of the CSCs and outgrowth of cells genetically and biologically divergent from the parental tumors. In contrast, the use of neurosphere assay, a serum-free culture for selection and propagation of CNS-derived stem cells, allows the selection of a subpopulation containing CSCs. Gliosarcoma (GS), a morphological variant comprising approximately 2% of GBMs, present a biphasic growth pattern, composed of glial and metaplastic mesenchymal components. To assess whether the neurosphere assay would allow the amplification of a subpopulation of cells with “gliosarcoma stem cell” properties, capable of propagating both components of this malignancy, we have generated neurospheres and serum cultures from primary GS and GBM surgical specimens. Neurosphere cultures from GBM and GS samples expressed neural stem cell markers Sox2, Msi1 and Nestin. In contrast to the GBM neurosphere lines, the GS neurospheres were negative for the stem cell marker CD133. All neurosphere lines generated high grade invasive orthotopic tumor xenografts, with histological features strickingly similar to the parental tumors, demonstrating that these cultures indeed are enriched in CSCs. Remarkably, low passage GS serum culture retained the expression of stem cell markers, the ability to form neurospheres, and tumorigenicity. The GS experimental tumors phenocopied the parental tumor, exhibiting biphasic glial and mesenchymal components, constituting a clinically relevant model to investigate mesenchymal differentiation in GBMs.
Keywords: gliosarcoma, glioblastoma, neurospheres, cancer stem cells, mesenchymal differentiation
Introduction
Glioblastoma (GBM), classified as grade IV astrocytoma by the WHO, is the most aggressive and frequent type of glial neoplasia. Glioblastomas are heterogeneous tumors and are refractive to therapy. Recent global gene expression analysis defined a subtype of glioblastoma associated with a poor prognosis characterized by aberrant expression of mesenchymal genes 1, 2. Another example of a prominent mesenchymal phenotype in a central nervous system tumor is found in a morphological variant of GBM, termed gliosarcoma (GS), comprising approximately 2% of GBMs 3. GS presents a biphasic growth pattern, composed of glial and sarcomatous components, typically identified by GFAP immunohistochemical labeling and reticulin staining of the dense extracellular matrix, respectively 4. Gliosarcoma occurs de novo, as a primary tumor, or develops after radiotherapy of glioblastomas 5, 6. Gliosarcoma is indistinguishable from primary glioblastoma in relation to median patient age, cerebral hemisphere location, standard treatment, and clinical outcomes 5, 7–9. The mesenchymal component of GSs can present differentiation along fibroblastic, osteogenic, chondrogenic, adipogenic, smooth and skeletal muscle lineages, according to morphological appearance and expression of lineage specific markers 4. The sarcomatous component of gliosarcomas show signs of malignant transformation, such as nuclear atypia, mitotic activity and necrosis 4. Importantly, identical genetic abnormalities have been identified in both glial and mesenchymal components, suggesting the occurrence of a metaplastic mesenchymal differentiation from a common origin, during the progression of the astrocytic tumor 7, 10–13. The molecular mechanisms directing the mesenchymal differentiation in glioblastomas, whether in the mesenchymal subgroup of GBMs or in the biphasic GS subtype, and the significance of this plasticity for tumor progression and growth are poorly understood.
The remarkable heterogeneity of GBMs is also observed in the range of differentiation states of the neoplastic cells. Along with other solid and hematopoietic malignancies, it has been hypothesized that a subpopulation of GBM tumor cells present cancer stem cell (CSC) properties. These properties include the ability to self-renew and differentiate into cells composing the bulk of the tumor, and are thus implicated in post-treatment recurrence 14. Long term propagation of glioma cells in serum-containing medium results in loss of the CSC population and outgrowth of cells genetically and biologically divergent from their parental tumors 15. In contrast, culturing dissociated glioblastoma surgical specimens in a serum-free culture medium developed for propagation of neural stem cell (NSC) as floating multicellular spheroids, known as neurospheres 16, allows for the selection and expantion of a subpopulation of cells enriched in CSCs carrying patient-specific molecular fingerprints 15, 17–19. These CSC-enriched subpopulations are characterized by long term self-renewal in vitro, by the expression of NSC markers, such as Sox2, Nestin and CD133, and by the ability to differentiate into cells that form the bulk of the tumor, recapitulating the parental tumor in orthotopic xenografts 17–19. Long term culture in serum containing medium leads to differentiation of CSC accompanied by loss of NSC markers such as Sox2, Cd133 and Nestin 15. On the other hand, the recovery of cells with CSC properties from long term serum cultured human 20 and rat 21 GBM cell lines, as well as a rat gliosarcoma cell line 22, has been reported. Thus, neurosphere cultures from GBM surgical specimens as a stable source of cells carrying patient-specific molecular fingerprints have become an asset for pre-clinical studies. Here we investigate how culture conditions for GBM and GS tumor cells affect the potential for recapitulating the mesenchymal differentiation of the founder tumor in orthotopic xenografts. Given the proper stimulus, adult mouse neural stem cells differentiate along mesenchymal lines in vitro and in vivo 23. Likewise, modulation of mesenchymal differentiation of GBM stem cells by the microenvironment would be expected. A recent study shows that GBM neurospheres underwent chondroblastic and osteoblastic differentiation in subcutaneous tumor xenografts, but not in orthotopic xenografts 24. Here we show mesenchymal differentiation in immunocompromised rat brain by cultured GS cells, recapitulating the biphasic parental tumor phenotype. Furthermore, early passage GS1 serum cultured cells retained the expression of NSC markers, the ability to form neurospheres when transferred to serum free medium, and recapitulated the parental tumor in rats, in contrast to the GBM serum monolayers.
