

Abstract
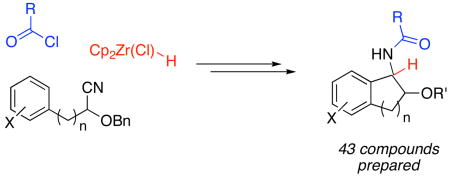
In this manuscript we describe the synthesis of a bicyclic β-benzyloxy and β-hydroxy amide library from cyanohydrin ethers. The benzyloxy amides were prepared through a one-pot sequence of hydrozirconation, acylation, and intramolecular Friedel-Crafts alkylation. Selected benzyl ethers were converted to alcohols by hydrogenolysis in a continuous flow reactor. Chemistry space BCUT metrics and 2D fingerprint similarity calculations showed that these compounds contribute chemical diversity value to existing chemical libraries.
Introduction
In this manuscript we report on our efforts to utilize nitriles as substrates for diversity oriented synthesis (DOS),1 in accord with our objective of devising new chemical reactions that can be applied to library preparation. The goal of these studies is to develop new structural motifs that can be screened for activity as probes in chemical genetics and as leads in drug discovery. New reaction design is directed toward creating protocols that provide opportunities for exploring substitutional (appendage), stereochemical, and skeletal diversity. The latter two areas can be explored most readily by developing reactions that rapidly increase the complexity of readily-available precursors through fragment coupling, ring formation, or stereocenter introduction, with multicomponent reactions2 being particularly desirable.
We have reported a multicomponent protocol for the conversion of nitriles to α-substituted amides through a sequence of nitrile hydrozirconation, acylation, and nucleophilic addition.3 This protocol was subsequently modified for the design of a new annulation reaction4 by tethering a nucleophilic arene to the nitrile and promoting the intramolecular addition by Lewis acid activation of the intermediate acylimine (Scheme 1). We reported several examples of this bicyclic amide construction to demonstrate its viability and scope. The reactions generally proceed efficiently and with good to excellent levels of diastereocontrol.
Scheme 1.

Bicyclic amide synthesis.
We have directed our efforts toward the synthesis of bicyclic amide libraries in consideration of the diverse range of biological responses that compounds with this basic core elicit. Representative literature examples of biologically active molecules that contain this motif and show including HIV protease inhibition (4),5 Factor Xa inhibition (5),6 and CB2 agonism (6)7 are shown in Figure 1. In this manuscript we report our applications of the sequence of nitrile hydrozirconation, acylation, and intramolecular nucleophilic addition to the formation of three bicyclic β-alkoxy amide libraries. We also report ether cleavage reactions to provide several bicyclic β-hydroxy amides in order to incorporate an additional hydrogen bond donor into the structures.
Figure 1.

Biologically active bicyclic amides and related structures.
Results and Discussion
Our previous studies4 demonstrated that cyclization efficiency is dramatically improved when cyanohydrin ethers rather than unsubstituted nitriles are used as reaction substrates because of the diminished tautomerization rates of the intermediate acylimines. Thus the alkoxy group can be employed to promote smooth reactivity and to introduce a point of variation in the library. Benzyl ethers are ideally suited for this purpose because they can be cleaved or modified through numerous procedures to enhance library size and structural diversity. Following established protocols, three cyanohydrin benzyl ethers were prepared in a convenient one-pot procedure by exposing the appropriate aldehydes to trimethylsilyl cyanide and BnOTMS in the presence of a catalytic amount of BiBr3 (Scheme 2).8 Thus substrates 7, 8, and 9 are available in sufficient quantities for library preparation with minimal effort.
Scheme 2.

Substrate preparation.
A group of 13 acid chloride components (Figure 2) was selected based on commercial availability and functional group content. This set included aliphatic, aromatic, and heteroaromatic acid chlorides. Specifically, aliphatic acid chlorides were chosen because of their hydrophobicity. Oxygen-containing and unsaturated acid chlorides were chosen because of their polarity and capacity for subsequent structural manipulation. Fluorine-containing acid chlorides were chosen because of the frequent presence of fluorine in medicinally relevant structures.9 Aromatic and heteroaromatic acid chlorides were also chosen because of their frequent occurrence in drugs and drug-like structures. The library was designed to contain products derived from each nitrile and a subset of these acyl groups.
Figure 2.

