

Summary
DnaA initiates chromosomal replication in E. coli at a well regulated time in the cell-cycle. To determine how the spatial distribution of DnaA is related to the location of chromosomal replication and other cell-cycle events, the localization of DnaA in living cells was visualized by confocal fluorescence microscopy. The gfp gene was randomly inserted into a dnaA-bearing plasmid via in vitro transposition to create a library that included internally GFP-tagged DnaA proteins. The library was screened for the ability to rescue dnaAts mutants, and a candidate gfp-dnaA was used to replace the dnaA gene of wild-type cells. The resulting cells produce close to physiological levels of GFP-DnaA from the endogenous promoter as their only source of DnaA and somewhat under-initiate replication with moderate asynchrony. Visualization of GFP-tagged DnaA in living cells revealed that DnaA adopts a helical pattern that spirals along the long axis of the cell, a pattern also seen in wild-type cells by immunofluorescence with affinity purified anti-DnaA antibody. Although the DnaA helices closely resemble the helices of the actin-analog MreB, co-visualization of GFP-tagged DnaA and RFP-tagged MreB demonstrates that DnaA and MreB adopt discrete helical structures along the length of the longitudinal cell axis.
Keywords: GFP, DNA replication, DnaA, fluorescence microscopy, MreB
Introduction
DnaA protein, the initiator of chromosomal replication in E. coli, exists in both an active ATP-bound form and an inactive ADP-bound form. ATP-DnaA, assisted by DiaA, binds DnaA binding sites, called R boxes and I sites, in the origin region with varied affinities. Complete occupation of the R boxes and I sites leads to localized unwinding of the A-T rich DNA unwinding element, recruitment of DnaB helicase, and assembly of the replisomes responsible for bi-directional DNA synthesis (Leonard and Grimwade, 2005; Kaguni 2006; Mott and Berger, 2007; Grimwade, et al, 2007; Keyamura et al, 2007; Ozaki, et al, 2008).
A wealth of biochemical data (Abe et al., 2007; Kubota et al., 2001; Marszalek et al., 1996; Messer et al., 1999; Weigel et al., 1999) coupled with recent structural studies (Erzberger et al., 2002; Abe et al., 2007; Fujikawa et al., 2003) has allowed certain replication functions of DnaA to be assigned to distinct regions of the protein. Briefly, DnaA protein is composed of an amino-terminal domain, domain I, involved in helicase recruitment (Abe et al., 2007) and with specific residues needed for oligomerization at oriC (Abe et al., 2007, Weigel et al., 1999), domain II that is variable among bacteria and perhaps dispensible, a core domain III that contains the nucleotide binding site (Kubota et al., 2001) and an oligomerization motif (Messer et al., 2001), and a carboxy-terminal DNA binding domain (Roth and Messer, 1995; Messer et al., 1999; Fujikawa et al., 2003).
In vitro and in vivo evidence suggests a close link between DnaA protein activity and cellular membrane lipids. DnaA-bound nucleotide can be released in vitro by treatment with acidic phospholipids in a fluid bilayer, and in the presence of physiological levels of ATP and oriC, this release can lead to an exchange of ATP for ADP, and thus, reactivation of ADP-DnaA to ATP-DnaA (Castuma et al., 1993; Crooke et al., 1992; Sekimizu and Kornberg, 1988; Yung and Kornberg, 1988). Analysis of functional fragments of DnaA and chemical crosslinking between DnaA and a photoactivated phospholipid analog identified a region of DnaA (an amphiphatic helix that joins domain III and the helix-turn-helix motif of domain IV) that interacts with acidic bilayers via electrostatic and hydrophobic interactions (Garner and Crooke, 1996; Garner et al., 1998; Kitchen et al., 1999). More recently, the crystal structure for a truncated form of Aquifex aeolicus DnaA was solved (Erzberger et al., 2002), which showed that this region is indeed an amphipathic helix (helix 12), as predicted earlier (Garner and Crooke, 1996)
A close in vivo link between chromosomal replication and membrane lipids was seen through the examination of cells deficient in acidic phospholipids. Repressed expression of phosphotidylglycerol synthase A, which catalyzes the committed step in the synthesis of the predominant acidic phospholipids, phosphatidylglycerol and cardiolipin, results in cell growth arrest after several generations (Heacock and Dowhan, 1989). However, the growth-arrest can be avoided if the cells are allowed to bypass normal oriC-dependent initiation by instead carrying out constitutive stable DNA replication (Xia and Dowhan, 1995). Moreover, arrested growth in acidic phospholipid-deficient cells that must initiate chromosomal replication from oriC is prevented if the cells express a form of DnaA that contains a leucine-to-lysine point mutation in the membrane binding region of DnaA (Zheng et al., 2001). Of interest, wild-type DnaA must also be present for DnaA(L366K) to efficiently initiate chromosomal replication in vivo and in vitro (Zheng et al., 2001), and purified DnaA(L366K), like wild-type DnaA, still requires acidic phospholipids for in vitro reactivation of ADP-DnaA (Li et al., 2005). While membrane-mediated nucleotide release from DnaA(L366K) requires a 1.5-fold lower concentration of acidic phospholipids than that for wild-type DnaA (Aranovich et al., 2007), this relatively small difference leaves open the possibility that there is at least one other function of DnaA, beyond nucleotide exchange, that requires acidic phospholipid membranes.