Materials and Methods
Primary tumor cell culture
Resected brain tumors were collected at Henry Ford Hospital (Detroit, MI) with written consent from patients in accordance with institutional guidelines, and graded pathologically according to the WHO criteria. Tumors were processed immediately after surgery, essentially as described 17, 19. Briefly, tumors were cut to 1 mm3 pieces, washed 3 times in DMEM/F-12 serum free medium (Invitrogen Life Technologies, Carlsbad, CA) and dissociated in cell dissociation buffer (Cognate Bioservices, Baltimore, MD). Digestion was stopped with 1 volume of DMEM/F-12 containing 0.7 mg/ml ovomucoid (Sigma-Aldrich, St. Louis, MO) and the tissue was triturated mechanically with fire narrowed Pasteur pipettes and filtrated through a 40 μm cell strainer (Becton Dickinson, Franklin Lakes, NJ). Red blood cells were removed by density centrifugation in Lympholyte-M (Cedarlane Laboratories, Burlington, NC). Nucleated cells were washed several times in DMEM/F-12. For regular monolayer cultures, cells were cultured in DMEM supplemented with 10% FBS (HyClone Laboratories, Logan, UT), 50 units/ml penicillin G sodium, 50 μg/ml streptomycin sulfate. For neurosphere cultures, cells were plated in T-25 flasks at a density of 2 × 104 cells/ml in neurosphere medium (NM): DMEM/F12 medium containing 2 mM L-glutamine and supplemented with N-2 (Gibco Invitrogen Cell Culture, Grand Island, NY), 0.05% BSA, 25 μg/ml gentamicin, 50 units/ml penicillin G sodium, 50 μg/ml streptomycin sulfate, 20 ng/ml EGF and 20 ng/ml bFGF (PeproTech, Rocky Hills, NJ). After 1 to 3 weeks, multicellular floating neurospheres formed and were dissociated in Mg2+-Ca2+-free PBS and replated in NM medium for the formation of secondary spheroids. The glioblastoma neurospheres used for this work were all below passage 10.
Neurosphere Assay for differentiated glioblastoma cells and 3D cell cultures
Neurosphere assay for differentiated GBM or GS monolayer cultures: dissociated tumors were first cultured in 10% FBS DMEM and passaged upon confluency. After 3 to 20 passages, as indicated, monolayer cells were harvested by Trypsin (Invitrogen) treatment, washed several times in serum-free DMEM/F-12 medium and plated at a density of 2 × 104 cells/ml in neurosphere medium (NM) in regular tissue cultured treated flasks. Neurosphere formation was monitored for up to 4 weeks.
To evaluate the differentiation potential, GS1 neurospheres were incubated in NM medium without EGF and bFGF for one week in a regular tissue culture treated flask. In parallel, neurospheres were dissociated and plated in 10% FBS DMEM in ultra-low attachment plates (Corning., Corning, NY), to preserve the 3D architecture, and incubated for 1 week under standard conditions. After one week in regular NM, NM without growth factors or serum medium, multicellular spheroids were harvested, fixed in formalin for 10 minutes and paraffin embedded for immunohistochemistry (IHC).
Experimental orthotopic tumor
Following IACUC guidelines in an institutionally approved animal use protocol, dissociated GBM neurosphere or serum cultured cells were inoculated intracranially in nude rats (RNU/RNU) Animals were anesthetized and immobilized in a small animal stereotactic device (Kopf, Cayunga, CA)., A Hamilton syringe containing the used to inject between 4× 104 to 4 × 105 cells, as indicated, through a hole 3 mm to the right of the bregma, at a depth of 2.5 mm, at a rate of 0.5 μL/10 sec. The surgical zone was flushed with sterile saline and the hole sealed with bone wax and the skin over the injection site sutured. Animals were monitored daily and sacrificed between 9 and 14 weeks post implantation.
Histology and Immunohistochemistry
Sections of formalin fixed, paraffin embedded human glioma surgical samples, tumor xenografts, or multicellular spheroids were deparaffinized with xylene and rehydrated through graded alcohol into in phosphate buffered saline. Antigens were unmasked by 10 min incubation in boiling in citrate buffer. Primary antibodies used were: mouse anti-human Nestin (Chemicon International/Millipore, Billerica, MA), mouse anti-human GFAP (Biocare Medical, Concord, CA), CD133 (Miltenyi, Auburn CA), mouse anti-human Vimentin (clone CM048) (Biocare Medical), mouse anti-human smooth muscle actin (α-SMA) clone HHF35 (DakoCytomation, Carpinteria, CA). After primary antibody incubation, slides were washed several times with PBS and incubated with appropriate biotinylated secondary antibody (Vector Laboratories, Burlingame, CA). After washes in PBS, sections are treated with streptavidin-peroxidase complex and incubated in diaminobenzidine tetrachloride (DAB), AEC+, or blue substrate-chromogen solutions (Dako Cytomation). Sections were counterstained with Mayer’s hematoxylin or nuclear fast red and mounted using Vectamount (Vector Laboratories). For antigen specificity control, pre-immune serum replaced the primary antibody. Images of the labeled sections were captured with an Olympus IX50 microscope, equipped with an SPOT Insight 4 camera. Reticulin was stained with an automated reticulin stain kit (Dako Cytomation). Images of the labeled sections were captured with an Olympus IX50 microscope, equipped with an SPOT Insight 4 camera.
RNA preparation, RT-PCR and quantitative real-time PCR analysis
Total RNA was isolated from cultured cells using RNeasy Miniprep kit (Qiagen, Valencia, CA). cDNA was prepared from 1 μg DNAseI-treated RNA using Superscript III and oligo dT (Invitrogen Life Technologies). Relative quantification of gene expression was performed by real time PCR with SybrGreen and ABIPrism 7000 sequence detection system (Applied Biosystems, Foster City, CA), according to the manufacturer instructions. PCR conditions were: 10 min at 95°C followed by 40 cycles of 15s at 95 °C and 1 min at 60 °C. Dissociation curves and agarose gel electrophoresis were used to verify primer specificity. β-actin was used as internal reference and relative mRNA levels were quantified by the 2(−ΔΔCt) method 25. Primer sets used were: CD105 sense 5′-ACAAGTCTTGCAGAAACAGTCC-3′, antisense 5′-GACCTGGCTAGTGGTATATGTCA; Sox2 sense 5′-TGGACAGTTACGCGCACAT-3′, anti-sense 5′-CGAGTAGGACATGCTGTAGGT3′; Msi1 sense 5′-TTCGGGTTTGTCACGTTTGAG-3′, anti-sense 5′-GGCCTGTATAACTCCGGCTG-3′; nestin sense 5′-ATCGCTCAGGTCCTGGAAGG-3′, anti-sense 5′-AAGCTGAGGGAAGTCTTGGAG-3′; β-actin sense 5′-CCGACAGGATGCAGAAGGAG-3′, anti-sense 5′-CATCTGCTGGAAGGTGGACA-3′.
Protein sample preparation and Western blot analysis
Tumor tissue: fresh tumor samples were snap frozen in liquid nitrogen. Frozen tissue was minced before being transferred to lysis buffer. The GS1 patient presented local progressive disease 88 days after the first surgery, with a pathology essentially identical to the primary tumor. Tissue from this recurrence (GS1R) was used for the Western Blot analysis, due to limited availability of the primary tumor.