Acid chlorides for library construction.
Our initial library construction was directed toward the preparation of tetrahydronaphthyl amides of the general structure types 23 and 24 from cyanohydrin ether 7 (Scheme 3 and Table 1). These reactions proceeded in modest to good yield to form diastereomeric product mixtures. In general reactions with aliphatic acid chlorides provided the best yields and reactions with aromatic acid chlorides proceeded less efficiently. Product stereoisomers were readily separated by preparative HPLC with mass-directed fractionation. The final purities of all compounds were determined to be ≥ 92% by LC analysis with UV detection at 214 nm. The structures of the stereoisomers were assigned by analyzing 1H NMR coupling constants and through analogy with previously characterized structures, with the syn-isomers being the major products in all reactions. This stereochemical outcome arises from the reactions proceeding preferentially through chelated transition state 25. The minor diastereomers arise from the reaction proceeding through a Felkin-type transition state. The moderate level of diastereocontrol and facile stereoisomer separations are desirable for the purposes of library synthesis since the transformations provided suitable amounts of each diastereomer for future screening experiments. No effort was made to optimize for the formation either stereoisomer, although this task could in principle be accomplished through Lewis acid screening.
Scheme 3.

Bicyclic amide formation from 7.
Table 1.
Synthesis of a tetrahydronaphthyl amide library
| entry | acid chloride | yield (%)a | drb | major product | purity (%)c | minor product | purity (%)c |
|---|---|---|---|---|---|---|---|
| 1 | 10 (R = C(H)Me2) | 52 | 3:1 | 23{1} | 100 | 24{1} | 100 |
| 2 | 12 (R = CH=CHMe) | 59 | 3:1 | 23{2} | 92 | 24{2} | 99 |
| 3 | 13 (R = CH2OMe) | 34 | 3:1 | 23{3} | 100 | 24{3} | 99 |
| 4 | 14 (R = CH2OPh) | 73 | 3:1 | 23{4} | 100 | 24{4} | 100 |
| 5 | 15 (R = CH2OBn) | 39 | 4:1 | 23{5} | 100 | 24{5} | 99 |
| 6 | 16 (R = CH2PhOMe) | 41 | 3:1 | 23{6} | 100 | 24{6} | 99 |
| 7 | 17 (R = CH2Ph) | 34 | 3:1 | 23{7} | 100 | 24{7} | 98 |
| 8 | 18 (R = CH2PhF) | 50 | 3:1 | 23{8} | 100 | 24{8} | 98 |
| 9 | 19 (R = CH2C4H3S) | 51 | 9:1 | 23{9} | 100 | 24{9} | 100 |
| 10 | 20 (R = CH2CH2Ph) | 54 | 3:1 | 23{10} | 100 | 24{10} | 100 |
| 11 | 21 (R = Ph-Ph) | 16 | 4:1 | 23{11} | 100 | 24{11} | 93 |
| 12 | 22 (R = PhCF3) | 28 | 3:1 | 23{12} | 100 | 24{12} | 100 |
Isolated yield.
dr = ratio of the diastereomers.
UV purities determined at 214 nm after preparative HPLC.
We prepared a set of indanyl amides of the general structure 26 by the hydrozirconation, acylation, and cyclization of cyanohydrin ether 8 (Scheme 4 and Table 2) to incorporate compounds with a different ring size into our library. Eight bicycles were obtained with high stereocontrol (>10:1 in all cases) and in modest to good isolated yields. The stereochemical outcome of these reactions is consistent with the reaction proceeding through transition state 27, in which chelation control is disfavored relative to the Felkin-Anh type model due to steric clashes that arise in the cyclization of the chelated intermediate.4 Cyclizations to form the indanyl core were slower than those that formed the tetrahydronaphthyl ring system, thereby mandating the presence of the electron donating methoxy groups in the substrate. In general cyclizations were more efficient with unfunctionalized acid chlorides, but suitable quantities of products could be isolated in all cases for subsequent screening experiments. The purities of all compounds in this set were ≥ 99% after recrystallization as determined by HPLC.
Scheme 4.

