

Abstract
Lipoxygenases catalyse the oxidation of polyunsaturated fatty acids and have been invoked in many diseases including cancer, atherosclerosis and Alzheimer’s disease. Currently, no X-ray structures are available with substrate or substrate analogues bound in a productive conformation. Such structures would be very useful for examining interactions between substrate and active site residues. Reported here are the syntheses of linoleic acid analogues containing a sulphur atom at the 11 or 14 positions. The key steps in the syntheses were the incorporation of sulphur using nucleophilic attack of metallated alkynes on electrophilic sulphur compounds and the subsequent stereospecific tantalum-mediated reduction of the alkynylsulphide to the cis-alkenylsulphide. Kinetic assays performed with soybean lipoxygenase-1 showed that both 11-thialinoleic acid and 14-thialinoleic acid were competitive inhibitors with respect to linoleic acid with Ki values of 22 and 35 µM, respectively. On the other hand, 11-thialinoleic acid was a noncompetitive inhibitor with respect to arachidonic acid with Kis and Kii values of 48 and 36 µM, respectively. 11-Thialinoleic acid was also a noncompetitive inhibitor of human 15-lipoxygenase-1 with arachidonic acid (Kis = 11.4 µM, Kii = 18.1 µM) or linoleic acid as substrate (Kis = 20.1 µM, Kii = 20.0 µM), and a competitive inhibitor of human 12-lipoxygenase with arachidonic acid as substrate (Ki = 2.5 µM). The presence of inhibitor did not change the regioselectivity of soybean lipoxygenase-1, human 12- or 15-lipoxygenase-1.
Introduction
Lipoxygenases catalyse the first committed step in one of the two major pathways leading from arachidonic acid to eicosanoids.1 They are non-heme iron proteins that abstract a hydrogen atom from a bisallylic position of unsaturated fatty acids followed by the addition of molecular oxygen to generate a hydroperoxide (eg, Figure 1A).2 The substrates for these enzymes are polyunsaturated fatty acids containing nonconjugated cis double bonds. The mammalian lipoxygenases catalyse key steps in the conversion of arachidonic acid (AA) to lipoxins and leukotrienes, which are mediators of inflammation and regulators of the immune system.3, 4 Several studies have suggested that these lipids may also be involved in a number of pathologies including cancer, 5, 6 atherosclerosis,7 and Alzheimer’s disease.8, 9 In plants, lipoxygenases convert linoleic acid (LA) into jasmonates and aldehydes, which are involved in signalling, germination and senescence.10 In mammals, the enzymes are named according to the position of arachidonic acid that reacts with molecular oxygen.11 Several human isozymes (5-, 12-, and 15-hLOs) have thus far been identified12, 13 with this study focusing on the latter two.
Fig. 1.

A. The reactions catalysed by sLO-1 and 15-hLO-1 with linoleic acid and arachidonic acid. B. Proposed catalytic cycle of lipoxygenases.
The majority of our understanding of lipoxygenase structure and mechanism comes from studies on soybean lipoxygenase-1 (sLO-1), which acts on polyunsaturated fatty acids in which a 1,4-diene unit is located six carbons away from the methyl terminus (ω-6 fatty acids).10, 14 Soybean lipoxygenase is relatively easy to purify, kinetically stable and it requires no cofactors or activating proteins like some mammalian lipoxygenases. Although the natural substrate of sLO-1 is LA whereas human lipoxygenases predominantly act on AA (Figure 1A), studies on sLO-1 have led to a better understanding of both classes of enzymes. The chemistry catalysed is the same, even though the substrates differ in chain length and the number of unsaturated bonds.
Lipoxygenases carry out oxidations in an unusual manner. Most oxidative enzymes first activate molecular oxygen by catalysing its reaction with a low valent transition metal and then transferring the activated oxygen species to the substrate, giving the oxidized product. In lipoxygenases, the fatty acid substrate is first activated by hydrogen atom removal to form a radical, which then reacts with molecular oxygen.15, 16 Substrate activation is accomplished by a non-heme ferric hydroxide (Figure 1B). In resting lipoxygenase, the iron is in the ferrous form and the enzyme is inactive.17 The iron must first be converted to the active ferric form by autooxidized compounds before the catalytic cycle can commence. Then, the formal hydrogen atom abstraction is thought to involve a proton-coupled electron transfer between the substrate and the ferric species forming an intermediate radical (R•) and a ferrous species.18 After stereoselective antarafacial reaction of the substrate radical with molecular oxygen, the peroxyl radical oxidizes the iron back to the active ferric state and the peroxide product (ROOH) is released from the enzyme. The sLO-1 products of linoleic acid and arachidonic acid are 13-hydroperoxy-octadecadienoic acid (13-HPODE) and 15-hydroperoxy-eicosatetraenoic acid (15-HPETE), respectively (Figure 1A).
The hydrogen abstraction step has received much interest since kinetic isotope effects (KIE) up to 80 have been reported in studies with linoleic acid and arachidonic acid.19–25 These observations have led to a model in which quantum mechanical tunneling20 is coupled to environmental motions governed by protein dynamics.26 Several X-ray structures of various lipoxygenases have been obtained.27–40 However, no structures of lipoxygenases with a bound substrate or substrate analogue have been reported, and thus relatively little structural information is available regarding the binding interactions between substrate and enzyme. Such structures are eagerly anticipated as they may provide insight into protein dynamics, the unusually large isotope effects observed, and the regioselectivity of catalysis.
In this work, sulphur-containing fatty acid analogues were evaluated as possible inhibitors. Previous studies have demonstrated that a variety of organosulphur compounds derived from garlic essential oil act as inhibitors of soybean lipoxygenase.41, 42 Sulphur-containing arachidonic acid analogues have also been described as inhibitors.43–46 Herein are described the syntheses of 11-thialinoleic acid (11-thiaLA) and 14-thialinoleic acid (14-thiaLA), two linoleic acid analogues containing sulphur at allylic positions. Both compounds were competitive inhibitors for the sLO-1-catalyzed oxidation of linoleic acid. 11-ThiaLA also behaved as a competitive inhibitor for the reaction of human platelet 12-lipoxygenase (12-hLO) with arachidonic acid, but as a noncompetitive inhibitor for oxidation of AA and LA by human reticulocyte 15-lipoxygenase-1 (15-hLO-1, also called 12/15-LO) and the oxidation of AA by sLO-1.
Results and Discussion
Synthesis of 11- and 14-thialinoleic acids
Recently, our laboratory reported the synthesis of 7-thiaarachidonic acid (1, Figure 2) for the purpose of identifying radical intermediates in the reaction of prostaglandin H synthase with arachidonic acid.47 Compound 1 was contructed by the preparation of a bis(alkynyl)sulphide and its subsequent stereoselective reduction to a (cis, cis)-bis(alkenyl)sulphide in the presence of tantalum(V) chloride.48 In this work, we have applied this methodology to the synthesis of linoleic acids with sulphur present at two of the three allylic positions of the fatty acid. These thialinoleic acids (2 and 3, Figure 2) contain sulphur either at the position of hydrogen atom abstraction (position 11) or at a position where it could potentially stabilize the radical intermediate formed upon hydrogen atom abstraction (position 14). Thus, they could either be substrate analogues or inhibitors that bind in the lipoxygenase active site.
Fig. 2.

Fatty acid analogues containing sulphur at allylic positions.
The synthesis of 11-thiaLA is outlined in Scheme 1. 8-Bromooctanoic acid (4) was first converted to the tert-butyl ester 5, which was then reacted with lithium (trimethylsilyl) acetylide. The silyl protecting group was subsequently removed with TBAF to yield the terminal alkyne 6. Deprotonation of a 1-to-3 mixture of alkyne 6 and 1-heptyne with n-butyllithium followed by slow addition of sulphur dichloride at −78 °C yielded the desired unsymmetrical bis(alkynyl)sulphide 7 along with minor amounts of the two symmetrical dialkyl sulphides. Selective reduction of 7 mediated by a low-valent tantalum species48 yielded the bis(alkenyl)sulphide 8. Addition of a small amount of 1-hexyne was needed to achieve a good yield, although the reason for this requirement is unknown.47 Only the cis, cis-isomer was observed by 1H NMR spectroscopy. Finally, the tert-butyl ester was hydrolyzed at 50 °C with a 1.0 M solution of lithium hydroxide in a water-DME mixture. 11-ThiaLA (2) was purified by reverse-phase HPLC and isolated as a single isomer.
Scheme 1.

Synthesis of 11-thiaLA. Reagents and conditions: (a) (CF3CO)2O, tert-BuOH, CH2Cl2, 0 °C, (b) lithium (trimethylsilyl)-acetylide, HMPA, THF, −78 °C, 3 h; TBAF, THF, 25 °C, 30 min, (c) 1-heptyne, n-BuLi, THF, −78 °C, 1 h; SCl2, THF, 2 h, (d) TaCl5, Zn, pyridine, DME, benzene, 1-hexyne, 25 °C, 1 h, (e) LiOH, water, DME, 50 °C, 40 h.
The synthesis of 14-thiaLA is outlined in Scheme 2. Azelaic monomethyl ester (9) was chemoselectively reduced with borane-THF to alcohol 10. Conversion of 10 to bromide 11 was achieved by treatment with bromine in pyridine. Subsequent refluxing of 11 in acetonitrile with triphenylphosphine yielded the phosphonium salt 12. The aldehyde partner 18 for the Wittig reaction of 12 was prepared by protection of 3-butynol (13) with triisopropylsilylchloride to afford 14. The terminal alkyne was deprotonated with n-butyllithium and reacted with dibutyl disulphide at −78 °C to yield alkynylsulphide 15. Selective reduction with low-valent tantalum provided the alkenylsulphide 16 in good yield. Deprotection of the silyl group with TBAF followed by oxidation of the alcohol with Dess-Martin periodinane afforded the aldehyde 18. Subsequent Wittig reaction between compounds 12 and 18 yielded the desired methyl 14-thialinoleate (19). Only the cis, cis-isomer was observed by 1H NMR spectroscopy. Finally, the methyl ester was hydrolyzed at 25 °C with a 1.0 M solution of lithium hydroxide in water-DME. 14-ThiaLA (3) was purified by reverse-phase HPLC and isolated as a single isomer.
Scheme 2.

Synthesis of 14-thiaLA. Reagents and conditions: (a) BH3-THF, 25 °C (b) PPh3, Br2, pyridine, CH2Cl2, 0 °C, 1 h (c) PPh3, acetonitrile, reflux, 24 h (d) TIPSCl, imidazole, CH2Cl2, 0 °C, 3 h, (e) n-BuLi, THF, −78 °C, 1 h; butyl disulphide, − 78 °C, 3 h, (f) TaCl5, Zn, pyridine, DME, benzene, rt, 1 h, (g) TBAF, THF, 25 °C, 1 h, (h) Dess-Martin periodinane, CH2Cl2, 0 °C, 1 h, (i) NaHMDS, THF, −78 °C, 1 h; 12, 25 °C, 12 h, (j) LiOH, water, DME, 25 °C, 24 h.
Inhibition of sLO-1
Lipoxygenase-catalysed hydrogen abstraction from an allylic position of a fatty acid substrate has been previously observed, but this reaction is much slower than at a bisallylic position.24 Thus, compound 2 was not expected to undergo oxidation by lipoxygenase, but compound 3 still contains a bisallylic methylene group and could be a substrate. However, analysis by thin-layer chromatography and UV-visible spectroscopy showed that incubation of 11-thiaLA or 14-thiaLA with sLO-1 did not result in any appreciable activity over the course of one hour. Next, the inhibitory behaviour of these compounds was assessed. The conversion of linoleic acid to 13-HPODE catalysed by sLO-1 was monitored by following the formation of product at 235 nm at pH 10.0, the pH optimum for this enzyme.49 The Km for linoleic acid with sLO-1 was 28 ± 5 µM, in good agreement with previous reports.22 The rate of reaction was examined in the presence of inhibitor at several different concentrations. Competitive inhibition was observed with both inhibitors as shown in Figures 3 and 4 (see also Dixon and Cornish-Bowden plots in the Supplementary Information†). The Ki of 11-thiaLA was 22 ± 5 µM, and the value for 14-thiaLA was 35 ± 5 µM. These are reasonably good inhibitors since the inhibitor Ki and the substrate Km are similar. Although competive inhibition visually appears to be the best model to fit the data, statistically noncompetitive inhibition with 11-thiaLA having a Kis of 36 µM for binding to the enzyme and a Kii of 115 ± 60 µM for binding to the enzyme-substrate complex (Figure 5) cannot be ruled out (see Electronic Supplementary Information). Due to the poor solubility of LA at pH 7.5, no quantitative inhibition parameters could be determined at this pH for this substrate.