Cell lysates: cell monolayers will be washed 3 times with phosphate-buffered saline and lysed with cell lysis buffer (150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.5% Triton X-100, 0.5 mM EDTA, 0.1% SDS, 50 mM Tris-HCl, pH 7.6), containing protease and phosphatase inhibitor cocktails (Calbiochem, San Diego, CA) supplemented with 17.4 μg/ml PMSF (Sigma-Aldrich). Proteins in the lysates were quantified by a modified Bradford Protein Assay (Bio-Rad Laboratories, Hercules, CA), denatured in SDS-gel loading buffer and frozen at −80°C.
Immunoblot Analysis: Thirty micrograms of total protein from cell lysates were separated by SDS-PAGE. Proteins were electrophoretically transferred to nitrocellulose membranes (Invitrogen Life Technologies) and probed with the following antibodies: Sox2 (Chemicon), anti-human specific Nestin (Chemicon), anti-human specific GFAP (Biocare Medical), CD133 (Cell Signalling), β-actin (Sigma), followed by secondary peroxidase conjugated antibodies (Jackson ImmunoResearch Laboratories). Protein bands were visualized using SuperSignal West Pico Chemoluminescent Substrate (Pierce, Rockford, IL).
In vivo MRI imaging studies
Animals underwent in vivo magnetic resonance imaging (MRI) at different time points after implantation of either cells grown as neurospheres or cells grown in monolayer. MRI was obtained with a 3 Tesla clinical system (Signa Excite, GE health) using 50 mm diameter × 108 mm RF rung length small animal imaging coil (Litzcage small animal imaging system, Doty Scientific Inc, Columbia, SC). Animals were anesthetized, positioned using a triplanar FLASH sequence, and MR studies were performed using three dimensional (3D) FIESTA sequence before and after gadolinium based contrast enhancement. The following parameters were used to acquire MRI: 3D FIESTA-c MR images were obtained with TR=12 msec, TE=2.8 msec, 40° flip angle, 60×50 FOV, 320×256 matrix, 32–40 slices with a slice thickness of 0.5 mm and NEX = 4. To delineate the margin of the tumor properly, gadolinium contrast agent was injected intravenously and post-contrast 3D FIESTA images were also acquired using above mentioned image parameters.
Molecular characterization
TP53 mutations. cDNA was obtained from total RNA extracted from tumor samples, neurosphere cultures and xenograft tumor, as described above. The p53 region corresponding to nucleotides 678–1176 (exons 5–8) of the human TP53 mRNA (NM_000546) was amplified by PCR and sequenced (DNA Sequencing Facility, Wayne State University, Detroit MI), using DNA oligo sequences for amplification and sequencing described elsewhere 26.
Loss of heterozygosity. Allelic deletions in the PTEN region of chromosome 10 were detected using the microsatellite markers D10S215, D10S574, D10S187 and D10S217, essentially as described 27.
Results
Plasticity of gliosarcoma serum cultured cells
Serum cultured glioma cells undergo a phenotypic alteration over time termed “mesenchymal drift”, the tendency to lose glial and acquire mesenchymal characteristics 28, 29. Culturing GBM surgical specimens in serum-free neurosphere medium (NM), on the other hand, allows the amplification of a cell subpopulation with neural stem/progenitor cell characteristics, namely long-term self-renewal ability, and the potential differentiate and to recapitulate the original tumor in orthotopic xenografts 15. Because the GBM variant gliosarcoma presents a metaplastic mesenchymal differentiation morphologically distinct from the astrocytic component, we tested how culture conditions would affect the amplification of cells with gliosarcoma stem cell characteristics, capable of recapitulating the biphasic nature of the tumor in vivo. Surgical GBM and GS tumor specimens were cultured immediately after surgical resection in NM supplemented with EGF and bFGF, or in medium supplemented with 10% FBS. First, we tested the recovery of cells with progenitor/stem cell characteristics from early passage serum cultures derived from 12 primary GBM and one GS surgical specimens, using the neurosphere assay and monitoring neurosphere formation for several weeks (as detailed in Materials and Methods). Whether a cell subpopulation capable of dedifferentiating to a progenitor/stem cell phenotype in vitro can be recovered from serum cultured human glioblastoma cells is an unsettled matter 15, 20. Passage 3 gliosarcoma (GS1) cells readily formed self-adherent multicellular spheroids when transferred from 10% FBS growth medium into serum-free NM (“GS1 P3”, Fig. 1A). The property of yielding neurospheres in vitro was lost after 15 passages (“GS1 P16”, Fig. 1A). None of the serum cultured GBM samples tested exhibited the neural stem cell growth phenotype in the neurosphere assay, even when kept in NM for 4 weeks, as shown for GBM2 (Fig. 1A), in agreement with previous reports 15. The GS1 neurospheres were dissociated and re-plated for de novo neurosphere formation and passaged for over 18 months (not shown), an indication of long term self-renewal potential. Because the property of dedifferentiating into long term self-renewing neurospheres was exclusive of low passage GS1-FBS, the neural stem cell markers expression in this population was compared against the higher GS1 – FBS passages and low GBM2-FBS-P3 passage, which did not dedifferentiate in vitro (Fig. 1A). GBM2 and GBM3 neurospheres were obtained from the dissociated tumor biopsies. All tumor neurospheres lines in this study were below passage 10, although self-renewal, tumorigenicity and NSC gene expression in higher passages cells maintained in NM remained stable (not shown), in agreement with previous work [15]. The NSC markers Sox2, Musashi1 and Nestin mRNA levels, determined by quantitative RT-PCR (qRTPCR), were downregulated in GBM2-FBS P3 cells in comparison to GBM2 neurospheres (Figure 1B). In contrast, the mesenchymal marker endoglin (CD105), expressed in a subgroup of GBMs30, is upregulated in GBM2 FBS – P3 in relation to the neurospheres (Fig. 1B). These results are in agreement with previously reported loss of stem cell marker expression in GBM cells cultured in the presence of serum 15. Conversely, GS1 FBS-P3 retained the expression of Sox2, Msi1 and Nestin (Fig. 1B), suggesting that the ability to grow as neuroprogenitor cells in NM described above (Fig 1A) is accompanied by the maintenance of the expression of neural stem cell markers, typically downregulated differentiated serum cultured GBM cells. Accordingly, the decreased developmental plasticity observed in higher passages serum cultured GS1 cells (Fig. 1A) was accompanied by transcriptional downregulation of the NSC markers Sox2, Musashi-1 (Msi1) and Nestin, while the mesenchymal marker CD105 was upregulated (P16 and P20, Fig. 1B).