Bicyclic amide formation from 8.
Table 2.
Synthesis of an indanyl amide library
| entry | acid chloride | yield (%)a | major product | purity (%)b |
|---|---|---|---|---|
| 1 | 10 (R = C(H)Me2) | 40 | 26{1} | 100 |
| 2 | 11 (R = C9H19) | 64 | 26{2} | 100 |
| 3 | 13 (R = CH2OMe) | 18 | 26{3} | 100 |
| 4 | 14 (R = CH2OPh) | 33 | 26{4} | 100 |
| 5 | 16 (R = CH2PhOMe) | 40 | 26{5} | 100 |
| 6 | 18 (R = CH2PhF) | 17 | 26{6} | 99 |
| 7 | 20 (R = CH2CH2Ph) | 54 | 26{7} | 100 |
| 8 | 22 (R = PhCF3) | 27 | 26{8} | 100 |
Isolated yield.
UV purities determined at 214 nm after recrystallization.
The third set of amides was based on the cyclizations of indolyl cyanohydrin ether 9 (Scheme 5 and Table 3). The resulting cyclopentaindolyl amides (28) were prepared with high stereocontrol with no diastereomer formation being observed and, while the yields of the reactions were not high, useful amounts of products could be isolated in high purity. Superior yields for a similar cyclization reaction were previously observed,4 indicating that these reactions could be optimized if the products show interesting activity in biological screens. A notable benefit to this substrate class is that the sulfonyl group on the indole offers an additional point for diversification for future library syntheses.
Scheme 5.

Cyclopentaindolyl amide formation from 9.
Table 3.
Synthesis of cyclopentaindolyl amides
| entry | acid chloride | yield (%)a | major product | purity (%)b |
|---|---|---|---|---|
| 1 | 10 (R = C(H)Me2) | 14 | 28{1} | 99 |
| 2 | 16 (R = CH2PhOMe) | 19 | 28{2} | 100 |
| 3 | 20 (R = CH2CH2Ph) | 14 | 28(3} | 96 |
Isolated yield.
UV purities determined at 214 nm after recrystallization.
Several compounds from the first and second compound collections were selected for benzyl ether hydrogenolysis to increase polarity and introduce a hydrogen bond donor in a subset of our library. These reactions were conducted using the H-Cube,10 a safe and convenient device that generates hydrogen by the electrolytic decomposition of water and enables catalytic hydrogenations in a continuous flow mode. As shown in Table 4, the benzyl ethers of four compounds from the first set of structures and four compounds from the second set of structures were cleaved in the H-Cube™ to provide the corresponding alcohols. Hydrogenolyses of the tetrahydronaphthyl amide series proceeded smoothly at 1 atm and 45 °C to provide the desired products in ≥86% yield and in >95% purity. The indanyl amides reacted more sluggishly, but efficient conversions could be obtained at elevated pressures (20 bar). Yields and product purities in this series were also high except for 26{7}, which provided the product of phenyl group over reduction in addition to the desired alcohol.
Table 4.
Synthesis of bicyclic hydroxy amides
| entry | amide | yield (%)a | major product | purity (%)b |
|---|---|---|---|---|
| 1 | 23{1} (R = C(H)Me2) | 86 | 29{1} | 100 |
| 2 | 23{4} (R = CH2OPh) | 92 | 29{2} | 96 |
| 3 | 23{6} (R = CH2PhOMe) | 92 | 29{3) | 95 |
| 4 | 23{8} (R = CH2PhF) | 91 | 29{4} | 97 |
| 5 | 26{1} (R = C(H)Me2) | 86 | 30{1} | 99 |
| 6 | 26{2} (R = C9H19) | 93 | 30{2} | 96 |
| 7 | 26{5} (R = CH2PhOMe) | 90 | 30{3} | 99 |
| 8 | 26{7} (R = CH2CH2Ph) | 42 | 30{4} | 100 |
Isolated yield.
UV purities determined at 214 nm after recrystallization.
We applied 3D chemistry space BCUT metrics11 and 2D fingerprint similarity calculations to analyze the structural diversity of the 43 compounds and to determine whether the library contributes chemical diversity value to the existing NIH small molecular repository (SMR) or the commercial Maybridge chemical library. The chemical library diversity analysis using cell-based chemistry space BCUT metrics calculations has been described in a recent publication.12
Multi-dimensional chemistry space coordinates were calculated for each compound in the library according to four main classes of atomic properties including atomic Gasteiger-Hückel charge, polarity, hydrogen-bond donor and hydrogen-bond acceptor attributes. The chemistry space for each compound was defined by the best three BCUT descriptors (hydrogen bond acceptor, polarity, and partial charge) and raw descriptor values were rescaled to range from 0 to 10 for visualization and dimension reduction purposes. 3D structures for each member of the library were generated with the Tripos Concord program.
Figure 3 shows a 3D chemistry space plot of the 43 compound library (red points) versus the 57,000 compound Maybridge library (gray points). Eight points representing compounds 23{3}, 24{3}, 23{6}, 24{6}, 23{7}, 24{7}, 23{8}, and 24{8} filled a void cell. No Maybridge structures were observed in this cell, showing that the diversity increase resulting from filling this void is significant. Structures of near neighbors to compounds 24{3} and 24{9} from the Maybridge collection are also shown in Figure 2. The Tanimoto coefficients (Tc, see below for discussion) for these pairs are quite low, indicating that these compounds are not highly similar.
Figure 3.