Fig. 3.
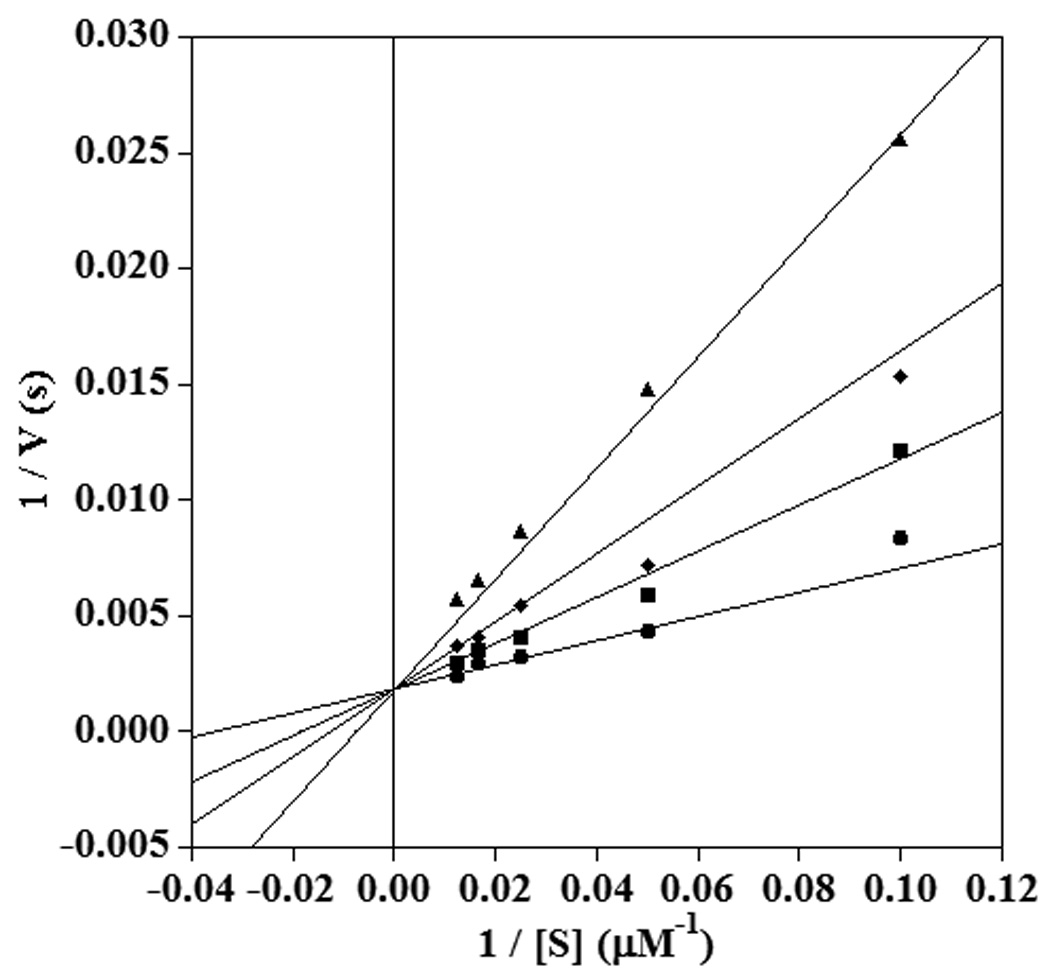
Double reciprocal plot showing competitive inhibition of sLO-1 by 11-thiaLA in the reaction with linoleic acid at pH 10.0. The inhibitor concentrations shown are 0 µM (circles), 20 µM (squares), 40 µM (diamonds), and 80 µM (triangles).
Fig. 4.
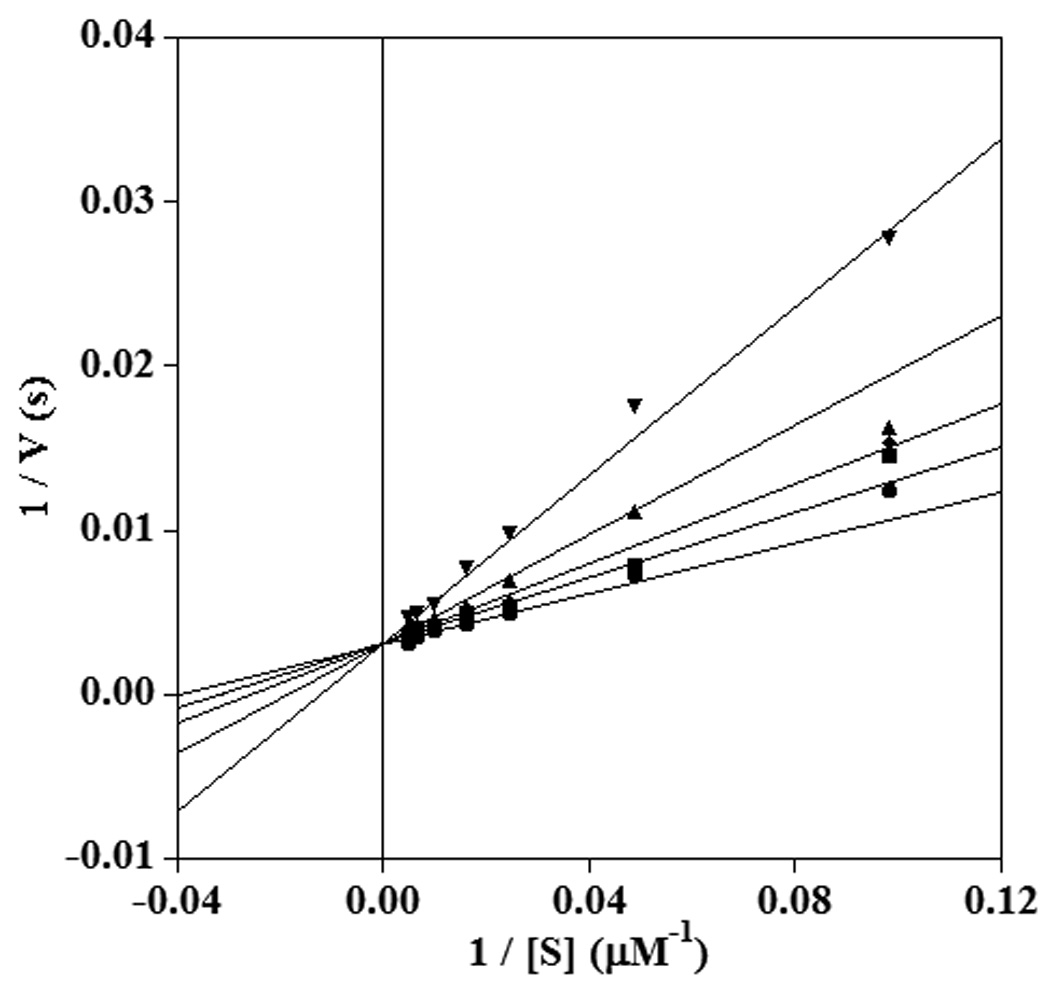
Double reciprocal plot showing competitive inhibition of sLO-1 by 14-thiaLA in the reaction with LA at pH 10.0. The inhibitor concentrations shown are 0 µM (circles), 20 µM (squares), 40 µM (diamonds), 80 µM (triangles), and 120 µM (inverted triangles).
Fig. 5.

Scheme describing noncompetitive inhibition. When Kis ≠ Kii, the term mixed inhibition is often used.
Next the inhibitory behaviour of 11-thiaLA was evaluated for the oxidation of AA by sLO-1. This transformation displayed a significant lag phase,50 which made rate measurements difficult. The lag phase is due to the slow conversion of the inactive ferrous form in which the enzyme is isolated to the active ferric form by autooxidized compounds in the assay. In order to obtain reliable initial rates, the enzyme was activated by adding a small amount of 13-HPODE before presenting substrate.25, 51 The addition of oxidant also helped eliminate substrate inhibition by AA.25
The addition of 13-HPODE raised a concern that this alkylperoxide might oxidize the thiaLAs and hence affect their inhibition behaviour. However, incubation of 11-thiaLA under turnover conditions with enzyme, AA, and 13-HPODE showed no discernable conversion of the inhibitor over a period of 1 hour as monitored by HPLC. On the other hand, 14-thiaLA was oxidized to a small extent over that time frame and was not investigated further as the oxidation products could affect the inhibition data. With 11-thiaLA noncompetitive inhibition was observed at both pH 10.0 and pH 7.5 (Figure 6 and Supplementary Information). Under these conditions, the Km of AA was 15.1 ± 1.5 µM at pH 10.0 compared to 31.7 ± 3.5 µM at pH 7.5, but the inhibition constants were similar under both conditions (pH 10.0, Kis = 48.5 ± 12.9 µM, Kii = 36.0 ± 4.0 µM; pH 7.5, Kis = 45.6 ± 11.1 µM, Kii = 38.2 ± 5.4 µM).
Fig. 6.
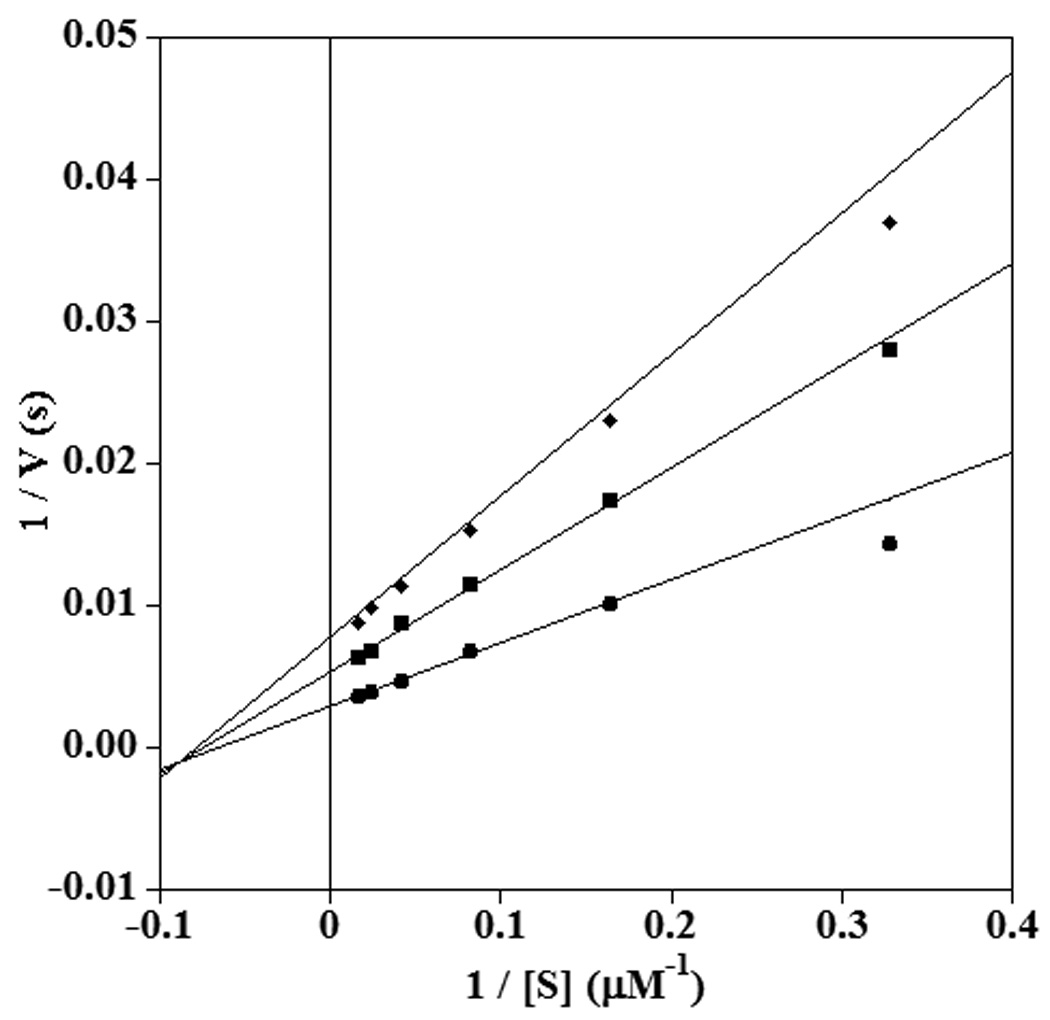
Double reciprocal plot showing noncompetitive inhibition of sLO-1 by 11-thiaLA in the reaction with AA at pH 10.0. The inhibitor concentrations shown are 0 µM (circles), 30 µM (squares) and 60 µM (diamonds).