Figure 1.

Neural stem cell properties of serum GS and GBM cultures. A) Neurosphere assay: low (P3) or higher (P16) passages GS1 monolayer cells cultured in 10% FBS DMEM medium (“day 0”), were harvested and seeded in serum-free NM. Neurosphere formation was monitored for 4 weeks. Phase contrast images shown for day 1 and day 14 are representative of 3 independent experiments. GBM2 P3 monolayer cells did not form neurospheres and this result is representative of observations made for 12 GBM samples tested.. B) Neural stem (Sox2, Msi1 and Nestin) and mesenchymal (CD105) cell markers expression in GBM2 and GS1 serum cultures of the indicated passage were compared by qRT-PCR. Values are expressed as fold increase in relation to mRNA levels in the corresponding neurosphere cultures. Results represent mean ± SE, for n=3 samples.
Sox2 protein was expressed in the GBM2 and GBM3 tumor samples and neurosphere cultures, and was not detected in the differentiated serum cultured monolayer cells (Fig. 2A). Conversely, Sox2 protein expression was observed in GS1 FBS-P3 cells, but downregulated in GS1 FBS-P20 (Fig. 2A), in agreement with the results above (Fig. 1B). The neural and hematopoietic stem/progenitor cell marker CD133, has been used to sort a subpopulation of GBM cells with CSC properties 19. Recently, it has been shown that CD133 expression is not a required property defining GBM CSC 31. CD133 was expressed in the tumor specimens and neurosphere cultures from GBM2 and GBM3, while no expression was observed for the differentiated cells (Fig. 2A). No CD133 expression was detected for GS1 neurosphere or serum cultures (Fig 2A). It has been reported that CD133 negative glioblastoma cells can give rise to CD133 positive cells in orthotopic xenografts 32. The orthotopic xenografts generated by the CD133 negative GS1 neurospheres did not contain CD133 positive cells (Fig. 2B). In contrast, CD133 positive GBM3 neurospheres gave rise to xenografts containing CD133 positive cells (Fig. 2B). Thus, CD133 expression in the tumor xenografts correlated with the CD133 status in the patient biopsy.
Figure 2.
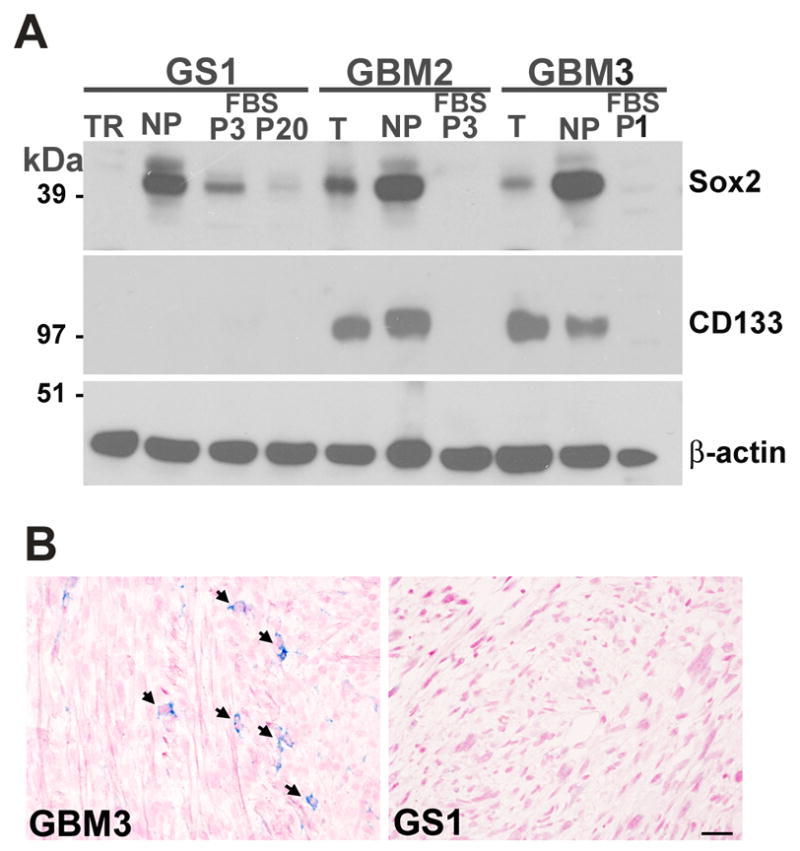
Sox2 and CD133 protein expression in GS1 and GBM samples. A) Sox2 and CD133 protein expression in tumors (T), derived neurospheres (NP) and serum cultures (FBS) was assessed by immunoblot; β-actin was used as loading control. B) CD133 expression (blue) in GBM3 and GS1 xenografts was analyzed by IHC of coronal brain sections; nuclei was counterstained with nuclear fast red. Scale, 20 μm.
Glioblastoma and gliosarcoma neurospheres form high grade tumors in rats
A defining property of cancer stem cells (CSCs) is the ability to phenocopy the original tumor in orthotopic xenografts. We tested whether the in vivo tumorigenic potential of the GS1 neurospheres, obtained from dedifferentiation of passage 3 monolayer cells (Fig. 1A) would be comparable to the GBM2 and GBM3 neurospheres lines, derived directly from dissociated tumor biopsies. Neurosphere cells were dissociated and implanted into the brains of immunodeficient rats. Tumors were established in 100% of animals implanted with the three neurosphere lines (Table 1). Animals were sacrificed 14 weeks post implantation and coronal brain sections were stained with eosin and hematoxylin. All three neurosphere lines initiated high grade invasive tumors (Fig. 3). Histological comparison with corresponding founder tumor biopsies revealed a remarkable similarity regarding cellular density and morphology (Fig. 3A–B, D–E, G–H). Pseudopalisading necrosis was observed in the GS1 patient biopsy (Fig. 3A) and xenografts (Fig. 3B). Clinical information is shown in Fig. 3J. Genetic abnormalities frequently associated with GBM, such as TP53 mutations and chromosome 10q losses were evaluated in the parental tumors, tumor neurospheres and derived xenografts. LOH on chromosome 10q was observed for all parental tumors, neurospheres and xenografts (Fig. 3J). Phosphatase and tensin homologue (PTEN) protein was not detected in any of the glioblastoma or gliosarcoma neurosphere lines (not shown). TP53 mutational status in the tumor neurospheres and derived xenografts was identical to the founder tumor (Fig. 3J). These results confirm the neoplastic origin of the neurosphere cultures and the preservation of the genetic abnormalities.
