

Abstract
Cloretazine [1, 2-bis(methylsulfonyl)-1-(2-chloroethyl)-2-[(methylamino)carbonyl]-hydrazine; VNP40101M; 101M] is a relatively new prodrug with activity in elderly acute myelogenous leukemia patients. Its therapeutic action is due largely to the production of 1-(3-cytosinyl),2-(1-guanyl)ethane cross-links (G-C ethane cross-links) in DNA. The number of cross-links produced in three experimental leukemia lines (L1210, U937 and HL-60) were fewer than 10 per genome at their respective LC50 concentrations. Only 1 in approximately 20,000 90CE molecules produce a cross-link in the AGT (O6-alkylguanine-DNA alkyltransferase) negative L1210 and U937 cell lines and 1 in 400,000 in the AGT positive HL-60 cell line.
Keywords: Cloretazine, 90CE, BCNU, leukemia cell lines, O6-alkylguanine-DNA alkyltransferase, G-C ethane cross-links
1. Introduction
Cloretazine [1,2-bis(methylsulfonyl)-1-(2-chloroethyl)-2-[(methylamino)carbonyl]-hydrazine; VNP40101M; 101M], is a relatively new alkylating agent designed and synthesized in our laboratory. It is a prodrug, which upon base catalyzed decomposition, generates 90CE [1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazine], a second short-lived prodrug (t1/2 ~ 30 s at pH 7.4 and 37°C) of hard chloroethylating intermediates [1]. The mechanism by which 90CE alkylates and cross-links DNA is illustrated in Figure 1A. Earlier studies in in vitro model systems indicated that only a very small fraction of the 90CE molecules interact with DNA in aqueous buffered solutions to generate cross-links [2]. This led us to speculate that very few cross-links would be generated at therapeutic drug levels in intact cells.
Figure 1.

Scheme illustrating the decomposition of 90CE in neutral aqueous medium to generate chloroethylating species which interact with water, biomolecules and DNA. A. Decomposition of 90CE to generate chloroethylating species. B. Generation of a G-C ethane cross-link from a normal GC base pair.
Alkylating drugs generate highly reactive electrophiles which covalently bind to electron rich biological nucleophiles. DNA is considered the target responsible for their antineoplastic activity [3,4]. Alkylating agents exhibit major differences in their preferred sites of attack and the degree of alkylation within DNA. These differences have resulted in additive and synergistic antineoplastic interactions between different antitumor alkylating agents [5], emphasizing the existence of significant differences in the modes of action of compounds of this class.
The lethality of unrepaired DNA modifications in neoplastic cells appears to be highly dependent upon the nature of the modification, with DNA single strand modifications generally being far less lethal than covalent DNA-DNA interstrand cross-links. The O-6 chloroethylation of DNA guanine leads to the eventual formation of a covalent DNA-DNA interstand cross-link [6]. After O-6 chloroethylation of guanine, an intramolecular nucleophilic substitution reaction results in the formation of N1,O6-ethanoguanine; this product then reacts with the N-3 position of an opposing cytosine to form a 1-(N3-deoxycytidinyl)-2-(N1-deoxyguanosinyl)ethane cross-link (G-C ethane cross-link). This appears to be the primary therapeutic lesion produced by Cloretazine [1]. This agent, on the basis of early indications of activity in relapsed and refractory acute myelogenous leukemia (AML) patients, has received fast track and orphan drug designations from the FDA.
The G-C ethane cross-link is the smallest known alkyl DNA interstrand cross-link, with the central ~3Å N-H···N hydrogen bond of the three involved in normal GC base pairs being replaced by a ~4.5Å (in linear configuration) -CH2-CH2- ethylene bridge. It is therefore possible that the small and internal nature of this cross-link may make it more difficult for cells to recognize and repair than cross-links involving more bulky and peripheral lesions between diagonally facing bases.
The repair protein AGT [O6-alkylguanine-DNA alkyltransferase] can react with both O6-(2-chloroethyl)guanine and N1,O6-ethanoguanine DNA cross-link precursor products of 90CE alkylations to prevent G-C ethane cross-link formation [7]. However, once the G-C ethane cross-link, is formed, repair by AGT cannot occur [8]. Each molecule of AGT can only repair a single O-6 guanine lesion, and decreased AGT activity is common in some tumor types where cytotoxicity differentials exist between the host and tumor due to reduced repair of O-6 guanine lesions in malignant cells [6].
90CE is arguably the best example of an agent that owes its cytotoxic activity to initial chloroethylation of the O-6 position of guanine, since a ~22-fold differential was observed between the clonogenic LC90 concentration in L1210 cell lines expressing and not expressing AGT; whereas, only a ~2-fold differential in the LC90 occurred with BCNU [9, 10], a representative of the nitrosoureas whose primary mechanism of action is also believed to be the chloroethylation of the O-6 position of guanine, even though in excess of 75% of the base alkylations produced by BCNU are on the N-7 position of guanine [9–11]. The ~30 s half-life of 90CE [12] indicates that the chloroethylation of DNA will be completed in cell lines before significant DNA-DNA cross-link formation, cellular intervention, or metabolism of this agent occurs. These properties make 90CE the chloroethylating agent of choice for estimating the number of G-C ethane cross-links required for lethality. Wild-type L1210 and U937 cells lack AGT [9, 13], while HL-60 cells express this protein [14]. Wild-type L1210 cells, however, are not considered defective in cross-link repair [15]. Never-the-less, the lack of AGT in L1210 and U937 cells should guarantee the unimpaired progression of any O6-chloroethylguanine lesion to form a cross-link. In HL-60 cells, the basal AGT level is sufficient to impart an approximate 18-fold resistance to 90CE in cell growth assays, based upon the sensitizing effects of O6-benzylguanine, a highly selective irreversible AGT inhibitor [7].