Inhibition of 15-hLO-1
Next, the behaviour with 15-hLO-1 was examined. While sLO-1 was stable during the course of the reaction, 15-hLO-1 underwent rapid self-inactivation, as described previously.52–56 In addition, 15-hLO-1 also displayed an initial lag phase. Once again, addition on of 13-HPODE helped eliminate the lag phase and also allowed the measurement of initial rates before enzyme inactivation.57
A kinetic assay performed with 11-thiaLA demonstrated that the compound was a noncompetitive inhibitor for 15-hLO-1 with respect to LA as the lines converge on the X-axis to the left of the origin (Figure 7, see also Supplementary information). The Kis and Kii values were 20.1 ± 1.6 and 20.0 ± 1.4 µM, respectively. 11-ThiaLA was also a noncompetitive inhibitor of 15-hLO-1 with AA as substrate with Kis and Kii values of 11.4 ± 2.0 and 18.1 ± 2.0 µM respectively (Supplementary Information). For comparison, the measured Km values of LA and AA with 15-hLO-1 were 6.2 ± 0.9 and 5.0 ± 0.5 µM, respectively.
Fig. 7.
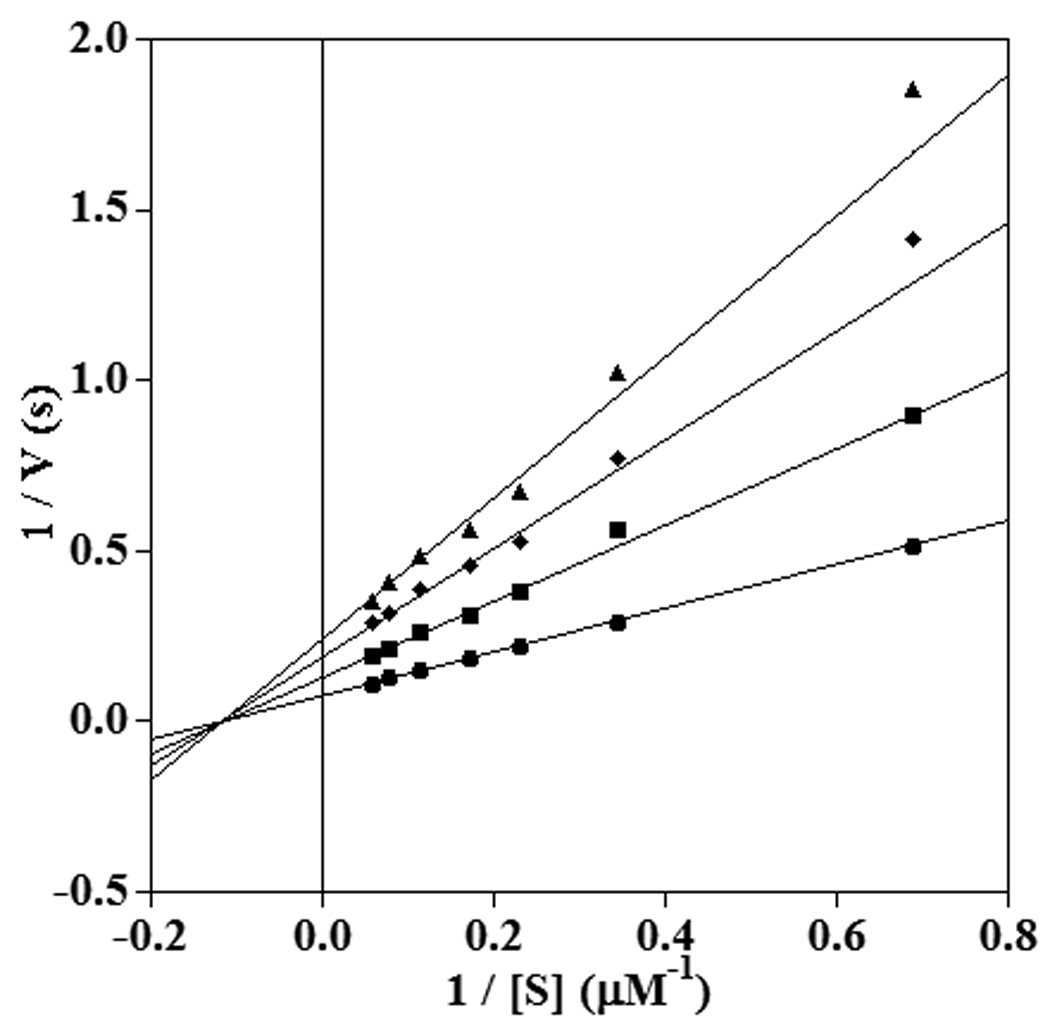
Double reciprocal plot showing noncompetitive inhibition of 15-hLO-1 by 11-thiaLA in the reaction with LA at pH 7.5. The inhibitor concentrations shown are 0 µM (circles), 15 µM (squares), 30 µM (diamonds), 45 µM (triangles).
Noncompetitive inhibition indicates a binding site away from the active site and suggests that the thiaLAs act as allosteric inhibitors of 15-hLO-1. Oleyl sulfate and oleic acid have been previously described as allosteric inhibitors for this enzyme.58, 59 In these previous reports, the ternary complex formed by enzyme, substrate, and inhibitor was catalytically active (kallo ≠ 0, Figure 5). The activity of the complex formed by 15-hLO-1, LA, and 11-thiaLA was investigated by measuring the kinetic isotope effect (KIE) on kcat/Km of the reaction of 15-hLO-1 with protiated and deuterated substrate in the presence of different concentrations of the inhibitor. The observed KIE on kcat/Km depends on the commitment of the reaction, which is the ratio of the forward rate of catalysis and the reverse rate of dissociation of the enzyme-substrate complex.60 The rate of product formation via the ternary complex, kallo, can affect this ratio (Figure 5). On the other hand, if the ternary complex is not active, the concentration of inhibitor affects neither the commitment to catalysis nor the KIE.61
If compound 2 formed an active complex with the enzyme and substrate, increasing inhibitor concentration would skew the reaction to proceed through the ternary complex which could change the observed KIE. For instance, a hyperbolic rise in the KIE with increasing concentration of oleylsulfates has been observed for 15-hLO-1 (Δ KIE on kcat/Km ≈ 40–60).58 This enzyme exhibits a much larger primary KIE on kcat/Km with 11-d2-LA (KIE on kcat/Km ≈ 50)23 than with 10,10,13,13-d4-AA (KIE on kcat/Km ≈ 7.5).62 Thus, a change in KIE was anticipated to be observable more readily with LA as substrate. The KIE on kcat/Km of the reaction of 15-hLO-1 with LA and 11-d2-LA was measured at a variety of concentrations of 2. The observed lack of change in the KIE on kcat/Km with increasing inhibitor concentration suggests the ternary complex is not catalytically active (Figure 8).
Fig. 8.
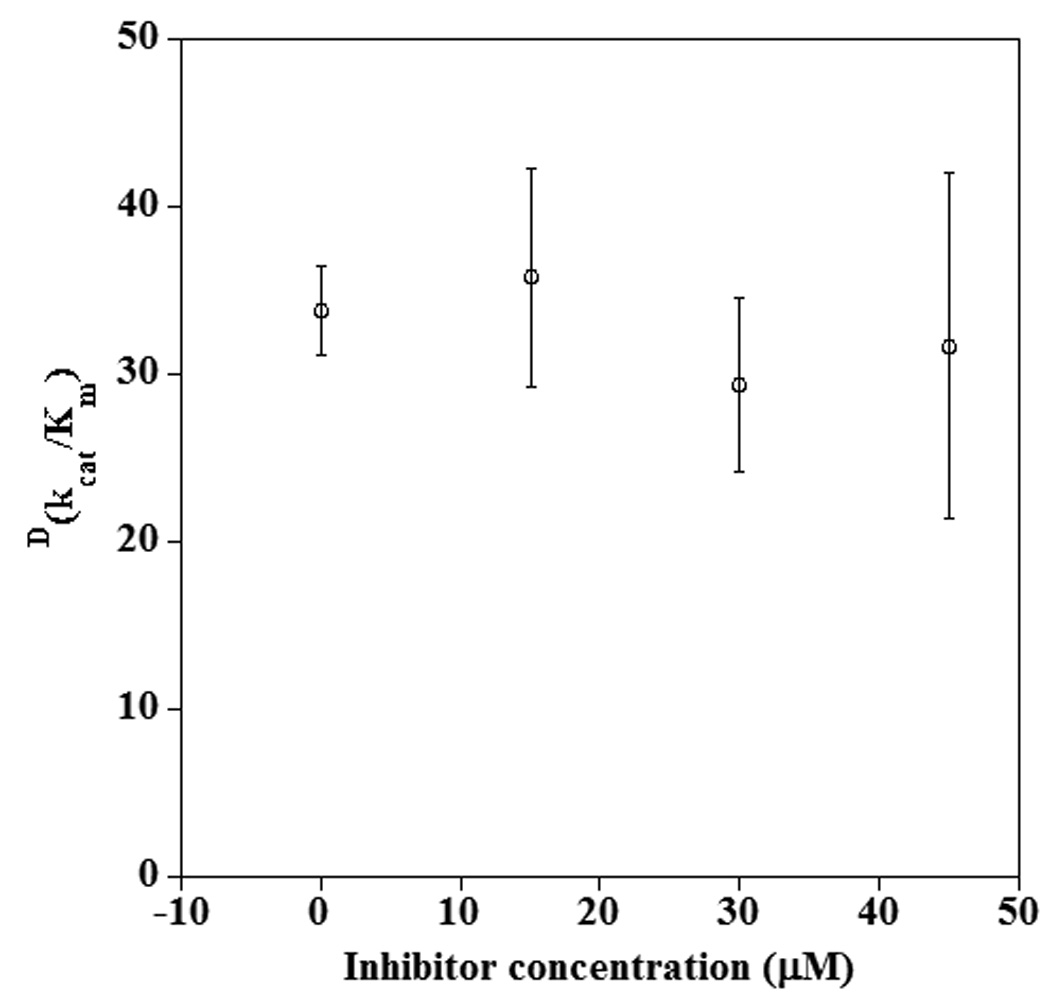
Dependence of the KIE on kcat/Km for the reaction of 15-hLO-1 with LA and 11-d2-LA in the presence of 11-thiaLA.