Table 1.
Percentage of rats developing tumors three months after intracranial implant
| Neurospheres | Serum monolayers | |||
|---|---|---|---|---|
| 2 – 4 × 105 cells | 4 × 104 cells | 4 × 105 cells | 4 × 104 cells | |
| GS1 | 100% (11/11) | 100% (4/4) | 100% (8/8) | 100% (4/4) |
| GBM2 | 100% (4/4) | 100% (4/4) | 0% (0/4) | ND |
| GBM3 | 100% (3/3) | 100% (6/6) | ND * | ND * |
Dissociated tumor cells did not grow well in serum containing medium after passage 2
Figure 3.

Orthotopic xenografts generated by GS1 and GBM neurospheres phenocopy the parental tumors. Tumor biopsy samples and rat brains implanted with the respective neurosphere cultures were formalin fixed and paraffin embedded, sectioned and stained with H&E. Representative images are shown for GS1 biopsy (A) and corresponding xenograft (B,C), where arrows point to pseudopalisading necrosis. Representative images are also shown for primary GBM samples GBM2 (D), and corresponding xenografts (E,F), and GBM3 (G) and corresponding xenografts (H,I). “T” indicates tumor in the 10× magnification images (C,F,I). Scale, 80 μm (A,B,D,E,G,H). J) Patient information and molecular analysis of the tumor biopsies.
GS1 neurospheres undergo astrocytic differentiation in vitro
Neural stem cells cultured in medium supplemented with FBS, or upon withdrawal of growth factors from the neurosphere medium, differentiate into GFAP expressing astrocytes 16, 33. To characterize the GS1 neurosphere differentiation potential in vitro, neurospheres with 30 to 50 μm diameter were divided into three groups in quadruplicates and cultured for 1 week in NM, in NM without growth factors, or in 10% FBS. Serum cultured cells were maintained in ultra-low attachment plates to preserve the 3D architecture, which is known to affect cell signaling and gene expression 34. Spheroids were collected and the expression of markers for cell proliferation (Ki-67), stem/progenitor cells (Nestin), and astrocytes (GFAP), were analyzed by immunocytochemistry (Fig. 4A). Ki-67 and GFAP positive cells were counted in four 400 × images, and results are expressed as percentage of total cells (Fig 4A). GS1 neurospheres cultured in NM medium were highly proliferative, exhibited widespread expression of nestin, and no expression of the astrocytic marker GFAP (Fig. 4A). The percentage of GFAP expressing cells significantly increased and the percentage of proliferating cells decreased upon growth factor withdrawal or in the presence of serum (fig. 4A). The intermediate filament protein Nestin is expressed in neural stem and precursor cells 35, as well as in a subpopulation of mesenchymal stem cells 36. Widespread Nestin expression was observed in GS1 neurospheres, and expression was retained upon growth factor withdrawal, or in the serum cultured spheroids, albeit at a lower intensity (Fig. 4A). In vivo expression pattern of Nestin, along with the cell proliferation marker Ki-67, was assessed by immunohistochemistry in xenograft tumors 9 weeks after GS1 neurosphere implantation, using human specific antibodies. Ubiquitous expression of Nestin and Ki-67 was observed in the tumor core and in invasive cells (Fig. 4B). Few cells positive for α-SMA and sporadic reticulin staining were observed in the GS1 neurospheres cultured for 1 week under the same differentiation conditions (Supplementary Fig. 1), suggesting in vitro mesenchymal differentiation potential. Vimentin expression was observed for neurospheres growing in NM and under differentiation conditions (Supplementary Fig. 1).
Figure 4.

Differentiation of GS1 neurospheres in vitro and ubiquitous expression of nestin in vitro and in vivo. A) GS1 neurospheres cultured in neurosphere medium (NM), for 7 days in the absence of growth factors (NM w/o GF), or for 7 days in 10% FBS medium (FBS) were collected fixed and embedded in paraffin. Representative images of 6 μm thick sections stained for the astrocytic marker GFAP, the proliferation marker Ki-67 and Nestin are shown. Nuclei were counterstained with hematoxylin. Ki-67 and GFAP positive cells were counted in four 400 × images and results are expressed as mean percentage of positive cells + SE. B) Representative image showing Nestin and Ki-67 expression in the GS1 tumor xenograft core (C) and invasive (I) cells, assessed by immunohistochemistry of coronal sections 9 weeks post implant of GS1 neurospheres (Magnification, 10× and 200×).
Serum cultured gliosarcoma cells are tumorigenic
It has been shown that patient derived serum-cultured glioblastoma cells are not tumorigenic at early passages 15. We compared the tumorigenic potential of neurosphere and serum monolayer cells to assess if the expression of NSC markers in the low passage GS1 serum cultures, along with the ability to form neurospheres when transferred to NM (Fig. 1A), correlates with tumorigenicity in vivo. The take rate for nude rats implanted with 2 – 4 × 105 GS1, GBM2 and GBM3 neurosphere cells was 100% (Table 1). The take rate remained 100% when the number of neurosphere cells implanted was reduced to 4 × 104 (Table 1). Low passage GBM2 serum cultured cells were not tumorigenic (Table 1), as expected from previous reports 15. In contrast, 100% take rate was observed for GS1 early passage serum cultured cells, when 4 × 105 or 4 × 104 cells were implanted (Table 1). Histological comparison of representative xenografts derived from 4 × 105 early passage serum (FBS P3) or neurosphere (NP) GS1 cells are shown in Figure 5A. At 9 weeks post implantation, the neurosphere tumors are more disperse diffusely along the white matter tract, followed by rapid growth resulting in a high grade tumor with necrotic areas at 11 weeks (Fig. 5A). By comparison, the FBS P3 tumors are larger and more circumscribed at 9 weeks, but also grow into a large necrotic tumor (Fig. 5A). Lee et al has observed that GBM cells cultured in serum undergo genomic rearrangements after passage 10, regaining the ability to form tumor xenografts, although such tumors are genetically and morphologically dissimilar to the original tumor, in contrast to xenografts generated from neurospheres15. Rats implanted with passage 20 serum cultured GS1 cells also developed tumors (Fig. 5A). To evaluate the GS1 CSCs long term self-renewal in vivo, we compared the tumorigenicity of neurospheres derived from excised xenograft tumors (GS1-XP1) with the original GS1 neurospheres cells. Nude rats were implanted with 4 × 104 GS1-XP1 or GS1 cells, 4 animals each, and sacrificed after 11 weeks. All animals developed tumors which were analyzed by histology, and found to be remarkably similar between the two groups (Fig. 5B). Tumor growth monitoring by MRI showed that rats implanted with GS1 FBS-P3 cells developed a detectable tumors earlier, while a longer lag time followed by a rapid growth was observed for NP tumors (Fig. 5C), consistent with the histological observations (Fig. 5A).