Recent evidence suggests that the bacterial membrane is dynamic yet structured. For instance, acidic phospholipids are not homogeneously distributed throughout the E. coli plasma membrane, but instead are found in enriched domains (Mileykovskaya and Dowhan, 2000; Vanounou et al., 2003) (Boeneman and Crooke, unpublished data), and the number and location of these domains changes as a cell progresses through its cell cycle (Boeneman and Crooke, unpublished data). Similar enrichment of acidic phospholipids in the polar and potential septal regions of cell membranes have also been observed in Bacillus subtilis (Kawai et al., 2004) and Pseudomonas putida (Bernal et al, 2007). Moreover, the lipid composition in E. coli alters in response to changes in growth phase or with certain mutations in replication components (Ichihashi et al., 2003; Daghfous et al., 2006; Wegrzyn et al., 1999). Thus, the dynamic yet structured characteristic of the membrane may be critical for key cellular processes, including proper temporal and spatial controlled chromosomal replication.
Because of this structured nature of the membrane and the known dynamics of components of chromosomal replication and segregation (Gordon et al., 1997; Lau et al., 2003; Woldringh and Nanninga, 2006) we have sought to visualize the cellular localization of DnaA in live cells. Earlier immunofluorescence and immunogold cryo-thin section electron microscopy studies of fixed cells found that DnaA is localized at or immediately adjacent to the membrane (Newman and Crooke, 2000). However, we also observed that the domains of acidic phospholipids were disrupted by our fixation process (Newman and Crooke, unpublished data). We have therefore sought to visualize DnaA in live cells and use improved microscopy technology to gain a better understanding of the membrane-associated cellular location of DnaA.
In this work, we have created an internal fusion of GFP in domain II of DnaA protein. Cells that express from the dnaA promoter at the dnaA locus wild-type levels of this GFP-DnaA protein as the sole source of DnaA are viable. When viewed by confocal fluorescence microscopy, DnaA in these cells was found, quite unexpectedly, to form helical structures that spiral along the periphery of the long axis of the cell. Immunofluorescence with improved microscopy instrumentation since our earlier studies (Newman and Crooke, 2000) showed similar results.
Because the cellular content of approximately 1000 DnaA molecules per cell is likely too low to form a helix along the length of the cell on its own, we wondered whether DnaA decorates existing helical structures. An obvious candidate is the bacterial actin analog, MreB, which forms a very similar helical pattern (van den Ent et al., 2001). A recently constructed RFP-tagged MreB, which has mCherry RFP sandwiched between helices 6 and 7 of MreB, gives rise to wild-type cell morphology, growth, and MreB helices when expressed under its native control (Bendezu et al., 2009). We tested whether DnaA helices colocalize with MreB helices by co-visualizing GFP-tagged DnaA and RFP-tagged MreB. Merged images show that DnaA and MreB adopt helical structures distinct from each other along the longitudinal axis of the cell.
Results
Creation of internal GFP-DnaA fusion proteins that can serve a sole source of DnaA activity
Initially, through homologous recombination and resolution of the resulting cointegrate, we attempted to replace the chromosomal copy of dnaA with a gene coding for GFP fused to the amino terminus of DnaA. Even though a single, integrated copy of this gfp-dnaA could rescue dnaAts strains at the non-permissive temperature, and plasmid-borne gfp-dnaA could support in vivo DnaA-dependent pSC101 initiation in a dnaA null strain (Sutton and Kaguni, 1995), cells clearly could not tolerate it as the sole allele for DnaA. Varying the length of the poly-alanine linker between GFP and DnaA failed to provide a completely functional fusion protein, as did fusing GFP to the carboxy-terminus of DnaA with numerous linkers of different sizes.