Inhibition of 12-hLO
Finally, the inhibition of 12-hLO was examined. A previous study showed that LA was a poor substrate for this enzyme,54 and hence the inhibition was evaluated only with AA as substrate. Michaelis-Menten kinetics were obtained without the addition of 13-HPODE as an activator, and thus it was possible to investigate both 11- and 14-thiaLAs as inhibitors. 11-ThiaLA was a competitive inhibitor of 12-hLO (Figure 9) with a Ki value of 2.5 ± 0.3 µM compared to a Km value of 0.3 ± 0.1 µM for arachidonic acid. Surprisingly, 14-thiaLA was neither a substrate nor an inhibitor.
Fig. 9.
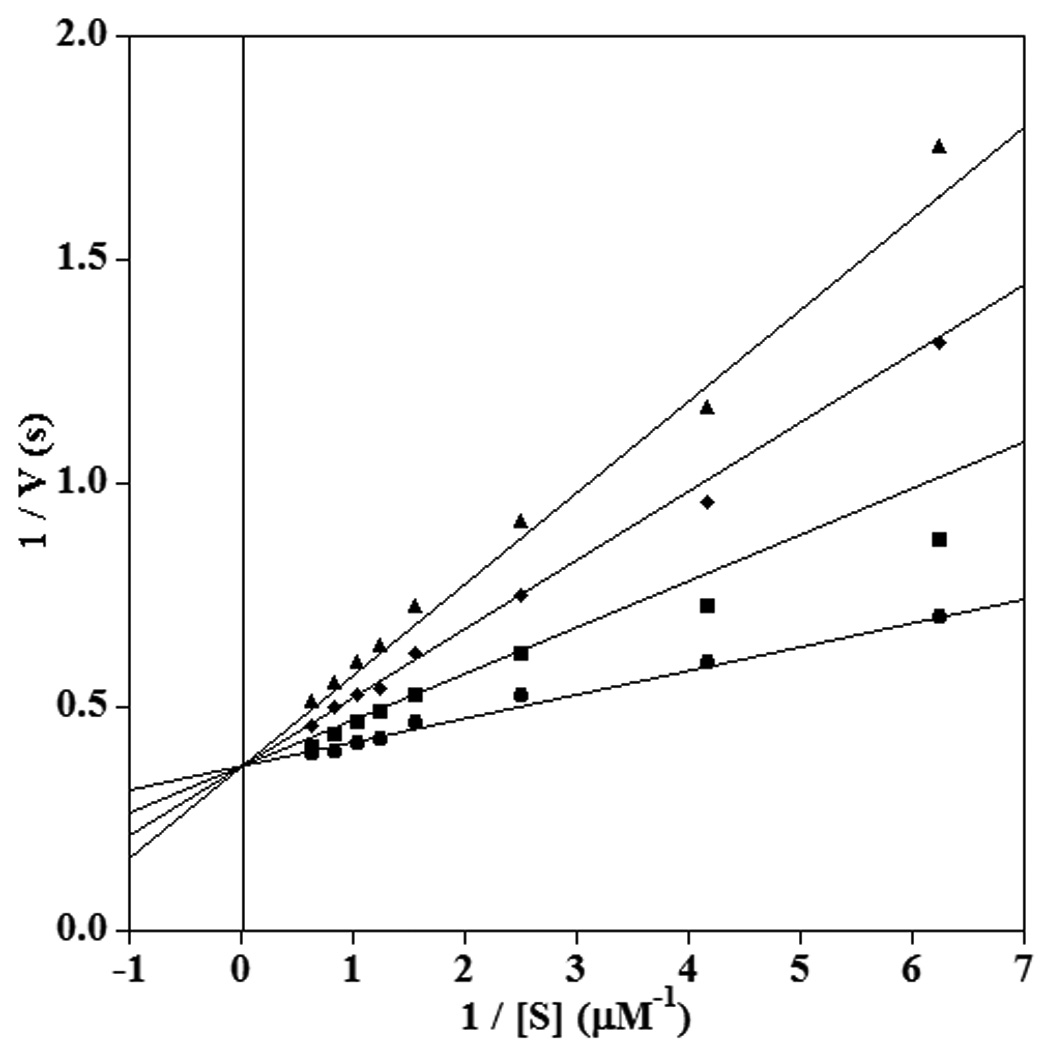
Double reciprocal plot showing competitive inhibition of 12-hLO by 11-thiaLA in the reaction with AA. The inhibitor concentrations shown are 0 µM (circles), 2.5 µM (squares), 5 µM (diamonds) and 7.5 µM (triangles).
One particular aspect of the inhibition assays that stands out is that all noncompetitive inhibition patterns were observed in experiments in which 13-HPODE was added to activate the enzyme. As mentioned, the oxidation of AA by 15-hLO-1 or sLO-1 requires this hydroperoxide in order to obtain reproducible kinetics without interference by a severe lag phase or substrate inhibition. However, the oxidation of AA by 12-hLO and LA by sLO-1 proceeds in the absence or presence of 13-HPODE. Thus, to investigate whether the presence of 13-HPODE might in some way be responsible for the observation of noncompetitive inhibition, the inhibition patterns for these two enzymes were monitored in the presence of activator. 11-ThiaLA displayed competitive inhibition in both cases (Supplementary Information), suggesting that the addition of activator does not affect the mode of inhibition.
Effect of Inhibitor on Product Distribution
A number of lipoxygenase isozymes have been isolated thus far that differ in the regioselectivity of oxidation of AA. Since an allosteric effector could change the regioselectivity, the product distribution was determined in the presence of 11-thiaLA. The hydroperoxide products (HPETEs) were reduced with NaBH4 to provide the corresponding alcohols (HETEs) and the relative amounts of isomers were determined by HPLC analysis. As shown in Table 1, the addition of inhibitor did not alter the product distribution for 12-hLO and 15-hLO-1. 11-ThiaLA also did not affect the product regioselectivity of sLO-1. In the presence and absence of inhibitor, the exclusive product was 13-HPODE with no 9-HPODE observed. Thus, both the KIE and product distribution studies suggest that the ternary complexes of the enzymes with substrate and 11-thiaLA are not active.
Table 1.
Effect of 11-thiaLA on human lipoxygenase product distributions. The error is shown in parentheses.
| Conditions | 15-HETE (%) | 11-HETE (%) | 8-/12-HETE (%) |
|---|---|---|---|
| 15-hLO-1 and AA | 80 (3) | 6 (1) | 14 (3) |
| 15-hLO-1, AA and 2 | 79 (3) | 5 (1) | 16 (3) |
| 12-hLO and AA | 5 (2) | 4 (2) | 91 (4) |
| 12-hLO, AA and 2 | 5 (1) | 2 (1) | 93 (1) |
Discussion
The three lipoxygenases investigated show different inhibition types, and one of the enzymes, sLO-1, displays a substrate dependence with respect to the observed inhibition type. With arachidonic acid and 11-thiaLA, noncompetitive inhibition was observed at both pH 7.5 and 10, showing the inhibitor can bind both to the catalytic site and a physically distinct site on the enzyme in the ES complex with similar affinities. Such allosteric sites have been previously documented for sLO-1.58, 59, 63, 64 However, with linoleic acid as substrate, the data can be fit about equally well using either the competitive inhibition model or by using noncompetitive inhibition with a weak affinity of the inhibitor for the ES complex. These observations suggest that the second site is less available for binding when linoleic acid occupies the active site than when arachidonic acid is bound to the active site. We note that both the Km values (28 µM for LA, 15 µM for AA) and kcat values (490 s−1 for LA, 340 s−1 for AA) were similar for the two substrates. Previous work on sLO-1 has shown that the allosteric site on the enzyme is sensitive to the chain length of inhibitors with long alkyl chains.63 To the best of our knowledge, this is the first report that the affinity of the inhibitor for this site (Kii, Figure 5) is dependent on the chain length of the substrate.
Human 15-lipoxygenase-1 also showed non-competitive inhibition by 11-thiaLA, regardless of whether the substrate was LA or AA. In both cases, the affinity of the inhibitor for the empty enzyme and ES complex is similar. Inhibition of 12-hLO by 11-thiaLA on the other hand indicates a much higher affinity for the catalytic site than for a potential allosteric site, with the data for AA only fit well with a competitive inhibition model. The inhibition constants are smaller for 12-hLO than those observed for sLO-1 and 15-hLO-1 but this enzyme also has signicantly lower Km values for its substrates. Hence, 11-thiaLA is not expected to show much selectivity in a physiological setting. In general, the affinity of the inhibitor for the catalytic site for these different lipoxygenases tracks well with the Km values for their substrates. The most obvious difference between the enzymes is the greatly different affinities for an allosteric site.
Conclusions
In summary, we have synthesized analogues of linoleic acids containing sulphur at the 11 and 14 allylic positions. The key steps in the syntheses were the incorporation of sulphur using nucleophilic attack of metallated alkynes on electrophilic sulphur compounds and the subsequent stereospecific tantalum-mediated reduction of the alkynylsulphide to the cis-alkenylsulphide. Both desired compounds were isolated as single isomers after purification by reverse phase HPLC.
Both compounds competitively inhibited the oxidation of LA by sLO-1 with about equal effectiveness, whereas only 11-thiaLA inhibited 12-hLO and with an approximately 10-fold smaller Ki. 11-ThiaLA was a noncompetitive inhibitor of the oxidation of AA by sLO-1 and 15-hLO-1, indicating it has an additional, different binding site from the fatty acid substrate. This different mode of inhibition is not due to the presence of the activator or to a pH effect. The location of this binding site and how binding in this site affords inhibition is currently not known. Future studies will attempt to answer these questions through crystallographic investigations.
Experimental Section
General
All reactions were performed in oven-dried or flame-dried glassware under an inert atmosphere of dry nitrogen. THF was distilled from Na metal and benzophenone. CH2Cl2 was distilled from CaH2. Benzene was distilled from Na metal. Brine refers to a saturated aqueous solution of NaCl. Commercial reagents were used as received without further purification. Analytical thin-layer chromatography was performed on Merck silica gel plates with QF-254 indicator. Visualization was performed with a KMnO4 solution or a UV light. Flash column chromatography was performed using 230–400 mesh silica gel purchased from EM Science.
1H NMR and 13C NMR spectra were recorded on 400 MHz and 500 MHz, 1H (100.6 MHz and 125.6 MHz, 13C) spectrometers in the VOICE laboratory at the University of Illinois, Urbana-Champaign. Spectra were referenced to chloroform-d1 as an internal standard (δ 7.26 ppm, 1H; δ 77.0 ppm, 13C). Chemical shifts are reported in ppm (δ) and peak multiplicities are labeled as s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Coupling constants are given in Hertz.