Chemistry space analysis vs the Maybridge library.
Figure 2 also illustrates that the chemistry space coordinates of compounds 23{3}, 23{6}, 23{7}, 23{8}, and 23{9} follow a pattern that that is similar but not identical to those of 24{3}, 24{6}, 24{7}, 24{8}, and 24{9}. Scaffolds 23 and 24 differ only in their relative stereochemistry, indicating that the molecular conformations generated by Concord is reflected in the properties of these compounds.
Pairwise Tanimoto coefficient (Tc) calculations were carried out based on 2D molecular fingerprints to evaluate the properties of the library relative to the 330,000 compound NIH Small Molecule Repository (SMR) database. In this analysis each molecule is represented by a string of bits (0’s or 1’s) with each bit indicating the absence or presence of the molecular features or particular fragments in the molecule, leading to a 2D fingerprint.13 Tanimoto coefficients have values between 0 and 1, with higher values indicating higher similarity. Compounds from the 23 and 24 series are indistinguishable to the 2D fingerprint algorithm since these fingerprints do not reflect chirality. Thus 31 distinct 2D structures were evaluated. Figure 4 shows a distribution of the Tanimoto coefficients for the 31 compounds in comparison with their most similar compounds from the SMR. This comparison shows a mean TC value of 0.77 and a standard deviation of 0.05. A TC value of 0.85 has been proposed as a boundry for establishing molecular similarity, with values of ≥0.85 indicating a high degree of similarity between two compounds.14 Only two examples were identified that possess a pair wise TC value that is equal to or higher than 0.85 in 2D molecular fingerprint similarity calculations of the two libraries. The structures of these compound pairs are illustrated in Figure 3. These results further confirmed that the newly synthesized compound library enhance the molecular diversity of existing libraries.
Figure 4.

Pairwise Tanimoto coefficients relative to the SMR library.
Conclusions
We have reported the synthesis of a library of 35 bicyclic β-benzyloxy amides through a one pot protocol that proceeds through nitrile hydrozirconation, acylation, and Lewis acid promoted Friedel-Crafts alkylation. From this collection an additional eight β-hydroxy amides were prepared through hydrogenolyses that were conducted in the H-Cube. All compounds were isolated as single diastereomers and were shown to be ≥ 92% pure by HPLC analysis. The combined 3D chemical space and 2D fingerprint pairwise Tc similarity calculations conclude that the library adds certain structural diversity value to the existing chemical libraries. These compounds will be screened for biological activity in accord with the objectives of the Center for Methodology and Library Development (CMLD) program at the University of Pittsburgh.
Supplementary Material
Scheme 6.