In this report we have measured the number and yield of G-C ethane cross-links produced per cell genome by 90CE, the initial active species generated by Cloretazine, in three pseudodiploid cell lines; wild-type L1210, U937 and HL-60 leukemia cells at their respective LC50 concentrations. This information is important to our understanding of the action of Cloretazine in AGT expressing and non-expressing cells. Furthermore, the 90CE moiety is a component of a number of targeted strategies [12], so that information concerning the yield and lethality of the delivered lesion is an important consideration. The findings are of relevance to other hard chloroethylating species prodrugs such as the chloroethylnitrosoureas which also generate the same ultimate lesion.
2. Materials and Methods
2.1. Chemicals
T7 DNA and other chemicals were purchased from the Sigma Chemical Company, St. Louis, MO, except where specified. Hoechst 33258 was obtained from Molecular Probes, Inc., Eugene, OR. T7 DNA was employed because it is a long linear dsDNA of known size (37,900 bp; molecular weight 25 × 106 daltons) devoid of single-chain interruptions [16]. 90CE was synthesized as previously described [17].
2.2. Growth of murine L1210 and human U937 and HL-60 cells
All leukemia cell lines were grown in suspension culture in RPMI 1640 medium supplemented with 10% FBS in air/5% CO2 at 37° C. The cells were subcultured as required every 2–3 days.
2.3. Treatment of L1210, U937 and HL-60 cells with 90CE and DNA isolation
Two × 106 L1210 cells were placed in a 1.5 ml tube and treated with the desired concentration of 90CE dissolved in DMSO (1 μl/ml of medium). Cells were incubated for 5 min at 37° C and DNA was then isolated using a Puregene DNA isolation kit (Gentra Systems, Minneapolis, MN) using procedures recommended by the manufacturer.
2.4. Cell growth inhibition assays
Cell growth assays were carried out in 24-well plates in a volume of 1 ml/well as previously described [10]. Briefly, cells were seeded at an initial density of 7 × 104 cells/ml and 1 μl of drug solution made in DMSO to give various concentrations was added to each well. The plate was shaken by hand at each addition to disperse the drug solution into the cell suspension. This practice was important to achieve consistent results because of the short half-life of 90CE. After incubation for 3 days, cell numbers were determined using a Coulter counter with a Multisizer II analyzer. DMSO at the final concentration of 0.1% used had no effect on cellular growth.
2.5. Determination of the L1210 leukemia clonogenic LC50 value for 90CE
For clonogenic assays, cells in the exponential phase of growth were concentrated to a density of 2 × 106 cells/ml in medium supplemented with 25 mM Hepes buffer, pH 7.4. One μl of 90CE containing solution was added to 1 ml of cell suspension and incubations were carried out in a water bath at 37oC. After exposure to 90CE, 2 to 250 μl of cell suspension containing the predetermined number of cells that yielded 10 to 60 colonies/well for each treatment were mixed with 20 ml of RPMI 1640 medium containing 0.8% methylcellulose and 15% FBS, divided into 3 wells in 6-well plates, and incubated for 8 days to allow for colony formation.
2.6. Treatment of isolated DNA with 90CE to generate mono-adduct cross-link precursors
To aliquots of T7 or leukemia cell DNA (25–100 μg/ml) in 10 mM Tris-HCl buffer containing 1 mM EDTA at pH 8.0, 10 μl/ml of the appropriate concentration of 90CE in DMSO was added with very rapid mixing. This mixture was then incubated for 5 min at 37°C to allow the chloroethylation reaction to go to completion.
2.7. Conversion of DNA mono-adducts to cross-links
DNA (T7 or leukemia cells), containing the cross-link precursor mono-adducts O6-(2-chloroethyl)guanine and N1,O6-ethanoguanine, was incubated in small sealed tubes for 12 to 15 h at ~37°C or 2 h at ~50°C to allow the mono-adducts to complete the intramolecular G-C cross-linking reaction.