Synthesis of 11-thialinoleic acid
8-Bromo-octanoic acid tert-butyl ester (5)
A dry flask was charged with 8-bromooctanoic acid (700 mg, 3.1 mmol) and purged with nitrogen. THF (30 mL) was added and the solution was cooled to 0 °C. Trifluoroacetic anhydride (0.96 mL, 6.9 mmol) was added dropwise. The reaction mixture was stirred at 0 °C for 2.5 h, then tert-butanol (1 mL, 11 mmol) was added slowly. After 1 h, the reaction was warmed to room temperature. The reaction mixture was stirred for an additional 12 h, quenched with water (20 mL) and extracted with diethyl ether (4 × 20 mL). The combined organic layers were washed with brine (2 × 20 mL), dried over MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (20% diethyl ether/pentane), yielding 5 (850 mg, 99%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ1.32 (m, 6 H), 1.45 (s, 9 H), 1.58 (m, 2 H), 1.85 (m, 2 H), 2.22 (t, J = 7.5 Hz, 2 H), 3.40 (t, J = 6.8 Hz, 2 H). 13C NMR (125.6 MHz, CDCl3) δ 25.2 (CH2), 28.2 (CH2), 28.3 (CH3), 28.7 (CH2), 29.1 (CH2), 32.9 (CH2), 34.2 (CH2), 35.7 (CH2), 80.2 (Cq), 173.4 (Cq). The spectroscopic data agreed with a previous report in the literature.65
Dec-9-ynoic acid tert-butyl ester (6)
A dry flask was charged with bromide 5 (300 mg, 1.1 mmol) and purged with nitrogen. A 4:1 THF/HMPA mixture (10 mL total) was added and the solution was cooled to −78 °C. A 0.5 M lithium(trimethylsilyl) acetylide solution in THF (2.6 mL, 1.3 mmol) was added and the reaction mixture was stirred at −78 °C for 1 h, then slowly warmed to room temperature. After an additional 2 h, hexane (25 mL) and saturated NH4Cl solution (25 mL) were added and the layers were separated. The aqueous layer was extracted with hexane (3 × 20 mL). The combined organic layers were washed with saturated NH4Cl solution (25 mL), dried over MgSO4, filtered and concentrated. The resulting crude material was redissolved in THF (15 mL). A 1 M solution of TBAF in THF (1.3 mL, 1.3 mmol) was added and the reaction mixture was stirred at room temperature for 30 min. The reaction mixture was diluted with diethyl ether (20 mL) and washed with water (20 mL). The aqueous layer was extracted with diethyl ether (2 × 20 mL). The combined organic layers were washed with brine (25 mL), dried over MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (20% diethyl ether/pentane), yielding 6 (155 mg, 66%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 1.32 (m, 6 H), 1.44 (s, 9 H), 1.48–1.60 (m, 4 H), 1.93 (t, J = 2.7 Hz, 1 H), 2.18 (m, 4 H). 13C NMR (125.6 MHz, CDCl3) δ 18.6 (CH2), 25.2 (CH2), 28.3 (CH3), 28.6 (CH2), 28.8 (CH2), 29.0 (CH2), 29.1 (CH2), 35.8 (CH2), 68.3 (CH), 80.1 (Cq), 84.9 (Cq), 173.5 (Cq). HRMS (CI, M+) for C14H24O2 calculated 224.1855, found 224.1859.
10-Hept-1-ynylsulfanyl-dec-9-ynoic acid tert-butyl ester (7)
A dry flask was charged with alkyne 6 (180 mg, 0.8 mmol) and purged with nitrogen. THF (20 mL) was added, followed by 1-heptyne (232 mg, 2.41 mmol) and the solution was cooled to −78 °C. A 1.6 M solution of n-butyllithium in THF (1.53 mL, 2.4 mmol) was added dropwise and the reaction mixture was stirred at −78 °C. After 30 min, sulphur dichloride (78 µL, 1.2 mmol) was added. The reaction mixture was stirred at −78 °C for 2 h then slowly warmed to room temperature. After an additional 3 h, the reaction was quenched with water (20 mL) and extracted with diethyl ether (4 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (5% diethyl ether/pentane), yielding 7 (113 mg, 40%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 7.2 Hz, 3 H), 1.26–1.38 (m, 10 H), 1.43 (s, 9 H), 1.46–1.60 (m, 6 H), 2.19 (t, J = 7.5 Hz, 2 H), 2.30 (t, J = 7.1 Hz, 4 H). HRMS (EI, M+) for C21H34O2S calculated 350.2280, found 350.2280.
10-Hept-1-enylsulfanyl-dec-9-enoic acid tert-butyl ester (8)
A dry flask was charged with tantalum pentachloride (820 mg, 2.28 mmol) and purged with nitrogen. Benzene (10 mL) and then dimethoxyethane (10 mL) were added, followed by zinc powder (224 mg, 3.42 mmol) in one portion. The solution was stirred at room temperature for 1 h. To the dark blue mixture was added pyridine (72 µL, 0.86 mmol) and the red mixture was stirred for 5 min. A solution of diyne 7 (100 mg, 0.29 mmol) and 1-hexyne (12 µL, 0.10 mmol) in a benzene:DME mixture (1:1 ratio, 8 mL) was added via cannulation. The reaction mixture was stirred at room temperature for 50 min, then additional pyridine (3 mL) was added. After 5 min, a 1 M solution of NaOH (5 mL) was added and the reaction mixture was vigorously stirred for 1 h. The mixture was diluted with water (20 mL) and extracted with diethyl ether (5 × 30 mL). The combined organic layers were washed with brine (2 × 25 mL), dried over MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (4% diethyl ether/pentane), yielding 8 (64 mg, 64%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 7.0 Hz, 3 H), 1.30 (m, 10 H), 1.40 (m, 4 H), 1.44 (s, 9 H), 1.57 (m, 2 H), 2.11 (m, 4 H), 2.20 (t, J = 7.5 Hz, 2 H), 5.58 (m, 2 H), 6.00 (m, 2 H). 13C NMR (125.6 MHz, CDCl3) δ 14.3 (CH3), 22.7 (CH2), 25.3 (CH2), 28.3 (CH3), 28.8 (CH2), 29.0 (CH2), 29.2 (CH2), 29.3 (CH2), 29.3 (CH2), 29.4 (CH2), 29.4 (CH2), 31.7 (CH2), 35.8 (CH2), 80.1 (Cq), 123.8 (CH), 123.9 (CH), 129.9 (CH), 130 (CH), 173.5 (Cq). HRMS (EI, M+) for C21H38O2S calculated 354.2593, found 354.2590.
11-Thialinoleic acid (2)
A flask was charged with tert-butyl ester 8 (20 mg) and purged with nitrogen. DME (2 mL) was added, followed by a 1 M LiOH solution (1 mL). The solution was stirred at 50 °C for 40 h, and then cooled to room temperature. The reaction mixture was acidified to a pH of 1 with 1 M HCl in water and extracted with diethyl ether (7 × 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (60% diethyl ether/pentane), yielding a clear oil.
The product was further purified by reverse-phase HPLC using a Varian Dynamax Microsorb 100-5 C18 column on a Rainin system (Dynamax model SD-200 pump and model UV-1 detector). Isocratic conditions were used as follows: 68% acetonitrile, 31.9% water, 0.1% acetic acid at a flow rate of 1 mL/min. Detection was performed at 210 nm. The desired product eluted at 40 min and after removal of the solvents, 2 (12 mg, 70%) was obtained as a clear oil. 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 7.0 Hz, 3 H), 1.29–1.44 (m, 14 H), 1.64 (m, 2 H), 2.12 (q, J = 7.1 Hz, 4 H), 2.35 (t, J = 7.6 Hz), 5.58 (m, 2 H), 6.00 (m, 2 H). HRMS (EI, M+) for C17H30O2S calculated 298.1967, found 298.1962. The purity of the compound was also checked using a reverse-phase silver-impregnated Varian ChromSpher 5 Lipids column (25 cm × 4.6 mm), which showed that only one isomer was present.
Synthesis of 14-thialinoleic acid
9-Hydroxy-nonanoic acid methyl ester (10)
A dry flask was charged with azelaic acid monomethyl ester, technical grade 85% (995.5 mg, 4.92 mmol) and purged with nitrogen. THF (30 mL) was added, and the solution cooled to −78 °C. A 1.0 M solution of BH3-THF in THF (5.4 mL, 5.41 mmol) was added dropwise. The reaction mixture was stirred at −78 °C for 30 min, and then warmed to room temperature. After an additional 2.5 h, the reaction was quenched with water and extracted with diethyl ether (5 × 30 mL). The combined organic layers were washed with water (1 × 30 mL), dried with MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (75% diethyl ether/pentane) yielding 10 (565 mg, 88%) as a clear oil. The yield was corrected based on the purity of the starting material. 1H NMR (400 MHz, CDCl3) 1.28–1.42 (m, 8 H), 1.52–1.64 (m, 4 H), 2.30 (t, J = 7.5 Hz, 2 H), 3.63 (t, J = 6.6 Hz, 2 H), 3.66 (s, 3 H). 13C NMR (125.6 MHz, CDCl3) 25.1 (CH2), 25.8 (CH2), 29.2 (CH2), 29.4 (CH2), 29.4 (CH2), 32.9 (CH2), 34.3 (CH2), 51.7 (CH2), 63.2 (CH2), 174.6 (Cq). HRMS (EI, M+) for C10H20O3 calculated 188.1413, found 188.1411. IR (cm−1): 3369 (broad), 2931, 2857, 1740, 1438.
9-Bromo-nonanoic acid methyl ester (11)
A dry flask was charged with triphenylphosphine (386.6 mg, 1.20 mmol) and purged with nitrogen. CH2Cl2 (10 mL) was added, and the solution cooled to 0 °C. Bromine (55 µL, 1.12 mmol) was added and the reaction mixture stirred for 20 min. Pyridine (150 µL, 1.36 mmol) was added and the reaction mixture was stirred an additional 20 min. A solution of the alcohol 10 (151.3 mg, 0.80 mmol) in CH2Cl2 (3 mL) was then added via cannulation. The reaction mixture was stirred at 0 °C for 2h, quenched with water and extracted with diethyl ether (5 × 30 mL). The combined organic layers were washed with water (1 × 20 mL), dried with MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (10% diethyl ether/pentane) yielding 11 (179 mg, 89%) as a clear oil. 1H NMR (500 MHz, CDCl3) 1.28–1.34 (m, 6 H), 1.38–1.45 (m, 2 H), 1.56–1.67 (m, 2 H), 1.81–1.87 (m, 2 H), 2.30 (t, J = 7.6 Hz, 2 H), 3.40 (t, J = 6.9 Hz, 2 H), 3.66 (s, 3 H). 13C NMR (125.6 MHz, CDCl3) 25.1 (CH2), 28.3 (CH2), 28.8 (CH2), 29.20 (CH2), 29.24 (CH2), 33.0 (CH2), 34.2 (CH2), 34.3 (CH2), 51.7 (CH3), 174.5 (Cq). HRMS (CI, M+) for C10H20O2Br calculated 251.0647, found 251.0645. IR (cm−1): 2931, 2856, 1740, 1461, 1436.
Phosphonium salt of 9-bromo-nonanoic acid methyl ester (12)
A dry flask was charged with bromide 11 (166.2 mg, 0.66 mol) and purged with nitrogen. Acetonitrile (4 mL) was added, followed by triphenylphosphine (521.7 mg, 1.98 mmol). The reaction was heated at 80 °C for 60 h, and then cooled to 25 °C. The solution was loaded directly onto a column impregnated with 50% diethyl ether/pentane to elute the excess triphenylphosphine. The mobile phase was then switched to 7% methanol/dichloromethane to elute 12 (314 mg, 95%) as a clear oil. 1H NMR (500 MHz, CDCl3) 1.10–1.22 (m, 6 H), 1.42–1.58 (m, 6 H), 2.16 (t, J = 7.6 Hz, 2 H), 3.54 (s, 3 H), 3.56–3.63 (m, 2 H), 7.60–7.65 (m, 6 H), 7.69–7.76 (m, 9 H). 13C NMR (125.6 MHz, CDCl3) 22.68 (CH2), 22.74 (CH2), 23.1 (CH2), 24.9 (CH2), 28.9 (CH2), 29.04 (CH2), 29.06 (CH2), 30.4 (CH2), 30.5 (CH2), 34.1 (CH2), 51.6 (CH3), 118.4 (d, J = 85.8 Hz, Cq), 130.7 (d, J = 12.5 Hz, CH), 133.8 (d, J = 9.7 Hz, CH), 135.3 (d, J = 2.8 Hz, CH), 174.4 (s, Cq). 31P NMR (202.3 MHz, CDCl3) 25.2 (s). HRMS (ESI, M-Br) for C28H34O2P calculated 433.2296, found 433.2297.