Benzyl ether hydrogenolysis.
Acknowledgments
This work was supported by the NIGMS (P50-GM067082). We thank Mr. Peter Chambers and Ms. Stephanie Nicolay for LC-MS/ELSD analyses, and Dr. Tom Jones and Lei Wang at Tripos Company for technical support.
Footnotes
Supporting Information Available. Experimental details and characterization of eight representative library members including 1H NMR, 13C NMR, IR and HRMS data. Details for chemistry space calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
References and notes
- 1.(a) Spandl RJ, Bender A, Spring DR. Org Biomol Chem. 2008;6:1149–1158. doi: 10.1039/b719372f. [DOI] [PubMed] [Google Scholar]; (b) Burke MD, Schreiber SL. Angew Chem, Int Ed. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]; (c) Burke MD, Berger EM, Schreiber SL. Science. 2003;302:613 – 618. doi: 10.1126/science.1089946. [DOI] [PubMed] [Google Scholar]
- 2.(a) Dömling A. Chem Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]; (b) Zhu J, Bienaymé H, editors. Multicomponent Reactions. Wiley; Weinheim: 2005. [Google Scholar]; (c) Ramón DJ, Yus M. Angew Chem, Int Ed. 2005;44:1602–1634. doi: 10.1002/anie.200460548. [DOI] [PubMed] [Google Scholar]; (d) Bienaymé H, Hulme C, Oddon G, Schmidt P. Chem Eur J. 2000;6:3321–3329. doi: 10.1002/1521-3765(20000915)6:18<3321::aid-chem3321>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 3.(a) Wan S, Green ME, Park JH, Floreancig PE. Org Lett. 2007;9:5385–5388. doi: 10.1021/ol702184n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DeBenedetto MV, Green ME, Wan S, Park JH, Floreancig PE. Org Lett. 2009;11:835–838. doi: 10.1021/ol802764j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao Q, Floreancig PE. Org Lett. 2008;10:1139–1142. doi: 10.1021/ol8000409. [DOI] [PubMed] [Google Scholar]
- 5.Dorsey BD, Levin RB, McDaniel SL, Vacca JP, Guare JP, Darke PL, Zugay JA, Emini EA, Schleif WA, Quintero JC, Lin JH, Chen IW, Holloway MK, Fitzgerald PMD, Axel MG, Ostovic D, Anderson PS, Huff JR. J Med Chem. 1994;37:3443–3451. doi: 10.1021/jm00047a001. [DOI] [PubMed] [Google Scholar]
- 6.Qiao JX, Wang TC, Wang GZ, Cheney DL, He K, Rendina AR, Xin B, Luettgen JM, Knabb RM, Wexler RR, Lam PYS. Bioorg Med Chem Lett. 2007;17:5041–5048. doi: 10.1016/j.bmcl.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 7.Omura H, Kawai M, Shima A, Iwata Y, Ito F, Masuda T, Ohta A, Makita N, Omoto K, Sugimoto H, Kikuchi A, Iwata H, Ando K. Bioorg Med Chem Lett. 2008;18:3310–3314. doi: 10.1016/j.bmcl.2008.04.032. [DOI] [PubMed] [Google Scholar]
- 8.(a) Komatsu N, Uda M, Suzuki H, Takahashi T, Domae T, Wada M. Tetrahedron Lett. 1997;38:7215–7218. [Google Scholar]; (b) Iwanami K, Oriyami T. Chem Lett. 2004;33:1324–1325. [Google Scholar]
- 9.Hiyama T, editor. Organofluorine Compounds. Chemistry and Applications. Springer-Verlag; New York: 2000. [Google Scholar]
- 10.Knudsen KR, Holden JJ, Ley SV, Ladlow M. Adv Synth Catal. 2007;349:535–538. [Google Scholar]
- 11.Pearlman RS, Smith KM. Persp Drug Disc Design. 1998;9:339–353. [Google Scholar]
- 12.Xie X, Chen J. J Chem Inf Model. 2008;48:465–475. doi: 10.1021/ci700193u. [DOI] [PubMed] [Google Scholar]
- 13.Wild DJ, Blankley CJ. J Chem Inf Comput Sci. 2000;40:155–162. doi: 10.1021/ci990086j. [DOI] [PubMed] [Google Scholar]
- 14.Matter H. J Med Chem. 1997;40:1219–1229. doi: 10.1021/jm960352+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.