2.8. Determination of DNA cross-links
The extent of DNA cross-linking was determined using a modification of the DNA renaturation assay previously described [18]. The assay is based upon the observation that under conditions of neutral or mildly alkaline pH snap cooling causes thermally denatured, covalently cross-linked DNA to rapidly renature, yielding a highly fluorescent complex with H33258 dye; whereas, DNA devoid of cross-links does not. A small aliquot (10 μl) of T7 or leukemia cells DNA (100 μg/ml unless otherwise stated) in 10 mM Tris-HCl buffer containing 1 mM EDTA at pH 8.0 was added to 1.5 ml of 5 mM Tris-HCl/1.0 mM EDTA/1.0 mM NaN3 buffer (pH 9.0) containing 0.1 μg/ml of H33258, heated to 100°C for 3 min, then plunged into a water bath at room temperature for 3 min. Fluorescence measurements were taken before the heating phase and after the 3 min chill using a Hoefer Scientific Instruments TKO 100 fluorometer, and the percentage of the DNA molecules that were cross-linked (i.e., that contained at least one cross-link per DNA molecule) was calculated as previously described [18]. The average number of cross-links per DNA molecule (A) was calculated from the cross-linked fraction, assuming a Poisson distribution such that A =-ln(1−X), where X = the cross-linked fraction. For a cross-linked fraction of 0.4 this calculates to be 0.51. The probability that a given DNA molecule has N cross-links equals e−A.AN/N!; where N! equals factorial N. In this case approximately 60% of the DNA molecules would have 0 cross-links, 30.6% with 1 cross-link; 7.8% with 2; 1.3% with 3; etc, to give a total of 40% of the DNA molecules containing one or more cross-links, with the average number of cross-links per DNA molecule being 0.51.
3. Results
3.1. Relationship between DNA cross-linking and concentrations of 90CE and DNA
Treatment of various concentrations (25 to 100 μg/ml) of T7 DNA (37.9 kbp; molecular weight 25 × 106 daltons; 48% GC [19]) with a range of concentrations of 90CE up to 200 μM gave a 90CE concentration dependent increase in the level of cross-linking (Figure 2A) that was independent of the concentration of DNA (Figure 2B). The molar yield of cross-links produced under these conditions was readily calculated from the fractional cross-linking value (i.e., the fraction of DNA molecules containing one or more cross-links per molecule) by assuming a Poisson distribution (Figure 2C, 2D). The relatively rapid reaction with water present at 55.5 M consumes the majority of the hard chloroethylating species and controls the concentration × time exposure of DNA molecules to chloroethylation. (‘Hard’ and ‘soft’ are chemical terms used in the description of electrophiles and nucleophiles. Although these are somewhat imprecise terms, the concept of “hard” versus “soft” is used to predict, on a qualitative level, how nucleophiles and electrophiles might interact with each other.) A linear dependency of the calculated yield of cross-links per unit length of DNA as a function of the concentration of 90CE was obtained (Figure 2D). This proportionality allowed the calculation of the yield of cross-links per unit length of DNA at low concentrations of 90CE where this could not be directly measured. From the gradient in Figure 2D, it can be seen that 90CE generated 0.0024 cross-links per T7 DNA molecule for each micromolar of 90CE concentration; this equates to 6.3 × 10−5 cross-links per kbp of DNA for each micromolar of 90CE concentration. The yield of cross-links would be expected to be dependent upon the GC content of DNA, since it is the GC concentration that is important, rather than the concentration of DNA per se. Since the GC content of mammalian DNA is marginally lower than that of T7 DNA (48% GC T7 DNA, ~ 42% GC murine and human DNA) [20–22], a reasonable figure to use as the cross-link yield in L1210, U937 and HL-60 cell DNA would be 5.5 × 10−5 cross-links per kbp for each micromolar of 90CE concentration.
Figure 2.
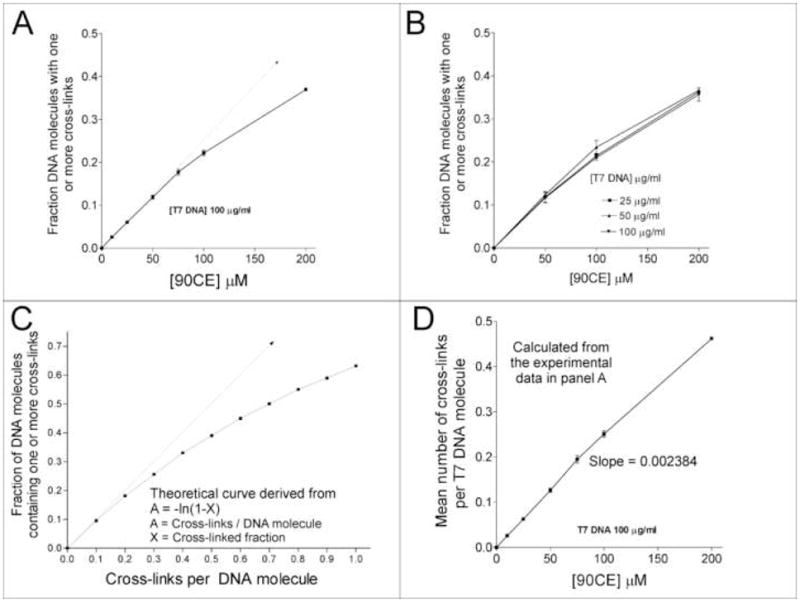
The relationships between the fractional cross-linking of a population of T7 DNA molecules, the number of cross-links per DNA molecule, and 90CE/DNA concentrations. A. The fractional cross-link of a population of T7 DNA molecules versus the concentration of 90CE. The dotted arrow is provided to highlight the expected divergence from linearity. B. Independence of the fractional cross-linking of a 90CE treated population of T7 DNA molecules on the T7 DNA concentration. C. The theoretical relationship between the cross-linked fraction of a population of DNA molecules versus the average number of cross-links per DNA molecule assuming a Poisson distribution. At higher levels of cross-linking the population of molecules with multiple cross-links increases, resulting in a divergence from the initial linear relationship between the cross-linked fraction and the number of cross-links per DNA molecule. D. The calculated mean number of cross-links per T7 DNA molecule versus the concentration of 90CE based on the data in panel A.