But-3-ynyloxy-triisopropyl-silane (14)
A flask was charged with 3-butynol (0.8 g, 11.4 mmol) and purged with nitrogen. Dichloromethane (20 mL) was added and the reaction was cooled to 0 °C. Imidazole (860 mg, 12.6 mmol) was added, followed by TIPSCl (2.42 g, 12.6 mmol). The reaction mixture was stirred at 0 °C for 3 h then slowly warmed to room temperature. The solution was washed with water (2 × 20 mL) and the aqueous layer was extracted with dichloromethane (2 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (10% diethyl ether/pentane) to yield 14 (2.45 g, 95%) as a clear oil. 1H NMR (500 MHz, CDCl3) δ 1.05 (m, 21 H), 1.95 (t, J = 2.7 Hz, 1 H), 2.43 (td, J = 2.7, 7.4 Hz, 2 H), 3.82 (t, J = 7.4 Hz, 2 H). 13C NMR (100.6 MHz, CDCl3) δ 12.2 (CH), 18.2 (CH3), 23.1 (CH2), 62.2 (CH2), 69.5 (CH), 81.7 (Cq). HRMS (EI, M+) for C13H27OSi calculated 227.1831, found 227.1829.
(4-Butylsulfanyl-but-3-ynyloxy)-triisopropyl-silane (15)
A flask was charged with protected alcohol 14 (497.5 mg, 2.19 mmol) and purged with nitrogen. THF (20 mL) was added and the reaction was cooled to −78 °C. A 1.6 M solution of n-butyllithium in THF (1.5 mL, 2.42 mmol) was added dropwise and the reaction mixture was stirred for 30 min. Butyl disulphide (490 µL, 2.74 mmol) was then added dropwise. The reaction mixture was stirred at −78 °C for 20 min then slowly warmed to room temperature. After an additional 19 h, the reaction was quenched with water and extracted with diethyl ether (4 × 30 mL). The combined organic layers were washed with water (1 × 30 mL), dried with MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (gradient from 100% pentane to 5% diethyl ether/pentane) yielding 15 (656 mg, 94%) as a clear oil. 1H NMR (500 MHz, CDCl3) δ 0.93 (t, J = 7.3 Hz, 3 H), 1.03–1.13 (m, 21 H), 1.42 (sextet, J = 7.4 Hz, 2 H), 1.66–1.72 (m, 2 H), 2.55 (t, J = 7.2 Hz, 2 H), 2.66 (t, J = 7.3 Hz, 2 H), 3.79 (t, J = 7.3 Hz, 2 H). 13C NMR (100.6 MHz, CDCl3) δ 12.2 (CH), 13.9 (CH3), 18.2 (CH3), 21.7 (CH2), 24.8 (CH2), 31.5 (CH2), 35.3 (CH2), 62.4 (CH2), 70.0 (Cq), 91.2 (Cq). HRMS (CI, M+) for C17H35OSiS calculated 315.2178, found 315.2179.
(4-Butylsulfanyl-but-3-enyloxy)-triisopropyl-silane (16)
A dry flask was charged with tantalum pentachloride (1.10 g, 3.13 mmol) under a nitrogen atmosphere. Benzene (10 mL) was added, followed by anhydrous dimethoxyethane (10 mL). Zinc powder (301.2 mg, 4.70 mmol) was added in one portion to the yellow solution and the reaction mixture was stirred at room temperature for 45 min. The reaction mixture turned a dark purple colour, and then pyridine (140 µL, 1.25 mmol) was added. After 5 min, the reaction mixture turned slightly red, and a solution of the alkyne 15 in a benzene:DME mixture (1:1 ratio, 6 mL) was added via cannulation. The reaction mixture was stirred under nitrogen for 1 h, and then pyridine (2.3 mL, 20.8 mmol) was added and stirred for 5 min. The reaction was quenched with 1 M NaOH in water (8 mL) and stirred vigorously for 25 min. The mixture was extracted with diethyl ether (4 × 50 mL), and then the combined organic layers were washed with water (1 × 20 mL), dried with MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (2% diethyl ether/pentane) yielding 16 (287 mg, 87%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, J = 7.3 Hz, 3 H), 1.03–1.12 (m, 21 H), 1.36–1.45 (m, 2 H), 1.56–1.63 (m, 2 H), 2.38 (dq, J = 1.3 Hz, 6.9 Hz, 2 H), 2.65 (t, J = 7.4 Hz, 2 H), 3.72 (t, J = 6.8 Hz, 2 H), 5.62 (td, J = 7.2 Hz, 9.5 Hz, 1 H), 5.98 (td, J = 1.4 Hz, 9.5 Hz, 1 H). 13C NMR (100.6 MHz, CDCl3) δ 12.2 (CH), 13.9 (CH3), 18.3 (CH3), 21.9 (CH2), 32.7 (CH2), 33.2 (CH2), 33.8 (CH2), 62.6 (CH2), 125.9 (CH), 126.9 (CH). HRMS (EI, M+) for C17H36OSiS calculated 316.2256, found 316.2263. IR (cm−1): 2944, 2866, 1727, 1716, 1464.
4-Butylsulfanyl-but-3-en-1-ol (17)
A dry flask was charged with protected alcohol 16 (106.6 mg, 0.34 mmol) and purged with nitrogen. THF (6 mL) was added, and the reaction was cooled to 0 °C. A 1.0 M solution of TBAF in THF (440 µL, 0.44 mmol) was added dropwise and the reaction mixture was stirred at 0 °C. After 90 min, the solvent was removed under vacuum and the resulting oil purified by flash chromatography (50% diethyl ether/pentane) yielding 17 (49 mg, 91%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, J = 7.3 Hz, 3 H), 1.36–1.45 (m, 3 H), 1.56–1.64 (m, 2 H), 2.41 (dq, J = 1.4 Hz, 6.5 Hz, 2 H), 2.67 (t, J = 7.4 Hz, 2 H), 3.70 (q, J = 6.3 Hz, 2 H), 5.58 (td, J = 7.3 Hz, 9.5 Hz, 1 H), 6.09 (td, J = 1.3 Hz, 9.5 Hz, 1 H). 13C NMR (100.6 MHz. CDCl3) δ 13.9 (CH3), 21.9 (CH2), 32.6 (CH2), 32.9 (CH2), 33.8 (CH2), 62.1 (CH2), 124.7 (CH), 128.7 (CH). HRMS (EI, M+) for C8H16OS calculated 160.0922, found 160.0924. IR (cm−1): 3350 (broad), 2958, 2929, 2874, 1608, 1465.
4-Butylsulfanyl-but-3-enal (18)
A dry flask was charged with Dess-Martin periodinane (75.5 mg, 0.18 mmol) and purged with nitrogen. CH2Cl2 (6 mL) was added, and the solution was cooled to 0 °C. A solution of alcohol 17 (28.1 mg, 0.175 mmol) in CH2Cl2 (2 mL) was added via cannulation. After 10 min, the reaction was slowly warmed to 25 °C and stirred for an additional 30 min. The reaction mixture was then diluted with pentane (75 mL) and washed with water (4 × 15 mL). The organic layer was dried with MgSO4, filtered and concentrated. The resulting oil 18 was used without further purification. 1H NMR (500 MHz, CDCl3) δ 0.91 (t. J = 7.3 Hz. 3 H), 1.36–1.44 (m, 2 H), 1.57–1.63 (m, 2 H), 2.70 (t, J = 7.3 Hz, 2 H), 3.28 (td, J = 1.5 Hz, 7.0 Hz, 2 H), 5.72 (td, J = 7.0 Hz, 9.5 Hz, 1 H), 6.26 (td, J = 1.4 Hz, 9.5 Hz, 1 H), 9.66 (t, J = 1.6 Hz, 1 H). 13C NMR (125.6 MHz, CDCl3) δ 13.8 (CH3), 21.8 (CH2), 32.6 (CH2), 33.9 (CH2), 44.1 (CH2), 117.5 (CH), 131.1 (CH), 200.0 (Cq). HRMS (EI, M+) for C8H14OS calculated 158.0765, found 158.0764.
Methyl 14-thialinoleate (19)
A dry flask was charged with phosphonium salt 12 (113.5 mg, 0.225 mmol) and purged with nitrogen. THF (10 mL) was added and the solution cooled to −78 °C. A 1.0 M solution of NaHMDS in THF (210 µL, 0.21 mmol) was added dropwise and the reaction mixture stirred for 1 h at −78°C. A solution of aldehyde 18 in THF (1 mL) was then added dropwise over 30 min. The reaction mixture was stirred at −78 °C for 2 h, then slowly warmed to 25 °C. After stirring an additional 16 h, the reaction was quenched with water (30 mL) and extracted with diethyl ether (4 × 40 mL). The combined organic layers were washed with water (1 × 20 mL), dried with MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (10% diethyl ether/pentane) yielding 19 (32 mg, 58% over 2 steps) as a clear oil. 1H NMR (500 MHz, CDCl3) δ 0.92 (t, J = 7.4 Hz, 3 H), 1.24–1.44 (m, 14 H), 1.52–1.64 (m, 6 H), 2.06 (q, J = 6.8 Hz, 2 H), 2.30 (t, J = 7.5 Hz, 2 H), 2.66 (t, J = 7.4 Hz, 2 H), 2.85 (t, J = 6.8 Hz, 2 H), 3.66 (s, 3 H), 5.32–5.43 (m, 2 H), 5.50 (td, J = 7.1 Hz, 9.3 Hz, 1 H), 5.92 (td, J = 1.5 Hz, 9.3 Hz, 1 H). 13C NMR (125.6 MHz, CDCl3) δ 13.9 (CH3), 21.9 (CH2), 25.1 (CH2), 27.4 (CH2), 27.7 (CH2), 29.27 (CH2), 29.32 (CH2), 29.35 (CH2), 29.8 (CH2), 32.6 (CH2), 33.8 (CH2), 34.3 (CH2), 51.7 (CH3), 125.6 (CH), 127.0 (CH), 127.7 (CH), 131.1 (CH), 174.6 (Cq). HRMS (EI, M+) for C18H32O2S calculated 312.2123, found 312.2124.
14-Thialinoleic acid (3)
A flask was charged with methyl ester 19 (7 mg, 0.02 mmol) and purged with nitrogen. DME (2 mL) was added, followed by a 1 M solution of LiOH in water (1 mL). The solution was stirred at room temperature for 40 h. The reaction mixture was acidified to a pH of 1 by adding 1 M HCl in water and extracted with diethyl ether (7 × 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated. The resulting oil was purified by flash chromatography (60% diethyl ether/pentane) to yield a clear oil.
The product was further purified by reverse-phase HPLC using a Varian Dynamax Microsorb 100-5 C18 column on a Rainin system (Dynamax model SD-200 pump and model UV-1 detector). Isocratic conditions were as follows: 70% acetonitrile, 29.9% water, 0.1% acetic acid at a flow rate of 1 mL / min. Detection was performed at 210 nm. The desired product eluted at 24 min and after removal of the solvents, 3 (4 mg, 60%) was obtained as a clear oil. 1H NMR (400 MHz, CDCl3) δ 0.91 (t, J = 7.3 Hz, 3 H), 1.25–1.45 (m, 10 H), 1.55–1.65 (m, 4 H), 2.06 (q, J = 6.5 Hz, 2 H), 2.35 (t, J = 7.6 Hz, 2 H), 2.66 (t, J = 7.5 Hz, 2 H), 2.85 (t, J = 6.9 Hz, 2 H), 5.33–5.44 (m, 2 H), 5.50 (td, J = 7.1, 9.4 Hz, 1 H), 5.93 (td, J = 1.5 Hz, 9.3 Hz, 1 H). HRMS (EI, M+) for C17H30O2S calculated 298.1967, found 298.1962. The purity of the compound was also checked using a reverse-phase silver-impregnated Varian ChromSpher 5 Lipids column (25 cm × 4.6 mm), which showed that only one isomer was present.