Figure 5.

Orthotopic tumor growth and morphology in nude rats implanted with GS1 cells. A) Rats implanted with equal number of GS1 cells from neurosphere cultures (NP), low passage serum (FBS P3) and higher passage serum (FBS P20) were sacrificed at 9 and 11 weeks, and tumor (T) was monitored by histology. B) Similar tumors were observed when 4 × 104 NP cells derived from surgical specimens (GS1) or from a GS1 tumor xenograft (GS1-XP1) were implanted in nude rats, demonstrating long term self-renewal of the GS1 CSCs by serial transplantation. C) MRI images were obtained at the indicated time points for a rat implanted with GS1 neurospheres (NP) and a rat implanted with low passage serum cultured GS1 cells (FBS P3). Axial plane images are shown, arrow head indicates the injection site for the 7 weeks NP rat and arrows point to the tumor in the other images.
Gliosarcoma phenotype in xenografts generated by neurospheres and low passage serum cultures
To probe if the GS1 cultured cells would retain their gliosarcoma multi-lineage differentiation potential in vivo, we compared the expression of glial and mesenchymal components of xenografts derived from neurosphere and serum GS1 cultured cells with the parental tumor by reticulin staining and IHC. Representative images of at least 3 samples per group for the GS1 biopsy (Fig. 6A–E), GS1 NP xenografts (Fig. 6F–G), GS1 FBS-P3 xenografts (Fig. 6H–L), GS1 FBS-P20 xenografts (Fig. 6P–T), and GBM3 NP xenografts (Fig. 6U–Y), are shown. The astrocytic and the sarcomatous components of a gliosarcoma can be geographically distinct or intermingled, and are typically identified by GFAP and reticulin staining, respectively 4. Both the sarcomatous component and GFAP positive cells observed in the GS1 biopsy (Fig. 6A,B) were present in the GS1 NP xenografts (Fig. 6F,G). The xenografts derived from GS1 FBS P3 cells presented extensive areas positive for reticulin staining and GFAP expression pattern similar to the patient biopsy (Fig. 6K,L). The GS1 parental tumor and all GS1 xenografts were positive for the intermediate filament Vimentin, found in both mesenchymal and neural progenitor cells 33 (Fig. 6D, I N,S). Nestin was widely expressed in the GS1 NP xenografts (Fig. 6H and Fig. 4B), and expressed in discrete regions of the GS1 FBS P3 xenografts (Fig. 6M), in a pattern similar to the GS1 patient biopsy (Fig. 6C). Reticulin and GFAP weakly positive regions were still observed in the GS1 FBS P20 xenografts (Fig. 6P,Q). Vimentin expression was maintained in GS1FBS P20 tumors (Fig. 6S,T), while nestin was down regulated in the FBS P20 xenografts (Fig. 6R), in agreement with the downregulation of the nestin transcript in vitro in higher passages (Fig. 1B). Glioblastomas frequently present gain of additional mesenchymal lineage markers. To further characterize the sarcomatous component of GS1 biopsy and xenografts, muscle, epithelial and neuroectodermal markers were analyzed by immunohistochemistry. Expression of α-SMA was observed in the patient biopsy and in discrete regions of all GS1 xenografts (Fig. 6E,J,O,T). Smooth muscle differentiation in gliosarcoma has been previously reported 5, 37. The GS1 specimen and xenografts were negative for the epithelial markers epithelial membrane antigen (EMA) and cytokeratin, and the neuroectodermal marker synaptophysin (not shown). The GBM3 xenograft was positive for GFAP and nestin (Fig. 6V,W), negative for the mesenchymal markers reticulin (Fig. 6U) and α-SMA (Fig. 6Y), and weakly positive for Vimentin (Fig. 6X). The lack of reticulin staining and α-SMA positive neoplastic cells shown for GBM3 is representative of observations for xenografts derived from 4 other primary GBM neurosphere lines, while variable levels of Vimentin expression was observed for all GBM xenografts and biopsies tested (not sown). These results demonstrate that GS cells cultured in neurosphere medium can recapitulate not only the astrocytic component of the tumor, but also the mesenchymal differentiation in vivo. The differentiated low passage GS1 serum cultured cells also retained the expression of neural stem cell markers and phenocopied the parental tumor in rats.
Figure 6.

Gliosarcoma phenotype is maintained in the GS1 xenografts. Expression of neural (GFAP and Nestin), mesenchymal (reticulin and α-SMA) and a marker for both lineages (Vimentin) in founder GS1 tumor (A-E), and in xenografts generated by neurosphere (NP, F-J), low passage (FBS P3, K-O), and high passage (FBS P20, P-T) serum cultured cells. Reticulin staining (r) was observed for all GS1 samples (A,F,K,P). GBM3 xenograft was included as a control (U-Y), and did not stain for the mesenchymal markers (U,Y). Arrows in E,J, O and T point to individual GS1 cells expressing α-SMA. Bar, 20 μm (C-E, H-J, M-O, R-T, W-Y) and 40 μm (A,B,F,G,K,L,P,Q,U,V).
Discussion
Cell culture medium containing fetal bovine serum (FBS) causes irreversible differentiation of neural stem cells (NSCs) 16. Likewise, it has been shown that serum cultured primary glioma cells undergo changes that cannot be subsequently reversed following transition to neurosphere medium, for example loss of NSC markers Sox2, Cd133 and Nestin 15. On the other hand, the recovery of cells with CSC properties from long term serum cultured human GBM cell lines 20 and rat gliosarcoma 9L cells has been reported 22. Here we report that low passage GS1 serum cultured cells retained the expression of NSC markers, the neural stem/progenitor cell property of growing as floating multicellular spheroids in NM, and the ability to recapitulate the parental tumor in experimental models. The latter are attributes defining CSCs. In contrast, no CSCs were recovered from the low passage GBM serum cultures tested. It has been suggested that in vitro neurosphere formation correlates with clinical outcome for gliomas 38. The tumor neurospheres used for this study were obtained from patients that presented a wide range of progression-free survival, 88, 196 and 648 days for GS1, GBM2 and GBM3, respectively (Fig. 3J). Whether the poor clinical outcome for the GS1 case is associated with the increased plasticity observed in low passage serum culture remains to be verified by the study of a larger number of samples.
