

Abstract
An efficient, second-generation synthesis of the signature dioxabicyclo[3.2.1]octane core of (+)-sorangicin A (1), in conjunction with an effective, stereocontrolled protocol to arrive at the requisite Z,Z,E triene acid system has been developed. Highlights of the core construction entail a three-component union, a KHMDS-promoted epoxide ring formation-ring opening cascade, a Takai olefination and a chemoselective Sharpless dihydroxylation. Assembly of the triene acid system was then achieved via Stille cross-coupling with the ethyl ester of (Z,Z)-5-tributylstannyl-2,4- pentadienoic acid, followed by mild hydrolysis preserving the triene configuration.
The sorangicins comprise a family of architecturally complex macrolide antibiotics isolated from a fermentation broth of the myxobacteria Sorangium cellulosum (strain So ce 12). 1 The most potent and prevalent congener, (+)-sorangicin A (1), was found to be highly effective against a spectrum of both Gram-positive (MIC 0.01–0.3 μg/mL) and Gram-negative bacteria (MIC 3–25 μg/mL). Subsequent studies revealed that (+)- sorangicin A (1) inhibits bacterial RNA-polymerase in both E. coli and S. aureus, while not affecting eukaryotic cells.2
The structure of (+)-sorangicin A (1),3 endowed with a highly unsaturated 31-membered macrolactone, a rare (Z,Z,E)-trienoate linkage, and the signature dioxabicyclo- [3.2.1]octane, in conjunction with the important biological properties, has engendered considerable interest within the synthetic and biomedical communities.4 Indeed, significant progress toward the total synthesis of (+)-sorangicin A has been recorded by the Schinzer5 and Crimmins6 groups, in addition to our laboratory.7
From the outset, our synthetic analysis of (+)-sorangicin A (1) called for disconnections at the macrocyclic lactone, the C(38–39) σ-bond, and both the C(15–16) and C(29–30) trans-disubstituted olefins to yield three advanced subtargets: bicyclic ether (−)-2, tetrahydropyran (−)-3 and dihydropyran 4 (Scheme 1).7 To construct the dioxabicyclo[3.2.1]octane core of (−)-2, our first-generation route featured an acid-promoted intramolecular cascade of epoxide openings, the first facilitated and controlled chemoselectively by a Co2(CO)6-alkyne complex of bis-epoxide (+)-5 and the second mediated by BF3•OEt2.7a Although effective, the route was not highly efficient vis-à-vis material advancement. We now report a second-generation synthesis of (−)-2, in conjunction with the development of an effective, highly stereocontrolled protocol to elaborate the C(37–43) (Z,Z,E)-triene acid unit.
Scheme 1.

Reanalysis of the structure of (−)-2 led to the observation that disconnection of the bicyclic ether fragment at the C(36)–O bond would lead to a tetrahydropyran,8 sharing the same 2,6-trans-relationship as 4, and thus potentially available via a similar substrate-controlled stereoselective conjugate addition of a Michael donor to a similar dihydropyrone as employed to construct 4.7b
Toward this end, dihydropyrone (−)-6 was readily prepared in 86% yield (33:1 dr) via a hetero Diels-Alder (HDA) reaction between the Danishefsky diene and aldehyde (−)-8, 9 catalyzed by the chromium(III)-Schiff base 9, the same Jacobsen catalyst employed for our earlier synthesis of dihydropyrone (−)-7 (Scheme 2).10
Scheme 2.