Lipoxygenase protein purification
For sLO-1, the expression and purification procedure developed by Holman and coworkers66 was used with modifications. Cells of BL21 Escherichia coli carrying the plasmid PT7-7/L1 VT-SLO-11 were grown in Luria-Bertani broth at 37 °C. When the OD600 reached 1, ethanol was added to give a 3% solution and the cells were cooled to 15 °C and shaken overnight. The cells were harvested and resuspended in 50 mL of 50 mM Bis-Tris buffer (pH 7.5) containing 10% glycerol, 0.1% Triton X-100, and 500 mM NaCl. The cells were lysed by sonication at 4 °C. The resulting mixture was centrifuged at 20000g for 20 min to remove cell remains. The supernatant was kept and dialyzed for 2 h versus 20 mM Bis-Tris buffer (pH 6.0). The solution was loaded onto an equilibrated SP-Sephadex column (120 mL), and the column was washed with buffer until the eluting solution did not contain any protein (A280 < 0.05). sLO-1 was then eluted with 500 mM NaCl in Bis-Tris buffer (pH 6.0). Activity assays were performed by adding aliquots of the fractions to LA and monitoring product formation at 235 nm. Fractions showing activity were combined and concentrated in an Amicon pressure concentrator to 35 mL. The solution was dialyzed versus 3.5 L of 20 mM Bis-Tris buffer (pH 6.0) overnight and centrifuged at 12000g for 15 min to remove a white precipitate that formed. The supernatant was loaded onto an equilibrated Macro-Prep column (25 mL). The column was washed with starting buffer and the protein was eluted with a step gradient of 500 mM NaCl and 20 mM Bis-Tris (pH 6.0). Activity assays were repeated as above and SDS-PAGE was performed to confirm the presence of a 94 kDa band. The fractions showing activity and containing purified protein were pooled and concentrated in an Amicon pressure concentration to a volume of 5 mL. The solution was diluted in borate buffer (100 mM, pH 10.0) with 30% glycerol before being concentrated again. The final solution was frozen in liquid nitrogen and stored at −80 °C. SDS-PAGE gel indicated greater than 90% purity.
12-hLO and 15-hLO-1 were expressed and purified as described previously.67 Briefly, these two enzymes contained hexa-His tags, and were purified in one step by Ni2+ affinity chromatography. The iron content of the lipoxygenase enzymes were determined on a Finnegan inductively coupled plasma mass spectrometer, using internal standards of cobalt(II)-EDTA, and the data were compared with those of standardized iron solutions. The two isozymes yielded approximately 50 mg/L of purified protein and were greater than 90% pure as judged by SDS-PAGE. As isolated, the human enzymes had the following iron content: 0.33 ± 0.02 equiv per 15-hLO-1, and 0.28 ± 0.03 equiv per 12-hLO. The enzymes were frozen at −80 °C, with glycerol added (20% for 12-hLO and 10% for 15-hLO-1) to prevent inactivation. All kinetic data reported herein were corrected for iron content by adjusting enzyme concentrations to reflect the active fraction containing metal.
HPLC purification of arachidonic and linoleic acids
Arachidonic acid and linoleic acid were purchased from Sigma-Aldrich (Milwaukee, WI). 11, 11-d2-Linoleic acid was synthesized as previously described.68, 69 All fatty acids were repurified by reverse phase HPLC on a Varian Dynamax Microsorb 100-5 C18 column (25 cm × 10 mm) prior to use in kinetic experiments to ensure that no auto-oxidation products were present. Solution A was 99.9% acetonitrile and 0.1% acetic acid, while solution B was 99.9% water and 0.1% acetic acid. For arachidonic acid, an isocratic elution of 82% A and 18% B was used; for linoleic acid and 11, 11-d2-linoleic acid, an isocratic elution of 88% A and 12% B was used. Both runs were monitored at 210 nm and the elution time was 14 min for arachidonic acid and 11 min for linoleic acid. In order to make sure that only the all-cis isomers were present, the purified compounds were analyzed on a silver-impregnated Varian ChromSpher 5 Lipids column (25 cm × 4.6 mm) at 210 nm using a solvent of 99% hexane and 1% acetonitrile. In all cases, only one peak was observed. The fatty acids were dissolved in ethanol and stored under argon at −80 °C.
Kinetic inhibition studies with thialinoleic acids
Kinetic assays were performed in 1 mL of borate buffer (100 mM, pH 10.0) or HEPES buffer (25 mM, pH 7.5). Both buffers were saturated with oxygen before the assays were performed. The concentrations of fatty acid solutions were determined by incubating the substrate with commercial soybean lipoxygenase (Sigma-Aldrich Chemical Company) and measuring the absorbance at 235 nm after the reaction was complete (ε = 23000 L mol−1 cm−1 for 13-HPODE and 27000 L mol−1 cm−1 for 15-HPETE).70, 71
The 13-HPODE solution used to activate sLO-1 and 15-hLO-1 was prepared by mixing linoleic acid (350 µM) and sLO-1 (10 nM) and allowing to react for 30 min at 0 °C. The solution was then filtered through a YM-30 Centricon microconcentrator (Millipore Corporation) to remove sLO-1. The concentration was determined by measuring the absorbance at 235 nm. The solution was checked for any remaining LA by adding fresh sLO-1 and monitoring at 235 nm to see if any more product was generated.
For sLO-1, the inhibitor and linoleic acid were combined in the buffer and the reaction was initiated by the addition of enzyme (4 nM). For sLO-1 reactions with arachidonic acid, 13-HPODE was added to a final concentration of 16 µM before the addition of enzyme. For 15-hLO-1, kinetic assays were performed for the reactions with linoleic acid and arachidonic acid. Substrate, inhibitor and 13-HPODE (7 µM) were combined in the buffer, then the reaction was initiated by the addition of enzyme (16 nM). Finally, for 12-hLO, the inhibitor and arachidonic acid were combined in the buffer and the reaction was initiated by the addition of enzyme (2 nM). The 12-hLO reaction was also monitored in the presence of 13-HPODE (16 µM) to see if the inhibition mode was altered.
Initial rates were measured on a Cary 100 Bio UV-visible spectrophotometer (Varian Inc.) by following product formation at 235 nm at room temperature. All kinetic parameters were determined by fitting the data to the Michaelis-Menten equation using KaleidaGraph 4.0 (Synergy Software). Double-reciprocal plots were fit to competitive and non-competitive models to obtain inhibition parameters using a Fortran program designed by W. W. Cleland.72 The parameters thus obtained for each inhibition model are shown in the Supplementary information.
KIE measurement of 15-hLO-1 in the presence of inhibitor
The kinetic isotope effect on kcat/Km was determined for the reaction of 15-hLO-1 with linoleic acid. Steady-state rates using protiated LA and 11, 11-d2-LA were compared at a variety of concentrations of 11-thiaLA. The concentrations of solutions of linoleic acid and 11, 11-d2-LA were determined by incubating the substrate with commercial sLO (Sigma-Aldrich Chemical Company) and measuring the absorbance at 235 nm after the reaction was complete. Assays were performed in 1 mL of O2-saturated HEPES buffer (25 mM, pH 7.5). The LA and 11, 11-d2-LA concentrations ranged from 1 to 12 µM, while the concentrations of 11-thiaLA used were 0, 15, 30 and 45 µM. The reaction was initiated by addition of 15-hLO-1 (22 nM for protiated substrate and 220 nM for deuterated substrate) and initial rates were measured. All kinetic parameters were determined by fitting the data to the Michaelis-Menten equation using KaleidaGraph 4.0 (Synergy Software).
HPLC analysis of product distributions
Authentic standards of 9-HODE, 13-HODE, 8-HETE, 11-HETE, 1 2-HETE and 15-HETE standards in ethanol were purchased from Cayman Chemical Company. Using an isocratic elution of 55% A/45% B afforded the separation of commercially obtained standards of 13-HODE, 15-HETE, 11-HETE, 8-HETE, 12-HETE. Under all the conditions tested, 8- and 12-HETE coeluted. The elution times were 35, 40, 45, and 50 min, respectively, for 13-HODE, 15-HETE, 11-HETE, and 8-HETE/12-HETE. 13-HODE was the corresponding alcohol to 13-HPODE, which was added as an activator in the kinetic experiments involving 15-hLO-1. For experiments on the regioselectivity of sLO-1, the separation of 9-HODE and 13-HODE required an isocratic elution of 52% A/48% B. In this case, the elution times were 82 and 84 minutes for 9-HODE and 13-HODE, respectively.
For each experiment, substrate (LA or AA; 15 µM) was combined with enzyme (12-hLO, 15-hLO-1 or sLO-1; 50 nM) in 1 mL of HEPES buffer (25 mM, pH 7.5). Assays were performed in the absence or presence of 11-thialinoleic acid (concentration of 5 × Ki). The reaction was monitored at 235 nm using a Cary UV-Vis spectrophotometer and reaction products were isolated after 15 min. When the reaction stopped, the substrate was not always entirely consumed in the case of 15-hLO-1 due to enzyme autoinactivation as previously reported.54 However, since HPLC analysis was performed at 235 nm, unreacted substrate did not interfere with product analysis because it did not absorb at that wavelength. In general, three 1 mL assays were performed; the products were pooled, acidified to pH 1 using 1 N HCl in water and extracted with dichloromethane (5 × 2 mL). The combined organic layers were dried by passing through a Pasteur pipette containing MgSO4 and concentrated under a steam of nitrogen gas. The HPODE or HPETE residue was reconstituted in dry tetrahydrofuran (120 µL).
The enzymes synthesize hydroperoxides, but only a subset of these are commercially available, whereas all of the corresponding alcohol standards are available. Therefore, the enzymatic products were reduced with sodium borohydride prior to analyzing the product mixture. An aliquot of hydroperoxide solution (20 µL) was reduced by adding a 0.1 M NaBH4 solution in dry tetrahydrofuran (10 µL) and allowing the reaction to proceed for 5 min at 0 °C. The reaction was quenched by adding 1 N HCl in water (10 µL) and waiting until gas evolution had ceased. The solution was analyzed by HPLC at 235 nm and the composition of the mixture was determined by comparison of peak areas.
To evaluate the potential consumption of the inhibitors, the mobile phase was switched to 80% A/20% B at 60 min when all of the HETE compounds had eluted. Under these conditions, a peak eluted at 72 min and was identified as 11-thiaLA based on co-injection with pure inhibitor.
Supplementary Material
Acknowledgements
This work was supported by pre-doctoral fellowships to CMM and CJ from the American Heart Association (#0310006Z and #0615604Z) and by the National Institutes of Health (GM44911).
