
Figure 1. Tissue factor, blood coagulation cascade, inflammation and the Tissue Factor Model for human malaria pathogenesis.
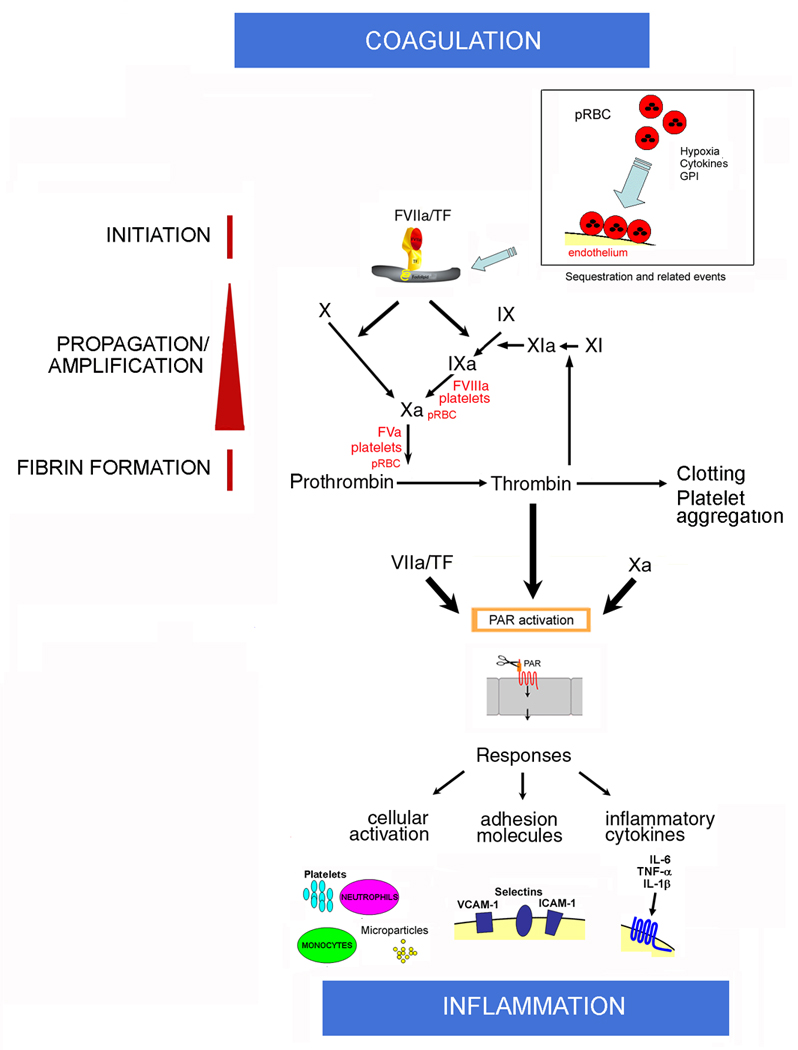
Initiation: Normal, quiescent endothelium does not express functional TF. According to this model, sequestration and sequestration-associated events is associated with EC activation and TF expression in the microvessels of the brain and in other vascular beds. Cytokines, fibrin, hypoxia, apoptosis, proinflammatory molecules released by pRBC (e.g. GPI, hemozoin, oxidized heme) or platelets, and monocytes might also contribute to TF expression (not shown) at sequestration sites and/or paracrinally. Mechanistically, TF initiates the coagulation cascade through binding to coagulation FVIIa and the substrates FIX and FX (extrinsic Xase) in the presence of Ca2+. In this ternary initiation complex, FIXa and FXa are generated.
Amplification: FXa, FVa, and prothrombin assemble in the pRBC surface and/or activated platelets in the presence of Ca2+, with formation of the prothrombinase complex leading to explosive thrombin formation and amplification of the coagulation cascade. Thrombin thus formed promotes fibrin deposition and induces platelet aggregation. Thrombin also activates FXI to FXIa, which activates FIX to FIXa. Factors IXa, FVIIIa, and FX assemble in the membrane of activated platelets and/or pRBC with formation of the intrinsic Xnase complex required for production of additional FXa owing to feedback inhibition of the FVIIa/TF complex by TFPI (not shown). Therefore, the Tissue Factor Model (TFM) for human malaria pathogenesis proposes that both initiation of blood coagulation by TF expression and amplification phase supported by pRBC and/or activated platelets—particularly at sequestration sites—are critical steps for pathogenesis of the disease.
Coagulation–Inflammation: Activated coagulation factors FVIIa/TF, FXa, and thrombin activate PAR in different cell types including mononuclear cells and EC at sequestration sites or paracrinally. PAR activation is accompanied by upregulation of molecules (e.g., ICAM-1, VCAM-1, E-selectin, COX-2, NO synthase) and production of proinflammatory cytokines (e.g., TNF-α, IL-1β, IL-6) that have reportedly been found in CM. Cytokines bind to specific receptors and, together with coagulation factors (and vice versa), perpetuate the inflammatory response, which promotes increased interaction of activated monocytes, elastase- and ROS-producing neutrophils, activated platelets, and/or GPI-containing pRBC with activated EC. The result is a convergence of signals leading to exacerbated TF expression that sustains the coagulation-inflammation cycle in different vascular beds. The cycle is potentially modulated by physiological inhibitors such as NO, prostacyclin, and anticoagulants tissue factor pathway inhibitor, thrombomodulin/protein C pathway, and antithrombin (not shown). As a result of pro- and anti-inflammatory events, a potentially systemic inflammation ensues, leading to metabolic stress, apoptosis, and production of TF-containing microparticles. In extreme cases, organ dysfunction and coma occur.