3.2. The effects of the intra-cellular environment on DNA cross-linking by 90CE
To obtain an approximate value for the cross-link yield in nuclear DNA of intact leukemia cells following treatment with 90CE, the degree of cross-linking achieved in DNA in solution isolated from L1210, U937 and HL60 cells, treated in the same buffer and under the same conditions of concentration used for T7 DNA, was compared to the level of cross-linking found when intact cells in culture medium were exposed to an identical concentration of 90CE for 5 min at 37°C. DNA from each of these leukemia cell lines was then isolated and incubated at 50°C for 2 h to convert cross-link precursors to G-C cross-links. In these experiments, the short t1/2 of 90CE at 37°C was important, since it allows the extraction of the cellular DNA promptly after drug treatment. The rapid isolation (within 5 min of exposure to 90CE) ensures that the DNA was not significantly modified by DNA damage surveillance and repair enzyme complexes. Upon isolation, under the conditions employed, the DNA contained essentially only mono-adduct cross-link precursors. Therefore, the DNA was incubated for 2 h at 50°C, which was equivalent to that of DNA exposed to 90CE following isolation, to allow the cross-link precursors contained in the DNA to complete the formation of the G-C cross-links. These experiments in essence measured the shielding of the DNA by cellular components, allowing the factoring of variables that would otherwise be difficult to quantify, as a single correction factor. The “cellular shielding factor” was very similar for all three leukemia cell lines, amounting to a 7- to 8-fold decrease in DNA cross-link precursor formation within intact cells compared to naked DNA under our standard conditions (Table 1).
Table 1.
Cell shielding factors for L1210, HL-60 and U937 leukemia cells.
| Leukemia | Genome size (number of GC base pairs) | Cellular shielding factor |
|---|---|---|
| L1210 | 2.5 × 109 | 7.4 |
| U937 | 3.2 × 109 | 8.0 |
| HL-60 | 3.2 × 109 | 6.9 |
The cellular shielding factor is the ratio of the G-C ethane cross-link yield in nuclear DNA of intact leukemia cells treated with 90CE to the G-C ethane cross-linking achieved in isolated L1210, U937 and HL-60 DNA in free solution under defined conditions.
3.3. Number of G-C ethane cross-links in L1210 leukemia cells required to produce lethality from 90CE
The LC50 value of 90CE measured by clonogenic assay using murine L1210 cells was determined to be 0.41 μM (Table 2); at this concentration 3.0 × 10−6 G-C ethane cross-links were estimated to be generated per kbp of cellular L1210 DNA. The murine genome is approximately 2.5 × 106 kbp [22]; therefore, only 7.5 G-C ethane cross-links would be expected to be generated per L1210 cell DNA genome following exposure to the LC50 of 90CE. The overall yield of cross-links generated in the nuclear DNA for all three cell lines is given in Table 3.
Table 2.
Properties of the three leukemia cell lines.
| Cell type | GC content | AGT status | 90CE IC50 value (Growth inhibition at 3 days) | 90CE LC50 value |
|---|---|---|---|---|
| L1210 | 42% | 0 | 6.5 μM | 0.41 μM |
| U937 | 42% | 0 | 5 μM | †0.32 μM |
| HL-60 | 42% | + | 91 μM, *5 μM in presence of O6BG |
†5.74 μM †0.32 μM in presence of O6BG |
The clonal LC50 value cannot be measured directly for HL-60 and U937 cells so it was calculated, assuming that HL-60 and U937 cell lines display the same relationship between their IC50 values and their clonogenic LC50 values for 90CE as L1210 leukemia cells.
Pretreatment with 20 μM O6BG (O6-benzylguanine) for 1 hour was used to inhibit the AGT present in HL-60 leukemia cells.
Table 3.
The calculated number of G-C ethane cross-links generated per genome of DNA in each cell line at concentrations of 90CE corresponding to their respective LC50 values.
| Leukemia | Average number of G-C ethane cross-links per cell genome at 90CE LC50 values |
|---|---|
| L1210 | 7–8 |
| U937 | 6–8 |
| HL-60 | 7–9 |
3.4. Number of G-C ethane cross-links in HL-60 and U937 leukemia cells required to produce lethality from 90CE
Unlike the L1210 leukemia, the human U937 and HL-60 leukemias did not form colonies in soft agar; as a consequence it was not possible to perform clonogenic assays directly on these cell lines to determine their LC50 values. Therefore, to estimate the clonogenic LC50 value for these cells, their 50% cell growth inhibition values (IC50) were measured; it was then assumed that these cell lines would have displayed the same relationship between their IC50 values and, if measurable, clonogenic LC50 values for 90CE, as did L1210 leukemia cells. The calculated value for an estimated clonogenic LC50 value was 0.32 μM for both HL-60 in the presence of O6BG (O6-benzylguanine) (pretreatment with 20 μM O6BG for 1 hour) to irreversibly inhibit the AGT activity and U937 cells. Using a value of 3.2 × 106 kbp for the human genome [20] and shielding factors of 6.9 and 8.0 for HL-60 and U937 cells, respectively, 8.2 and 7.1 cross-links per genome were calculated to be generated on average in HL-60 and U937 cells, respectively, at their estimated LC50 values.