Therefore, a strategy of introducing GFP to an internal site within DnaA was pursued. Instead of site-directed insertions, gfp was randomly inserted into an ampR, arabinose-inducible dnaA-bearing plasmid via in vitro transposition (Sheridan et al., 2002; Zheng et al., 2001) to create a library of GFP-tagged DnaA proteins (Figure 1). With this procedure, the low probability of in vitro transposition necessitates that an antibiotic resistance marker (kanR) be added to the transposon, which can be removed later by digestion at flanking restriction sites. Cells were transformed with the transposition products, plated on arabinose-containing medium, and thus selected for the plasmid (amp) and transposon (kan). Resulting colonies were screened for green fluorescence under UV light (365nm). Colonies expressing dual antibiotic resistance and showing arabinose-dependent green fluorescence were further screened by PCR to confirm proper orientation of gfp inserted within the dnaA coding region. Restriction digestion removed kanR cassettes from such plasmids, and subsequent ligation created genes encoding full-length DnaA proteins with GFP fused internally (Figure 1).
Figure 1.

Schematic of gfp-dnaA Construction (A–F). (A) The transposons were amplified and moved into DnaA expression plasmid pZL606 by a transposition reaction. (B) Candidate colonies were selected by ampicillin and kanamycin resistance and for fluorescence under UV light illumination. (C) Candidates were then screened for GFP insertion by digestion with BglII and HindIII and by PCR. Arrows denote primer locations. (D) KanR was removed by SmaI digestion and the plasmid was re-ligated. (E) Candidates were selected and screened for DnaA function by rescue experiments with DnaA temperature-sensitive mutants. F) Schematic of the insertion of GFP into DnaA in candidate plasmid pYYH327. GFP follows residue 118 of DnaA, and at the carboxy end of GFP, DnaA resumes beginning with residue 116. This candidate showed the best results in the temperature sensitive rescue assays and was selected for allelic replacement and use in the remainder of this paper.
The library of fusion proteins was screened for the ability to rescue three different dnaAts alleles at the non-permissive temperature: MM294 (dnaA5), CM742 (dnaA46), and CM746 (dnaA204) (Hansen et al., 1992). Of those able to do so, sequencing revealed that three of the four insertions of GFP fell within Domain II of DnaA, a domain that is variable in length and sequence among bacteria, and is thought to be flexible (Schaper and Messer, 1997). The fourth was at the carboxy end of domain I. The construct that was able to rescue at the lowest levels of arabinose induction, pYYH327, was chosen to replace the chromosomally encoded dnaA gene (Table 1). The fusion protein has GFP inserted into domain II of DnaA after amino acid 118, and DnaA resumes with residues 116 to 467 fused to the carboxy-terminus of GFP (Figure 1F).
Table 1.
| Plasmid | Insertion Point | Linker | Domain |
|---|---|---|---|
| pYYH303 | 97 | 95–97 | II |
| pYYH326 | 101 | 99–101 | II |
| pYYH327 | 118 | 116–118 | II |
| pYYH446 | 80 | 78–80 | I |
Four gfp-dnaA containing plasmids were sequenced to determine where GFP had inserted into DnaA protein. As a result of the insertions, the three amino acids prior to the insertion sites were repeated after the insertions, serving as linkers between the carboxy terminus of GFP and DnaA. Three of the candidates had GFP inserted into domain II of DnaA, the fourth was within domain I.
Gfp-dnaA from pYYH327 was moved via the pBIP system (Slater and Maurer, 1993), which utilizes both positive (kanamycin) and negative (5% sucrose) selection, into the wild-type strain W1485 and into a tna::Tn10 bearing strain of W1485 background, WZ52, to facilitate transfer of the fusion to other strains. The cell lines were named YYH605 and KB13, respectively. PCR analysis and genomic sequencing (data not shown) confirmed that internally fused gfp-dnaA at the dnaA locus and driven by the dnaA promoter had successfully replaced dnaA as the sole dnaA allele on the chromosome.
Cells expressing GFP-tagged DnaA as the sole form of DnaA are viable
Western blot analysis with anti-DnaA antiserum reveals that YYH605 cells no longer express non-tagged DnaA, but do produce a DnaA-containing protein of predicted molecular weight of approximately 82 kD, consistent with the size of a GFP-DnaA fusion (Figure 2A). The 82 kD band was also detected by a commercially available anti-GFP antibody (Clontech) in YYH605 and KB13 cells and not wild-type W1485 and WZ52 cells (data not shown). The level of GFP-DnaA in YYH605 is 0.43-fold that of DnaA in the parental wild-type cells, based on triplicate quantitative immunoblots using pure DnaA as a standard (data not shown).
Figure 2.