Footnotes
Electronic Supplementary Information (ESI) available: See DOI: 10.1039/b000000x/
Notes and references
- 1.Funk CD. Nat. Rev. Drug. Disc. 2005;4:664–672. doi: 10.1038/nrd1796. [DOI] [PubMed] [Google Scholar]
- 2.Brash AR. J. Biol. Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- 3.Ford-Hutchinson AW, Gresser M, Young RN. Annu. Rev. Biochem. 1994;63:383–347. doi: 10.1146/annurev.bi.63.070194.002123. [DOI] [PubMed] [Google Scholar]
- 4.Kuehn H, O'Donnell VB. Prog. Lipid Res. 2006;45:334–356. doi: 10.1016/j.plipres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Connolly JM, Rose DP. Cancer Lett. 1998;132:107–112. doi: 10.1016/s0304-3835(98)00171-2. [DOI] [PubMed] [Google Scholar]
- 6.Kamitani H, Geller M, Eling T. J. Biol. Chem. 1998;273:21569–21577. doi: 10.1074/jbc.273.34.21569. [DOI] [PubMed] [Google Scholar]
- 7.Harats D, Shaish A, George J, Mulkins M, Kurihara H, Levkovitz H, Sigal E. Arterioscl., Thromb., Vasc. Biol. 2000;20:2100–2105. doi: 10.1161/01.atv.20.9.2100. [DOI] [PubMed] [Google Scholar]
- 8.Pratico D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ, Lee VMY. Am. J. Pathology. 2004;164:1655–1662. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao Y, Clark CM, Trojanowski JQ, Lee VMY, Pratico D. Ann. Neurol. 2005;58:623–626. doi: 10.1002/ana.20558. [DOI] [PubMed] [Google Scholar]
- 10.Gardner HW. Biochimica et Biophysica Acta. 1991;1084:221–239. doi: 10.1016/0005-2760(91)90063-n. [DOI] [PubMed] [Google Scholar]
- 11.Kuehn H, Schewe T, Rapoport SM. Adv. Enzymol. Rel. Areas Mol. Biol. 1986;58:273–311. doi: 10.1002/9780470123041.ch7. [DOI] [PubMed] [Google Scholar]
- 12.Kuehn H, Barnett J, Grunberger D, Baecker P, Chow J, Nguyen B, Bursztyn-Pettegrew H, Chan H, Sigal E. Biochim. Biophys. Acta. 1993;1169:80–89. doi: 10.1016/0005-2760(93)90085-n. [DOI] [PubMed] [Google Scholar]
- 13.Kuehn H. Prostaglandins Other Lipid Mediat. 2000;62:255–270. doi: 10.1016/s0090-6980(00)00084-8. [DOI] [PubMed] [Google Scholar]
- 14.Siedow JN. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1991;42:145–188. [Google Scholar]
- 15.McGinley CM, van der Donk WA. Chem. Commun. 2003:2843–2846. doi: 10.1039/b311008g. [DOI] [PubMed] [Google Scholar]
- 16.Nagel ZD, Klinman JP. Chem. Rev. 2006;106:3095–3118. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- 17.Schilstra MJ, Veldink GA, Vliegenthart JFG. Biochemistry. 1994;33:3974–3979. doi: 10.1021/bi00179a025. [DOI] [PubMed] [Google Scholar]
- 18.Lehnert N, Solomon EI. J. Biol. Inorg. Chem. 2003;8:294–305. doi: 10.1007/s00775-002-0415-6. [DOI] [PubMed] [Google Scholar]
- 19.Glickman MH, Wiseman JS, Klinman JP. J. Am. Chem. Soc. 1994;116:793–794. [Google Scholar]
- 20.Jonsson T, Glickman MH, Sun S, Klinman JP. J. Am. Chem. Soc. 1996;118:10319–10320. [Google Scholar]
- 21.Hwang C-C, Grissom CB. J. Am. Chem. Soc. 1994;116:795–796. [Google Scholar]
- 22.Glickman MH, Klinman JP. Biochemistry. 1995;34:14077–14092. doi: 10.1021/bi00043a013. [DOI] [PubMed] [Google Scholar]
- 23.Lewis ER, Johansen E, Holman TR. J. Am. Chem. Soc. 1999;121:1395–1396. [Google Scholar]
- 24.Clapp CH, Senchak SE, Stover TJ, Potter TC, Findeis PM, Novak MJ. J. Am. Chem. Soc. 2001;123:747–748. doi: 10.1021/ja0032071. [DOI] [PubMed] [Google Scholar]
- 25.Peng S, van der Donk WA. J. Am. Chem. Soc. 2003;125:8988–8989. doi: 10.1021/ja035977t. [DOI] [PubMed] [Google Scholar]
- 26.Knapp MJ, Klinman JP. Eur. J. Biochem. 2002;269:3113–3121. doi: 10.1046/j.1432-1033.2002.03022.x. [DOI] [PubMed] [Google Scholar]
- 27.Boyington JC, Gaffney BJ, Amzel LM. Science. 1993;260:1482–1486. doi: 10.1126/science.8502991. [DOI] [PubMed] [Google Scholar]
- 28.Steczko J, Minor W, Stojanoff V, Axelrod B. Protein Science. 1995;4:1233–1235. doi: 10.1002/pro.5560040620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prigge ST, Boyington JC, Gaffney BJ, Amzel LM. Proteins. 1996;24:275–291. doi: 10.1002/(SICI)1097-0134(199603)24:3<275::AID-PROT1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 30.Minor W, Steczko J, Stec B, Otwinowski Z, Bolin JT, Walter R, Axelrod B. Biochemistry. 1996;35:10687–10701. doi: 10.1021/bi960576u. [DOI] [PubMed] [Google Scholar]
- 31.Boyington JC, Gaffney BJ, Amzel LM. Adv Exp Med Biol. 1997;400A:133–138. doi: 10.1007/978-1-4615-5325-0_19. [DOI] [PubMed] [Google Scholar]
- 32.Gillmor SA, Villasenor A, Fletterick R, Sigal E, Browner MF. Nat. Struct. Biol. 1997;4:1003–1009. doi: 10.1038/nsb1297-1003. [DOI] [PubMed] [Google Scholar]
- 33.Skrzypczak-Jankun E, Amzel LM, Kroa BA, Funk MO., Jr Proteins. 1997;29:15–31. [PubMed] [Google Scholar]
- 34.Skrzypczak-Jankun E, Bross RA, Carroll RT, Dunham WR, Funk MO., Jr J. Am. Chem. Soc. 2001;123:10814–10820. doi: 10.1021/ja011759t. [DOI] [PubMed] [Google Scholar]
- 35.Skrzypczak-Jankun E, Borbulevych OY, Jankun J. Acta Crystallogr., Sect D: Biol. Crystallogr. 2004;D60:613–615. doi: 10.1107/S0907444904000861. [DOI] [PubMed] [Google Scholar]
- 36.Oldham ML, Brash AR, Newcomer ME. J. Biol. Chem. 2005;280:39545–39552. doi: 10.1074/jbc.M506675200. [DOI] [PubMed] [Google Scholar]
- 37.Segraves EN, Chruszcz M, Neidig ML, Ruddat V, Zhou J, Wecksler AT, Minor W, Solomon EI, Holman TR. Biochemistry. 2006;45:10233–10242. doi: 10.1021/bi060577e. [DOI] [PubMed] [Google Scholar]
- 38.Youn B, Sellhorn GE, Mirchel RJ, Gaffney BJ, Grimes HD, Kang C. Proteins. 2006;65:1008–1020. doi: 10.1002/prot.21182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neau DB, Gilbert NC, Bartlett SG, Dassey A, Newcomer ME. Acta Crystallogr., Sect F: Struct. Biol. Crystal. Commun. 2007;F63:972–975. doi: 10.1107/S1744309107050993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi J, Chon JK, Kim S, Shin W. Proteins. 2008;70:1023–1032. doi: 10.1002/prot.21590. [DOI] [PubMed] [Google Scholar]
- 41.Block E, Iyer R, Grisoni S, Saha C, Belman S, Lossing FP. J. Am. Chem. Soc. 1988;110:7813–7827. [Google Scholar]
- 42.Camargo AB, Marchevsky E, Luco JM. J. Agric. Food Chem. 2007;55:3096–3103. doi: 10.1021/jf063020e. [DOI] [PubMed] [Google Scholar]
- 43.Corey EJ, D'Alarcao M, Kyler KS. Tetrahedron Lett. 1985;26:3919–3922. [Google Scholar]
- 44.Corey EJ, D'Alarcao M, Matsuda SPT. Tetrahedron Lett. 1986;27:3585–3588. [Google Scholar]
- 45.Zhu Z, Funk MO., Jr Bioorg. Chem. 1996;24:95–109. [Google Scholar]
- 46.Flock S, Holmeide AK, Skattebol L. Synth. Commun. 2007;37:4005–4015. [Google Scholar]
- 47.McGinley CM, Jacquot C, van der Donk WA. Bioorg. Med. Chem. Lett. 2007;17:4049–4052. doi: 10.1016/j.bmcl.2007.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kataoka Y, Takai K, Oshima K, Utimoto K. Tetrahedron Lett. 1990;31:365–368. [Google Scholar]
- 49.Christopher J, Pistorius E, Axelrod B. Biochimica et Biophysica Acta, Enzymology. 1970;198:12–19. doi: 10.1016/0005-2744(70)90028-8. [DOI] [PubMed] [Google Scholar]
- 50.Jacquot C, Peng S, van der Donk WA. Bioorg. Med. Chem. Lett. 2008 doi: 10.1016/j.bmcl.2008.08.108. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haining JL, Axelrod B. J. Biol. Chem. 1958;232:193–202. [PubMed] [Google Scholar]
- 52.Haertel B, Ludwig P, Schewe T, Rapoport SM. Eur. J. Biochem. 1982;126:353–357. doi: 10.1111/j.1432-1033.1982.tb06787.x. [DOI] [PubMed] [Google Scholar]
- 53.Rapoport S, Haertel B, Schewe T, Kuehn H. FEBS Letters. 1986;202:202–206. doi: 10.1016/0014-5793(86)80687-1. [DOI] [PubMed] [Google Scholar]
- 54.Segraves EN, Holman TR. Biochemistry. 2003;42:5236–5243. doi: 10.1021/bi0273462. [DOI] [PubMed] [Google Scholar]
- 55.Vahedi-Faridi A, Brault P-A, Shah P, Kim Y-W, Dunham WR, Funk MO., Jr J. Am. Chem. Soc. 2004. 126:2006–2015. doi: 10.1021/ja0390855. [DOI] [PubMed] [Google Scholar]
- 56.Gan Q-F, Browner MF, Sloane DL, Sigal E. J. Biol. Chem. 1996;271:25412–25418. doi: 10.1074/jbc.271.41.25412. [DOI] [PubMed] [Google Scholar]
- 57.Schilstra MJ, Veldink GA, Verhagen J, Vliegenthart JFG. Biochemistry. 1992;31:7692–7699. doi: 10.1021/bi00148a033. [DOI] [PubMed] [Google Scholar]
- 58.Mogul R, Johansen E, Holman TR. Biochemistry. 2000;39:4801–4807. doi: 10.1021/bi992805t. [DOI] [PubMed] [Google Scholar]
- 59.Mogul R, Holman TR. Biochemistry. 2001;40:4391–4397. doi: 10.1021/bi002581a. [DOI] [PubMed] [Google Scholar]
- 60.Northrop DB. Annu. Rev. Biochem. 1981;50:103–131. doi: 10.1146/annurev.bi.50.070181.000535. [DOI] [PubMed] [Google Scholar]
- 61.Northrop DB. Biochemistry. 1975;14:2644–2651. doi: 10.1021/bi00683a013. [DOI] [PubMed] [Google Scholar]
- 62.Jacquot C, Wecksler AT, McGinley CM, Segraves EN, Holman TR, van der Donk WA. Biochemistry. 2008;47:7295–7303. doi: 10.1021/bi800308q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruddat VC, Whitman S, Holman TR, Bernasconi CF. Biochemistry. 2003;42:4172–4178. doi: 10.1021/bi020698o. [DOI] [PubMed] [Google Scholar]
- 64.Wang ZX, Killilea SD, Srivastava DK. Biochemistry. 1993;32:1500–1509. doi: 10.1021/bi00057a014. [DOI] [PubMed] [Google Scholar]
- 65.Henschke JP, Rickards RW. J. Label. Compd. Radiopharm. 1998;41:211–220. [Google Scholar]
- 66.Holman TR, Zhou J, Solomon EI. J. Am. Chem. Soc. 1998;120:12564–12572. [Google Scholar]
- 67.Amagata T, Whitman S, Johnson TA, Stessman CC, Loo CP, Lobkovsky E, Clardy J, Crews P, Holman TR. J. Nat. Prod. 2003;66:230–235. doi: 10.1021/np020462l. [DOI] [PubMed] [Google Scholar]
- 68.Tucker WP, Tove SB, Kepler CR. J. Labelled Compd. 1970;7:11–15. [Google Scholar]
- 69.Peng S, McGinley CM, van der Donk WA. Org. Lett. 2004;6:349–352. doi: 10.1021/ol0361711. [DOI] [PubMed] [Google Scholar]
- 70.Gibian MJ, Vandenberg P. Anal. Biochem. 1987;163:343–349. doi: 10.1016/0003-2697(87)90234-x. [DOI] [PubMed] [Google Scholar]
- 71.Guajardo MH, Terrasa AM, Catala A. Biochim. Biophys. Acta. 2002;1581:65–74. doi: 10.1016/s1388-1981(02)00121-x. [DOI] [PubMed] [Google Scholar]
- 72.Cleland WW. Meth. Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.