3.5. Calculation of the yield of G-C ethane cross-links from 90CE within intact L1210 cells
If the cellular volume of a typical L1210 cell (10.45 μm cellular diameter) is 6.0 × 10−13 liters [23], at the LC50 of 0.41 μM 90CE (assuming full equilibration of 90CE where the LC50 concentration equals the intracellular concentration), there would be 1.5 × 105 90CE molecules in this volume. Therefore, within an L1210 cell only one in 20,000 90CE molecules will give rise to a G-C ethane cross-link. It is possible, however, that the short t1/2 of 90CE results in insufficient time for equilibrium of intracellular and extracellular concentrations of this agent to occur. Poor equilibration would, result in an over-estimate of the number of 90CE molecules required to generate a single G-C ethane cross-link within each cell, since this calculation assumes that the intracellular level of 90CE is equivalent to the external concentration. However, this factor would not result in errors in the calculation of the number of G-C ethane cross-links required for lethality, since this is taken into account in the “cellular shielding factor”. Since, similar cellular dimensions have been reported for HL-60 and U937 cells [24, 25] and these cell lines have similar estimated LC50 values and calculated cross-link numbers at the LC50 values (Tables 2 and 3), similar low cross-link yields would also occur in these cell lines.
4. Discussion
A variety of antineoplastic agents are capable of targeting the O-6 position of DNA guanine, and this action is considered to be primarily responsible for their anticancer effects. They include the 1,2-bis(sulfonyl)hydrazines, exemplified by Cloretazine, and the nitrosoureas, carmustine (BCNU) and lomustine (CCNU), which produce 2-chloroethylation of the O-6 position of DNA guanine, as well as temozolomide, procarbazine and dacarbazine (DTIC), which methylate the O-6 position of guanine in DNA. The biochemical consequences of methylation and 2-chloroethylation of the O-6 position of DNA guanine differ in important aspects. The O6-methylguanine lesion does not undergo spontaneous rearrangements and it reacts relatively rapidly with AGT, resulting in mutual titration [26]. Therefore, the O6-methylguanine lesion is likely to persist in DNA only if the number of methylations exceeds those of AGT molecules. If unrepaired, methylation lethality appears to be largely due to the “mismatched repair machinery”, which initiates apoptosis due to failed repair [27]. Thus, if a cell is “mismatch repair deficient” or somehow replication proceeds without repair, a point G to A mutation can result [28]. Point mutations are unlikely to be lethal unless a large number occur [29]. In contrast, O6-chloroethylguanine undergoes a spontaneous rearrangement to produce the highly lethal G-C ethane cross-link which cannot be repaired by AGT [11]. Furthermore, the initial lesion generated by 90CE is likely to be a poorer substrate for AGT than the O6-methylguanine lesion [30], which has an in vivo t1/2 in rat liver of 47 min [26]. Thus, for a cell to survive, the AGT level must be large enough to produce a repair rate that clears the G-C ethane cross-link precursor lesions before their transition to a lethal number of cross-links (<10). With methylation, mutual titration presumably occurs with the manifestation of cytotoxicity being coupled to exceeding the exhaustion of AGT by the lethal number of methyl lesions, which can amount to thousands of additional lesions [29]. In contrast, for resistance to 2-chloroethylation of the O-6 position of DNA guanine to occur, an excess of AGT relative to the number of O-6 guanine lesions is required to produce the repair rate necessary to prevent the accumulation of the relatively few G-C ethane cross-links required for lethality. In essence, two competing reactions for the fate of the initial alkylation lesions occur, progression to cross-links, the rate of which is determined by chemical kinetics, and AGT mediated repair, the rate of which is determined by the AGT concentration. Thus, chloroethylation should be lethal, despite a cell having many more AGT molecules than O-6 guanine lesions; furthermore, little net AGT depletion may occur at lethal 90CE concentrations. In support of this contention, HL-60 cells contain approximately 20,000 AGT molecules per genome of DNA (11 fmol of AGT/μg of cellular DNA) [14], yet would have fewer than 200 O-6 guanine position G-C ethane cross-link precursors per genome, based upon our calculations, after exposure to an LC50 concentration of 90CE (~6 μM). This level of AGT imparts an 18-fold level of resistance to 90CE, presumably by intercepting 95% of the precursor lesions before they have progressed to G-C ethane cross-links. Under such conditions only one net G-C ethane cross-link would be produced per ~400,000 90CE molecules entering the cell. These predictions and observations are consistent with findings in patients receiving therapeutic doses of Cloretazine, where no measured depletion in AGT activity occurred at any dose level of this agent [31], yet low levels of AGT were predictive of response [32]. These findings contrast with those of the guanine O-6 methylating drug temozolomide, where large decreases in the activity of AGT were observed at therapeutic doses [33]. The requirement to exceed the AGT molar activity by a significant excess of the number of DNA methylations to produce cytotoxicity in the case of the methylating agents, while cytotoxicity can be produced by the generation of O-6 guanine chloroethylations equivalent to a small fraction of the AGT molar activity (~1% in the case of HL-60 cells) may have important therapeutic consequences with respect to dose modulation and sequence when patients are co-treated with both types of agent.