The neural stem cell markers Sox2, Msi1 and nestin are expressed in high grade gliomas 39, and their expression was preserved in the GBM and GS neurosphere cultures reported here. CD133, a hematopoietic and neural stem cell marker, is also expressed in high grade gliomas 39, and has been proposed as a marker for GBM CSC 40. In contrast to the GBM neurosphere lines, the GS1 was CD133 negative. Recently it has been shown that GBM CSCs negative for CD133 are tumorigenic 31, 41. Our results support the concept that although frequently expressed in GBM neurospheres, CD133 expression is not a requirement to define CSCs. Sox-2, a master regulator of embryonic stem cells and neurogenesis 42, is upregulated in glioblastomas in comparison to non-tumor brain tissues 30, 43. It also has functions in the proliferation of glioblastoma CSC population and tumorigenesis 44. We speculate that Sox2 expression in low passage serum GS1 may have a role in the singular plasticity and tumorigenicity observed for this cell population.
The most frequent genetic alterations in gliosarcomas are gains on chromosome 7 and losses on chromosome 10, which include the PTEN locus 13. Similarly, these genetic alterations also characterize GBMs belonging to the mesenchymal subgroup, according to molecular profiling of glioblastomas 1. The GS1 tumor belongs to the mesenchymal subgroup, characterized by poor prognosis (Ken Adalpe, personal communication). Whether all gliosarcomas belong to the mesenchymal subgroup is currently unknown. In vitro transdifferentiation potential of neural stem cells along mesenchymal lineages has been observed 45. The ability of established serum cultured glioblastoma cell lines to transdifferentiate along mesenchymal lineages in vitro30, and the “mesenchymal drift” observed as a result of long term serum culture of glioblastomas 28, 29, have long been known, and are generally considered a consequence of genetic aberrations acquired under these conditions 15. Recently, it has been reported that GBM neurospheres can differentiate along mesenchymal lineages in vitro when the proper stimuli are present 24. Glioblastomas with chondroblastic and osteoblastic differentiation was observed in subcutaneous tumor xenografts from two glioblastoma CSC/neurosphere lines, but not in orthotopic xenografts 24. Here we show that high grade invasive experimental tumors were initiated by all glioblastoma neurosphere lines, as expected from a population containing CSCs. Furthermore, our results indicate that GS1 neurospheres and low passage serum cultures exhibited “gliosarcoma stem cell” properties, as metaplastic mesenchymal differentiation in orthotopic tumors was observed specifically for the GS1 cells, and not for the GBM neurospheres.
Analysis of additional GS samples will be necessary to clarify whether the in vitro phenotypic plasticity and in vivo recapitulation of the the biphasic tumor observed for the GS1 model are general characteristics of this glioblastoma variant. Mechanisms leading to mesenchymal differentiation in gliosarcomas, its pathological significance, or whether the same mechanisms are responsible for the mesenchymal phenotype observed for a subgroup of GBMs 1 are not currently understood. The human gliosarcoma model presented here will be useful in future investigations addressing these important questions.
Supplementary Material
Acknowledgments
The authors would like to thank Mr. Ali Rad for assistance with the MRI images, Mr. Enoch Carlton for histology work, Ms. Kathleen Roszka for expert immunohistochemistry work, and Ms. Laura Hasselbach for LOH analysis. Drs. Jacques Baudier and Laurent Balenci provided helpful advice at the earlier stages of this work. This work was supported in part by funds from the Hermelin Brain Tumor Center, Henry Ford Hospital, to S.K. and T.M., and by the NIH grant R01CA122031 to A.S.A.
Funding: Hermelin Brain Tumor Center and NIH grant R01CA122031
Footnotes
Author Contributions: Ana C. deCarvalho: Conception and design; data collection, analysis and interpretation, manuscript writing, final approval of manuscript
Kevin Nelson:Collection, assembly and analysis of data
Nancy Lemke: Collection, assembly and analysis of data
Norman L. Lehman: Data analysis and interpretation, manuscript writing, final approval of manuscript
Ali S. Arbab: Collection, assembly and analysis of data
Steven Kalkanis: Financial support, provision of patient material, final approval of manuscript
Tom Mikkelsen: Conception, financial support, provision of patient material, final approval of manuscript
References
- 1.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 2.Freije WA, Castro-Vargas FE, Fang Z, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64:6503–6510. doi: 10.1158/0008-5472.CAN-04-0452. [DOI] [PubMed] [Google Scholar]
- 3.Meis JM, Martz KL, Nelson JS. Mixed glioblastoma multiforme and sarcoma. A clinicopathologic study of 26 radiation therapy oncology group cases. Cancer. 1991;67:2342–2349. doi: 10.1002/1097-0142(19910501)67:9<2342::aid-cncr2820670922>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 4.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO Classification of Tumours of the Central Nervous System. 4. Lyon, France: IARC; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perry JR, Ang LC, Bilbao JM, et al. Clinicopathologic features of primary and postirradiation cerebral gliosarcoma. Cancer. 1995;75:2910–2918. doi: 10.1002/1097-0142(19950615)75:12<2910::aid-cncr2820751219>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 6.Lieberman KA, Fuller CE, Caruso RD, et al. Postradiation gliosarcoma with osteosarcomatous components. Neuroradiology. 2001;43:555–558. doi: 10.1007/s002340000531. [DOI] [PubMed] [Google Scholar]
- 7.Reis RM, Konu-Lebleblicioglu D, Lopes JM, et al. Genetic profile of gliosarcomas. The American journal of pathology. 2000;156:425–432. doi: 10.1016/S0002-9440(10)64746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lutterbach J, Guttenberger R, Pagenstecher A. Gliosarcoma: a clinical study. Radiother Oncol. 2001;61:57–64. doi: 10.1016/s0167-8140(01)00415-7. [DOI] [PubMed] [Google Scholar]
- 9.Galanis E, Buckner JC, Dinapoli RP, et al. Clinical outcome of gliosarcoma compared with glioblastoma multiforme: North Central Cancer Treatment Group results. Journal of neurosurgery. 1998;89:425–430. doi: 10.3171/jns.1998.89.3.0425. [DOI] [PubMed] [Google Scholar]