Attention next turned to the three-component union of dihydropyrone (−)-6 with MeI and a suitable Michael donor, the latter corresponding to a surrogate aldehyde. The literature however is not rich with such examples, due presumably to deactivation of the enone by the ring oxygen.11,7b In fact, dihydropyrone (−)-6 proved to be a reluctant Michael acceptor. For example, use of the cuprate derived from BnOCH2SnBu3 displayed no reactivity. This result may however be a donor problem, given the low reactivity of this type of organometallic addend towards Michael addition as observed by Fuchs et al.12
We turned next to the commercially available β-bromostyrene (10) as a prospective nucleophile progenitor, with a view to achieving olefin cleavage at a later stage to access the C(30) aldehyde. Application of the Noyori three-component prostaglandin coupling protocol,13 involving Li halogen exchange of the bromine in 10 with t-BuLi at −78 °C, 14 followed in turn by addition of Me2Zn, warming to 0 °C to furnish a mixed zincate, and then addition of dihydropyrone (−)-6 at −78 °C effectively led to conjugate addition. 15 Although forcing conditions (ca. 10 equiv. MeI and HMPA at −40 °C) were required to quench the resultant enolate (11), a single diastereomer (+)-12 was obtained in modest yield (51%), along with the formation of a significant amount of α,α′-bismethylated product (+)-13 (20%). This result is not without precedent. Alexakis et al. observed unusual reactivity of a Zn-methyl group with an enolate similar to 11 upon trapping with allyl bromide.16 We reasoned that during the slow enolate capture process, 11 possessing the Zn-methyl group, is sufficiently basic in the presence of excess HMPA to deprotonate (+)-12, and in turn lead via methylation to (+)-13. Lowering the alkylation temperature from −40 °C to −60 °C only led to longer reaction times and an increase of (+)-13 (38%). Higher temperature (−20 °C) however did have a beneficial effect on the yield of (+)-12; the same trend was observed by Alexakis et al. In the end, we discovered that the reactivity of the zinc enolate (11) could be successfully down-regulated by addition of CuI•PBu3 just prior to the addition of MeI, which led to a slower, but more selective reaction to furnish (+)-12 in 73% yield. Confirmation of the requisite 2,3,6-trans-cis-configuration was obtained by NOESY studies (Scheme 2).
Final elaboration to (−)-2 began with L-Selectride reduction of (+)-12 to furnish (−)-14 as a single diastereomer (Scheme 3); confirmation of the requisite configuration at C(33) was again achieved by NOESY correlations. The acetonide moiety was then removed with aqueous acetic acid to furnish triol (−)-15.
Scheme 3.

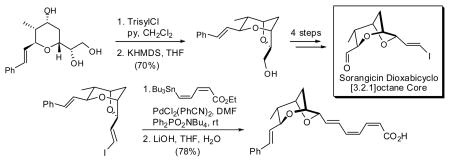
With (−)-15 in hand, we turned to the critical task of generating the two atom bridge. Triol (−)-15 was treated with KHMDS (1 equiv.), followed by slow addition of the bulky N-triisopropylbenzenesulfonylimidazole (Trisyl- Imid; 1 equiv.) to effect regioselective sulfonylation of the least hindered hydroxyl. In analogy with the work of Crimmins et al,6 treatment of the resultant trisylate (16) with an additional 2 equivalents of KHMDS then promoted a reaction cascade involving epoxide ring formation, followed by ring opening to generate the bridged bicycle. 17 Although this “one-pot” protocol delivered the desired product (−)-18, the yield was disappointing (ca. 33%), due to over-sulfonylation to form (−)-19 (ca. 36%). Lower reaction temperatures or the use of potassium tert-butoxide did not improve the situation. A less elegant, two-step protocol was thus explored. The primary hydroxyl of (−)-15 was first selectively sulfonylated with triisopropylbenzenesulfonyl chloride (TrisylCl) employing pyridine/CH2Cl2 (2:3) as solvent at room temperature.18 Under these conditions, sulfonylation of the secondary hydroxyl was suppressed; in addition the resultant sulfonate (−)-20 proved stable to purification and handling. The primary sulfonate was then treated with one equivalent of KHMDS to furnish bicyclic ether (−)-18 in high yield, possessing spectral data in complete accord with the data reported by the Crimmins laboratory.6 Bicycle (−)-18, comprising the signature dioxabicyclo- [3.2.1]octane core of (+)-sorangicin A (1), was thus available in 6 steps and 35% overall yield from (−)-8.