Speculation as to the number of BCNU derived G-C DNA cross-links required for lethality has been reported previously (see for example [6]); however, we could not find published details of how these estimates were obtained. The lethality of an interstrand G-C ethane cross-link was estimated in the present study using a fluorescence based assay developed in our laboratory [18]. A single interstrand G-C ethane cross-link in a DNA molecule results in rapid renaturation upon cooling of the entire molecule after thermal denaturation because the strands are covalently held in register. The yield of G-C ethane cross-links per kbp of DNA can then be calculated from the proportion of this rapidly renaturing sub-population if the test DNA molecule is of known length (assuming a Poisson distribution of G-C ethane cross-links). The experimentally determined relationship between the concentrations of DNA and 90CE for cross-link formation is shown in Figure 2 and closely match this model.
The method used to estimate the number of G-C ethane cross-links required for lethality in intact cells is based upon simple measurements and assumptions. Thus, it is assumed that 90CE reacts approximately equivalently with all “naked” DNA other than for a minor correction resulting in a 12% decrease in the final estimation being made for differences in the G-C content.
To relate the G-C ethane cross-linking yield under these defined conditions using naked DNA to those within intact leukemia cells in culture medium, the relative yield of G-C ethane cross-links generated in leukemia cell DNA under two sets of conditions was compared. While it is realized that the calculations presented are dependent upon a number of assumptions, they provide an indication of the high lethality of the G-C ethane cross-link and its initial precursor lesions in AGT deficient cells, where the repair of cross-link precursors is presumably negligible. It should be noted that the rapid isolation of DNA is expected to allow little AGT dependent repair of the initial cross-link precursor lesions in the AGT positive HL-60 cell line.
A primary assumption is that the major cytotoxic event is indeed the production of G-C ethane cross-links formed from the chloroethylation of the O-6 position of DNA guanine, and that chloroethylation of other molecules or other sites in DNA are relatively insignificant in terms of cytoxicity. This conclusion is reasonable, since the stable expression of AGT in L1210 leukemia cells, whose only known function is the repair of alkylations at the O-6 position of DNA guanine, results in a 22-fold increase in the degree of resistance of L1210 cells to 90CE [9]. In addition, the inhibition of AGT intrinsically expressed in HL-60 cells by O6-BG decreases its IC50 to 90CE by 18-fold (Table 2). Furthermore, the generation of G-C ethane cross-links appears to be the major cytotoxic lesion generated by BCNU, a less specific chloroethylating agent of the O-6 position of DNA guanine [8]. It is also assumed that the yield of G-C ethane cross-links from cross-link precursors is the same within intact cells and in in vitro treated isolated DNA. If the yield of G-C ethane cross-links is lower in the cellular environment, the number of precursor lesions required for lethality would be reduced by this factor.
A further assumption is that pretreatment with 90CE 5 min prior to DNA extraction does not grossly alter the size and size distribution of the extracted DNA. This is important because in the assay employed the entire DNA molecule renatures, binds dye and becomes fluorescent if it contains one or more cross-links. The larger the mean size of the DNA pieces, the greater the yield of double-stranded fluorescent DNA per cross-link upon renaturation. Thus, to accurately compare the relative levels of G-C ethane cross-linking of leukemia cell DNA treated with 90CE before extraction from cells with DNA samples treated after extraction by this technique, they are required to be of similar size and size distribution. This is a reasonable assumption because the DNA was extracted identically in all cases and very rapidly (5 min after 90CE treatment), which should preclude changes in the mean size of the DNA molecules as a result of the introduction of DNA nicks in response to DNA damage by the cell or by chemical nicking. It should also be noted that 90CE is both a poor chemical DNA nicking agent even over much longer time periods [1] and is a poor inducer of apoptosis, even at many times the lethal concentration of this agent [34, 35].
Acknowledgments
This research was supported in part by U.S. Public Health Service Grants CA-090671 and CA-122112 from the National Cancer Institute. Authors Penketh, Baumann, Ishiguro and Shyam contributed directly to the experimental work required for this manuscript. In addition authors Penketh, Baumann, Ishiguro, Shyam, Seow and Sartorelli contributed to the interpretation and discussion of the data and compilation of this manuscript. Dr. Alan C. Sartorelli is Director and Chairman of Vion’s Scientific Advisory Board. Furthermore, the following authors Penketh, Baumann, Shyam and Sartorelli currently own stock in Vion Pharmaceuticals. Cloretazine is currently being developed by Vion Pharmaceuticals for the treatment of AML.