- 10.Meis JM, Ho KL, Nelson JS. Gliosarcoma: a histologic and immunohistochemical reaffirmation. Mod Pathol. 1990;3:19–24. [PubMed] [Google Scholar]
- 11.Boerman RH, Anderl K, Herath J, et al. The glial and mesenchymal elements of gliosarcomas share similar genetic alterations. Journal of neuropathology and experimental neurology. 1996;55:973–981. doi: 10.1097/00005072-199609000-00004. [DOI] [PubMed] [Google Scholar]
- 12.Biernat W, Aguzzi A, Sure U, et al. Identical mutations of the p53 tumor suppressor gene in the gliomatous and the sarcomatous components of gliosarcomas suggest a common origin from glial cells. Journal of neuropathology and experimental neurology. 1995;54:651–656. doi: 10.1097/00005072-199509000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Actor B, Cobbers JM, Buschges R, et al. Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components. Genes, chromosomes & cancer. 2002;34:416–427. doi: 10.1002/gcc.10087. [DOI] [PubMed] [Google Scholar]
- 14.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 15.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 16.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science (New York, NY) 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 17.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 18.Ignatova TN, Kukekov VG, Laywell ED, et al. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 19.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 20.Yuan X, Curtin J, Xiong Y, et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23:9392–9400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 21.Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A. 2004;101:781–786. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghods AJ, Irvin D, Liu G, et al. Spheres isolated from 9L gliosarcoma rat cell line possess chemoresistant and aggressive cancer stem-like cells. Stem cells (Dayton, Ohio) 2007;25:1645–1653. doi: 10.1634/stemcells.2006-0624. [DOI] [PubMed] [Google Scholar]
- 23.Clarke DL, Johansson CB, Wilbertz J, et al. Generalized potential of adult neural stem cells. Science (New York, NY) 2000;288:1660–1663. doi: 10.1126/science.288.5471.1660. [DOI] [PubMed] [Google Scholar]
- 24.Ricci-Vitiani L, Pallini R, Larocca LM, et al. Mesenchymal differentiation of glioblastoma stem cells. Cell death and differentiation. 2008;15:1491–1498. doi: 10.1038/cdd.2008.72. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Szybka M, Zawlik I, Kulczycka D, et al. Elimination of wild-type P53 mRNA in glioblastomas showing heterozygous mutations of P53. British journal of cancer. 2008;98:1431–1433. doi: 10.1038/sj.bjc.6604258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mihaila D, Jankowski M, Gutierrez JA, et al. Meningiomas: loss of heterozygosity on chromosome 10 and marker-specific correlations with grade, recurrence, and survival. Clin Cancer Res. 2003;9:4443–4451. [PubMed] [Google Scholar]
- 28.McKeever PE, Varani J, Papadopoulos SM, et al. Products of cells from gliomas: IX. Evidence that two fundamentally different mechanisms change extracellular matrix expression by gliomas. J Neurooncol. 1995;24:267–280. doi: 10.1007/BF01052843. [DOI] [PubMed] [Google Scholar]
- 29.McKeever PE, Davenport RD, Shakui P. Patterns of antigenic expression of human glioma cells. Critical reviews in neurobiology. 1991;6:119–147. [PubMed] [Google Scholar]
- 30.Tso CL, Shintaku P, Chen J, et al. Primary glioblastomas express mesenchymal stem-like properties. Mol Cancer Res. 2006;4:607–619. doi: 10.1158/1541-7786.MCR-06-0005. [DOI] [PubMed] [Google Scholar]
- 31.Joo KM, Kim SY, Jin X, et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab Invest. 2008;88:808–815. doi: 10.1038/labinvest.2008.57. [DOI] [PubMed] [Google Scholar]
- 32.Wang J, Sakariassen PO, Tsinkalovsky O, et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. International journal of cancer. 2008;122:761–768. doi: 10.1002/ijc.23130. [DOI] [PubMed] [Google Scholar]
- 33.Gage FH, Ray J, Fisher LJ. Isolation, characterization, and use of stem cells from the CNS. Annual review of neuroscience. 1995;18:159–192. doi: 10.1146/annurev.ne.18.030195.001111. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh S, Spagnoli GC, Martin I, et al. Three-dimensional culture of melanoma cells profoundly affects gene expression profile: a high density oligonucleotide array study. J Cell Physiol. 2005;204:522–531. doi: 10.1002/jcp.20320. [DOI] [PubMed] [Google Scholar]
- 35.Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 36.Wislet-Gendebien S, Leprince P, Moonen G, et al. Regulation of neural markers nestin and GFAP expression by cultivated bone marrow stromal cells. J Cell Sci. 2003;116:3295–3302. doi: 10.1242/jcs.00639. [DOI] [PubMed] [Google Scholar]
- 37.Haddad SF, Moore SA, Schelper RL, et al. Smooth muscle can comprise the sarcomatous component of gliosarcomas. Journal of neuropathology and experimental neurology. 1992;51:493–498. doi: 10.1097/00005072-199209000-00003. [DOI] [PubMed] [Google Scholar]
- 38.Laks DR, Masterman-Smith M, Visnyei K, et al. Neurosphere Formation Is an Independent Predictor of Clinical Outcome in Malignant Glioma. Stem Cells. 2009;27:980–987. doi: 10.1002/stem.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma YH, Mentlein R, Knerlich F, et al. Expression of stem cell markers in human astrocytomas of different WHO grades. J Neurooncol. 2008;86:31–45. doi: 10.1007/s11060-007-9439-7. [DOI] [PubMed] [Google Scholar]
- 40.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 41.Beier D, Hau P, Proescholdt M, et al. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 42.Episkopou V. SOX2 functions in adult neural stem cells. Trends in neurosciences. 2005;28:219–221. doi: 10.1016/j.tins.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 43.Schmitz M, Temme A, Senner V, et al. Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. British journal of cancer. 2007;96:1293–1301. doi: 10.1038/sj.bjc.6603696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gangemi RM, Griffero F, Marubbi D, et al. SOX2 Silencing in Glioblastoma Tumor Initiating Cells Causes Stop of Proliferation and Loss of Tumorigenicity. Stem Cells. 2008 doi: 10.1634/stemcells.2008-0493. [DOI] [PubMed] [Google Scholar]
- 45.Galli R, Gritti A, Bonfanti L, et al. Neural stem cells: an overview. Circulation research. 2003;92:598–608. doi: 10.1161/01.RES.0000065580.02404.F4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.