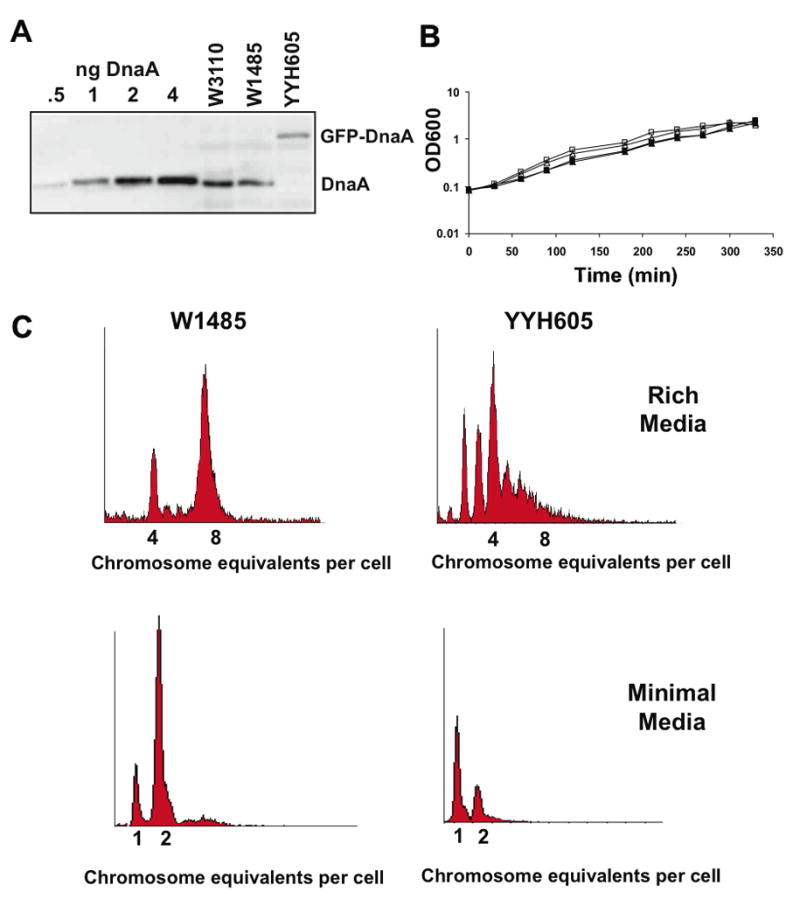
(A) Immunoblots of total cell protein prepared from wild-type strains (W1485 and WZ52) and a strain containing the gfp-dnaA allele (YYH605) grown at 30°C in LB medium were probed with DnaA anti-serum to detect DnaA and the GFP-tagged DnaA fusion protein. (B) Growth curves of parental strains W1485 (□) and WZ52 (△) and gfp-dnaA expressing strains YYH605 (■) and KB13 (▲) in LB medium at 30°C. (C) Flow cytometry of W1485 and YYH605 cells grown in LB medium (30°C) and in M9 + succinate medium with 0.2% succinate (23°C) and treated with rifampicin and cephalexin prior to analysis.
YYH605 and KB13 have similar growth rates as their isogenic parents in both rich (Figure 2B) and minimal (data not shown) media. Cells with GFP-tagged DnaA moderately underinitiate replication in an asynchronous fashion when grown in rich medium (Figure 2C). This is not unexpected given that the levels of GFP-tagged DnaA in these cells is approximately half that found in wild-type cells. When cultured under slow growth conditions (M9 + succinate medium), YYH605 cells also exhibit a moderate underinitiation phenotype, with a larger proportion of cells, compared to the wild type, containing one origin (Figure 2C). In agreement with the flow cytometry data, which indicates that the GFP-tagged DnaA is slightly compromised as an initiator, when compared to wild-type DnaA, YYH605 cells are temperature sensitive for growth at temperatures higher than 40°C.
GFP-DnaA forms a helical structure along the long axis of E. coli
To visualize the location of DnaA protein in living cells, exponentially growing YYH605 cells were examined by fluorescence microscopy. Briefly, the cells were cultured in M9 + succinate medium, harvested by centrifugation, resuspended in M9 medium containing 1% agarose, mounted on slides, and images collected as 0.2 micron z-stacks using a confocal microscope. A confocal field image revealed that the majority of cells in the population were slightly filamentous, but otherwise normal in appearance. Although the signal from GFP-tagged DnaA was weak due to the relatively low number of GFP-DnaA molecules present in each cell, clear images were obtained through two different systems of deconvolution to reduce the effects of light scattering. Metamorph software uses nearest neighbor analysis and DeltaVision software uses iterative deconvolution algorithims based on the specific point-spread function (PSF) of the objective used. The two programs gave comparable results. The unexpected, but most prominent impression from initial examination of the images was that GFP-tagged DnaA was seen as lines and spots that transversed the cell. Upon scrolling through the z-stack, the lines appeared to form a helical pattern spiralling around the periphery of the cell along its long axis (Figure 3A and C), reminiscent of those previously seen for MreB (van den Ent et al., 2001). This is also apparent upon viewing an animated partial 3-dimensional reconstitution generated with Fluoview software (see supplemental data). Similar helical banding of DnaA along the long axis of E. coli rods were seen at all phases of growth, including stationary phase, and in cells grown in rich medium and at different temperatures (data not shown).