Footnotes
Conflict of interest
Sartorelli, is a member of the Board of Directors and Chairman of the Scientific Advisory Board of Vion Pharmaceuticals and Penketh, Baumann, Shyam and Sartorelli currently own stock in Vion Pharmaceuticals. Cloretazine is currently being developed by Vion Pharmaceuticals for the treatment of AML.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Penketh PG, Shyam K, Sartorelli AC. Comparison of DNA lesions produced by tumor-inhibitory 1,2-bis(sulfonyl)hydrazines and chloroethylnitrosoureas. Biochem Pharmacol. 2000;59:283–291. doi: 10.1016/s0006-2952(99)00328-7. [DOI] [PubMed] [Google Scholar]
- 2.Penketh PG, Shyam K, Baumann RP, Remack JS, Brent TP, Sartorelli AC. 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-[(methylamino)carbonyl]hydrazine (VNP40101M): I. Direct inhibition of O6-alkylguanine-DNA alkyltransferase (AGT) by electrophilic species generated by decomposition. Cancer Chemother Pharmacol. 2004;53:279–287. doi: 10.1007/s00280-003-0740-7. [DOI] [PubMed] [Google Scholar]
- 3.Teicher BA. Antitumor alkylating agents. In: DeVita VT, Hellman S, Rosenburg SA, editors. Cancer: Principles & Practice of Oncology. 5. Lippincott-Raven; Philadelphia: 1997. pp. 405–418. [Google Scholar]
- 4.Lawley PD, Brooks P. Interstrand cross-linking of DNA by difunctional alkylating agents. J Mol Biol. 1967;25:143–160. doi: 10.1016/0022-2836(67)90285-9. [DOI] [PubMed] [Google Scholar]
- 5.Teicher BA. Preclinical models for high-dose therapy. In: Armitage JO, Antman KH, editors. High-dose Cancer Therapy: Pharmacology, Hematopoietins, Stem Cells. Williams and Wilkins; Philadelphia: 1995. p. 17. [Google Scholar]
- 6.Gerson SL. Clinical Relevance of MGMT in the Treatment of Cancer. J Clin Oncol. 2002;20:2388–2399. doi: 10.1200/JCO.2002.06.110. [DOI] [PubMed] [Google Scholar]
- 7.Pegg AE, Dolan ME, Moschel RC. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase. In: Cohn WE, Moldave K, editors. Progress in Nucleic Acid Research and Molecular Biology. Vol. 51. Academic Press; San Diego: 1995. pp. 167–223. [DOI] [PubMed] [Google Scholar]
- 8.Bodell WJ, Tokuda K, Ludlum DB. Differences in DNA alkylation products formed in sensitive and resistant human glioma cells treated with N-(2-chloroethyl)-N-nitrosourea. Cancer Res. 1988;48:4489–4492. [PubMed] [Google Scholar]
- 9.Ishiguro K, Seow HA, Penketh PG, Shyam K, Sartorelli AC. Mode of action of the chloroethylating and carbamoylating moieties of the prodrug cloretazine. Mol Cancer Ther. 2006;5:1–8. doi: 10.1158/1535-7163.MCT-05-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishiguro K, Shyam K, Penketh PG, Sartorelli AC. Role of O6-alkylguanine-DNA alkyltransferase in the cytotoxic activity of cloretazine. Mol Cancer Ther. 2005;4:1755–1763. doi: 10.1158/1535-7163.MCT-05-0169. [DOI] [PubMed] [Google Scholar]
- 11.Ludlum DB. The chloroethylnitrosoureas: sensitivity and resistance to cancer chemotherapy at the molecular level. Cancer Invest. 1997;15:588–598. doi: 10.3109/07357909709047601. [DOI] [PubMed] [Google Scholar]
- 12.Penketh PG, Shyam K, Sartorelli AC. Studies on the mechanism of decomposition and structural factors affecting the aqueous stability of 1,2-bis(sulfonyl)-1-alkylhydrazines. J Med Chem. 1994;37:2912–2917. doi: 10.1021/jm00044a012. [DOI] [PubMed] [Google Scholar]
- 13.Tentori L, Orlando L, Lacal PM, Benincasa E, Faraoni I, Bonmassar E, D’Atri S, Graziani G. Inhibition of O6-alkylguanine DNA-alkyltransferase or poly(ADP-ribose) polymerase increases susceptibility of leukemic cells to apoptosis induced by Temozolomide. Mol Pharmacol. 1997;52:249–258. doi: 10.1124/mol.52.2.249. [DOI] [PubMed] [Google Scholar]
- 14.Gerson SL, Trey JE, Miller K. Potentiation of nitrosourea cytotoxicity in human leukemic cells by inactivation of O6-alkylguanine-DNA alkyltransferase. Cancer Res. 1988;48:1521–1527. [PubMed] [Google Scholar]
- 15.Yajima N, Hochi S, Miyata N, Kishi T, Kawanishi G. DNA breaks and repair in the mouse leukemia L1210 cells exposed to three different types of interstrand DNA cross-linkers. Mutation Res. 1990;236:43–50. doi: 10.1016/0921-8777(90)90031-y. [DOI] [PubMed] [Google Scholar]
- 16.Thomas CA, Jr, MacHattie LA. The anatomy of viral DNA molecules. Ann Rev Biochem. 1967;36:485–518. doi: 10.1146/annurev.bi.36.070167.002413. [DOI] [PubMed] [Google Scholar]
- 17.Shyam K, Penketh PG, Divo AA, Loomis RH, Patton CL, Sartorelli AC. Synthesis and evaluation of 1,2,3-tris(sulfonyl)hydrazines as antineoplastic and trypanocidal agents. J Med Chem. 1990;33:2259–22640. doi: 10.1021/jm00170a033. [DOI] [PubMed] [Google Scholar]