To arrive at (−)-2 (Scheme 4), (−)-18 was oxidized employing Parikh-Doering conditions,19 and the resultant sensitive aldehyde 21 immediately subjected to Takai olefination without purification. 20 Initial experiments on small scale employing THF as solvent afforded an E/Z diastereomeric mixture (3.2:1); the olefin configurations were assigned respectively based on 1H NMR coupling constants (15.8 Hz vs. 8 Hz). 21 The observed low E/Z selectivity was unexpected given that α-alkoxy-aldehydes in general exhibit near complete (E)-selectivity.22 Larger-scale reactions also proved problematic, furnishing the vinyl iodides in significantly lower yield. Recourse to a mixture of dioxane/THF (4:1; v/v) as solvent system,23 although not significantly improving the selectivity, did improve the scale-up issue to furnish (−)-22 and (−)-23 in 52 and 16% respectively, on half gram reaction scale.
Scheme 4.

Required at this stage was differentiation of the two olefins present in (−)-22 to access aldehyde (−)-2. We reasoned that the electron withdrawing and donating biases respectively of the iodide and phenyl substituents would permit chemoselective functionalization of the more electron rich olefin. Gratifyingly, Sharpless dihydroxylation of (−)-22 at room temperature proceeded only at the styrene moiety to generate the corresponding diol, 24 which upon reaction with NaIO4 employing buffered conditions, furnished (−)-2 identical in all respects to material prepared previously in our laboratory.7a
Having achieved an effective, second-generation synthesis of (−)-2, we turned next to explore possible tactics to construct the sensitive (Z,Z,E)-triene acid fragment. Vinyl iodide (−)-22 was selected as a model system. Stille cross-coupling with known (Z,Z)-dienoate 24 led to (+)-25 (Scheme 5).25 Best results were obtained using bis(benzonitrile)-dichloropalladium(II) as catalyst in DMF, along with excess Ph2PO2NBu4 (6 equiv.) as a tin scavenger 26 to suppress Z/E isomerization. Under these conditions, (+)-25 was produced in 96% yield as a single isomer (>20:1). Correlations derived from NOESY studies, as well as coupling constants confirmed the desired (Z,Z,E)-configuration of (+)-25 (Scheme 5). Hydrolysis of trienoate (+)-25 was then achieved with LiOH in aqueous THF to furnish acid (+)-26 in 81% yield, with complete preservation of the olefin configuration.
Scheme 5.

In summary, an effective, scalable route to (−)-2 possessing the C(30–38) signature core of (+)-sorangicin A (1) has been achieved in 10 steps from (−)-8. In addition, an effective protocol has been developed for prospective elaboration of the C(37–43) (Z,Z,E)-triene acid functionality, required for any successful (+)- sorangicin A (1) endgame. Progress towards the total synthesis of (+)-sorangicin A (1) will be reported in due course.
Supplementary Material
Acknowledgments
Support was provided by the National Institutes of Health through Grant No. GM- 29028. We thank Drs. George Furst (University of Pennsylvania) and Rakesh Kohli (University of Pennsylvania) for assistance in obtaining NMR spectra and high-resolution mass spectra, respectively, and Dr. Kallol Basu (Schering-Plough Corporation) for the insightful discussions.
Footnotes
Supporting Information Available: Experimental procedures and full spectroscopic data are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Jansen R, Wray V, Irschik H, Reichenbach H, Höfle G. Tetrahedron Lett. 1985;26:6031. [Google Scholar]; (b) Jansen R, Irschik H, Reichenbach H, Schomburg D, Wray V, Höfle G. Liebigs Ann Chem. 1989:111. [Google Scholar]
- 2.Irschik H, Jansen R, Gerth K, Höfle G, Reichenbach H. J Antibiot. 1987;40:7. doi: 10.7164/antibiotics.40.7. [DOI] [PubMed] [Google Scholar]
- 3.The stereocenter at C(10) in (+)-sorangicin A, as confirmed by Professor R. Jansen (GBF, Braunschweig, Germany) is S, not R as depicted in reference 4b. We thank Prof. Jansen for this clarification.