- 18.Penketh PG, Shyam K, Sartorelli AC. Fluorometric assay for the determination of DNA-DNA cross-links utilizing Hoechst 33258 at neutral pH values. Anal Biochem. 1997;252:210–213. doi: 10.1006/abio.1997.9996. [DOI] [PubMed] [Google Scholar]
- 19.Dunn JJ, Studier FW. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J Mol Biol. 1983;166:477–535. doi: 10.1016/s0022-2836(83)80282-4. [DOI] [PubMed] [Google Scholar]
- 20.Green ED, Chakravarti A. The human genome sequence expedition: views from the “base camp. Genome Res. 2001;11:645–651. doi: 10.1101/gr.188701. [DOI] [PubMed] [Google Scholar]
- 21.Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002a;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 22.Mouse Genome Sequencing Consortium. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002b;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- 23.Schafer FQ, Buettner GR. Singlet oxygen toxicity is cell line-dependent: a study of lipid peroxidation in nine leukemia cell lines. Photochem Photobiol. 1999;70:858–867. [PubMed] [Google Scholar]
- 24.Hallows HR, Packman CH, Knauf PA. Acute cell volume changes in anisotonic media affect F-actin content of HL-60 cells. Am J Physiol. 1991;261:1154–1161. doi: 10.1152/ajpcell.1991.261.6.C1154. [DOI] [PubMed] [Google Scholar]
- 25.Arrebola F, Canizares J, Cubero MA, Crespo PV, Warley A, Fernandez-Segura E. Biphasic behavior of changes in elemental composition during staurosporine-induced apoptosis. Apoptosis. 2005;10:1317–1331. doi: 10.1007/s10495-005-2718-x. [DOI] [PubMed] [Google Scholar]
- 26.Pegg AE, Scicchitano D, Dolan ME. Comparison of the rates of repair of O6-alkylguanines in DNA by rat liver and bacterial O6-alkylguanine-DNA alkyltransferase. Cancer Res. 1984;44:3806–3811. [PubMed] [Google Scholar]
- 27.D’Atri S, Tentor L, Lacal PM, Graziani G, Pagani E, Benincasa E, Zambruno G, Bonmassar E, Jiricny J. Involvement of the mismatch repair system in temozolomide-induced apoptosis. Mol Pharmacol. 1998;54:334–341. doi: 10.1124/mol.54.2.334. [DOI] [PubMed] [Google Scholar]
- 28.Loveless A. Possible relevance of O-6 alkylation of deoxyguanosine to the mutagenicity and carcinogenicity of nitrosamines and nitrosamides. Nature. 1969;223:206–207. doi: 10.1038/223206a0. [DOI] [PubMed] [Google Scholar]
- 29.Rasouli-Nia A, Sibghat-Ullah, Mirzayans R, Paterson MC, Day RS., III On the quantitative relationship between O6-methylguanine residues in genomic DNA and production of sister-chromatid exchanges, mutations and lethal events in a Mer− human tumor cell line. Mutation Res. 1994;314:99–113. doi: 10.1016/0921-8777(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 30.Bishop RE, Dunn LL, Pauly GT, Dolan ME, Moschel RC. The role of O6 alkylguanine-DNA alkyltransferase in protecting Rat4 cells against the mutagenic effects of O6-substituted guanine residues incorporated in codon 12 of the H-ras gene. Carcinogenesis. 1993;4:593–598. doi: 10.1093/carcin/14.4.593. [DOI] [PubMed] [Google Scholar]
- 31.Murren J, Gerson S, Kummar S, Davies M, Remick S, Chu E, Karsten V, Sznol M. A phase I trial of the sulfonylhydrazine alkylator, VNP40101M (101M), administered weekly in patients with metastatic cancer. J Clin Oncol. 2004;22:206–207. [Google Scholar]
- 32.Giles F, Verstovsek S, Thomas D, Gerson S, Cortes J, Faderl S, Ferrajoli A, Ravandi F, Kornblau S, Garcia-Manero G, Jabbour E, O’Brien S, Karsten V, Cahill A, Yee K, Albitar M, Sznol M, Kantarjian H. Phase I study of cloretazine (VNP40101M), a novel sulfonylhydrazine alkylating agent, combined with cytarabine in patients with refractory leukemia. Clin Cancer Res. 2005;11:7817–7824. doi: 10.1158/1078-0432.CCR-05-1070. [DOI] [PubMed] [Google Scholar]
- 33.Spiro TP, Liu L, Majka S, Haaga J, Willson JKV, Gerson SL. Temozolomide: the effect of once- and twice-a-day dosing on tumor tissue levels of the DNA repair protein O6-alkylguanine-DNA-alkyltransferase. Clin Cancer Res. 2001;7:2309–2317. [PubMed] [Google Scholar]
- 34.Baumann RP, Shyam K, Penketh PG, Remack JS, Brent TP, Sartorelli AC. 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-[(methylamino)carbonyl]hydrazine (VNP40101M): II. Role of O6-alkylguanine-DNA alkyltransferase in cytotoxicity. Cancer Chemother Pharmacol. 2004;53:288–295. doi: 10.1007/s00280-003-0739-0. [DOI] [PubMed] [Google Scholar]
- 35.Baumann RP, Seow HA, Shyam K, Penketh PG, Sartorelli AC. The antineoplastic efficacy of the prodrug Cloretazine is produced by the synergistic interaction of carbamoylating and alkylating products of its activation. Oncology Res. 2005;15:313–325. doi: 10.3727/096504005776404553. [DOI] [PubMed] [Google Scholar]