- 4.(a) Jansen R, Schummer D, Irschik H, Höfle G. Liebigs Ann Chem. 1990:975. [Google Scholar]; (b) Schummer D, Irschik H, Höfle G. Liebigs Ann Chem. 1993:293. [Google Scholar]; (c) Campbell EA, Pavlova O, Zenkin N, Leon F, Irschik H, Jansen R, Severinov K, Darst SA. EMBO J. 2005;24:674. doi: 10.1038/sj.emboj.7600499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schinzer D, Schulz C, Krug O. Synlett. 2004;15:2689. [Google Scholar]
- 6.Crimmins MT, Haley MW. Org Lett. 2006;8:4223. doi: 10.1021/ol061339e. [DOI] [PubMed] [Google Scholar]
- 7.(a) Smith AB, III, Fox RJ. Org Lett. 2004;6:1477. doi: 10.1021/ol049644s. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Fox RJ, Vanecko JA. Org Lett. 2005;7:3099. doi: 10.1021/ol051119l. [DOI] [PubMed] [Google Scholar]
- 8.A similar disconnection was elegantly employed by the Crimmins laboratory in an efficient approach to (−)-18; see ref. 6.
- 9.Aldehyde (−)-8, although commercially available, was prepared in two steps from L-gulonic acid γ-lactone; see Hubschwerlen C, Specklin JL, Higelin J. Organic Syntheses. 1995;72:1.
- 10.Joly GD, Jacobsen EN. Org Lett. 2002;4:1795. doi: 10.1021/ol0258785. [DOI] [PubMed] [Google Scholar]
- 11.Paterson I, Steven A, Luckhurst CA. Org Biomol Chem. 2004;2:3026. doi: 10.1039/B407240E. [DOI] [PubMed] [Google Scholar]
- 12.Hutchinson DK, Fuchs PL. J Am Chem Soc. 1987;109:4930. [Google Scholar]
- 13.Suzuki M, Morita Y, Koyano H, Koga M, Noyori R. Tetrahedron. 1990;46:4809. [Google Scholar]
- 14.It is critical to add β-bromostyrene to t-BuLi; the inverse addition led to low conversion.
- 15.Commercial β-bromostyrene is a trans/cis mixture (ca. 9:1); interestingly only one geometric product was observed. This result could be attributed to unproductive 1,4-addition of the cis-isomer, cf.: Fürstner A, Grela K, Mathes C, Lehmann CW. J Am Chem Soc. 2000;122:11799.
- 16.Rathgeb X, March S, Alexakis A. J Org Chem. 2006;71:5737. doi: 10.1021/jo060814j. [DOI] [PubMed] [Google Scholar]
- 17.Dounay AB, Florence GJ, Saito A, Forsyth CJ. Tetrahedron. 2002;58:1865. [Google Scholar]
- 18.Kojima N, Maezaki N, Tominaga H, Asai M, Yanai M, Tanaka T. Chem Eur J. 2003;9:4980. doi: 10.1002/chem.200305185. [DOI] [PubMed] [Google Scholar]
- 19.Parikh J, Doering W. J Am Chem Soc. 1967;89:5505. [Google Scholar]
- 20.Takai K, Nitta K, Utimoto K. J Am Chem Soc. 1986;108:7408. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]
- 21.Z-Vinyl iodide (−)-23 could be useful for the synthesis of (+)- Srangicin A1.
- 22.Kende AS, DeVita RJ. Tetrahedron Lett. 1990;31:307. [Google Scholar]
- 23.Evans DA, Black WC. J Am Chem Soc. 1993;115:4497. [Google Scholar]
- 24.Sharpless KB, Amberg W, Bennani YL, Crispino GA, Hartung J, Jeong KS, Kwong HL, Morikawa K, Wang ZM, Xu D, Zhang XL. J Org Chem. 1992;57:2768. [Google Scholar]
- 25.Franci X, Martina SLX, McGrady JE, Webb MR, Donald C, Taylor RJK. Tetrahedron Lett. 2003;44:7735. [Google Scholar]
- 26.Srogl J, Allred GD, Liebeskind LS. J Am Chem Soc. 1997;119:12376. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.