Figure 3.

(A) Confocal fluorescence microscopy Z-stack images of YYH605 cells grown in M9 succinate to an OD600 of 0.3 were analyzed by three dimensional rendering with Fluoview software (v. 5.1). A representative angle from the 3D-rendering is shown here. An animated three dimensional reconstruction is available in the supplemental data. (B) YYH605 cells were treated with cephalexin (10 μg/ml) for approximately three doubling times, FM4-64 (30mM) for 30 min, and chloramphenical (200 μg/ml) and DAPI (1μg/ml) for 10 min prior to collecting Z-stack images by confocal microscopy with the appropriate filters. GFP-DnaA images were deconvolved using Metamorph nearest neighbor analysis. Maximum image projections of z-stacks from all channels are shown. From left to right: GFP-DnaA, DAPI, FM4-64, GFP-DnaA and FM4-64, and GFP-DnaA and DAPI. DAPI has been pseudocolored red in the GFP-DnaA DAPI merged field (right most panel) to allow for easy visualization of both fluorescent components. Calibration bar equals five microns. (C) Field images were deconvolved by Metamorph software and cross sections of individual cells in 0.2 micron increments were examined. Sectional images are shown from the bottom to top of the cell. Calibration bar equals two microns. (D) Schematic of DnaA helices in rod-shaped E. coli cells (not to scale).
To better determine whether DnaA is located in regions of the cell not occupied by the nucleoid, cells were treated with cephalexin to inhibit cell division and chloramphenical to condense the nucleoids prior to visualization. Additionally, the cells were stained with DAPI and the membrane dye FM4-64. While DnaA is enriched in the nucleoid region of the cell, DnaA is visible in the inter-nucleoid regions (Figure 3B), suggesting that DnaA can occupy cellular locations independent of its DNA binding activity.
Immunofluorescence reveals a similar helical pattern
DnaA visualized in log-phase W3110 cells by immunofluorescence microscopy with an affinity-purified DnaA antibody was punctate in nature, with the raw images suggesting a helical pattern similar to that seen with the GFP-tagged DnaA. The helical pattern in the immunofluorescent images became more apparent after deconvolution with Metamorph nearest neighbor software (Figure 4A). In contrast, a helical pattern was completely absent in immunofluorescent images of the integral membrane protein leader peptidase (Figure 4B), which yielded a more homogenous membrane staining pattern.
Figure 4.

(A) Immunofluorescent images of wild-type W3110 cells cultured in LB medium prior fixation and treatment with affinity-purified DnaA antibody and goat-anti-rabbit FITC conjugated secondary antibody. Images of cells are maximum image projections of 0.2 micron stacks taken on Olympus confocal microscope and deconvolved using Metamorph nearest neighbor software. Calibration bar equals 2 microns. (B) Immunofluorescent images of YYH605 cells treated with anti-leader peptidase antiseum and TRITC conjugated secondary antibody. Calibration bar equals 5 microns.
DnaA helices localize separately from MreB filaments
To determine whether the helical structure of DnaA resides along intact MreB filaments, we co-visualized GFP-tagged DnaA and RFP-tagged MreB in living cells. The gene encoding a functional RFP-MreB fusion protein, MreB-RFPsw, was transferred from FB76 (Bendezu et al., 2009) to YYH605 by P1 transduction to generate strain LBK2. Exponentially growing LBK2 cells were cultured in M9 + succinate medium, harvested and prepared for visualization, and examined by confocal fluorescence microscopy, as described above for YYH605 cells. The observed helical structure of DnaA in LBK2 cells (Figure 5) mirrors that seen in YYH605 cells and W3110 cells (Figures 3 and 4), and the helical pattern of MreB (Figure 5) are comparable to those described earlier (Bendezu et al., 2009). The merged images of GFP-tagged DnaA and RFP-tagged MreB in individual LBK2 cells demonstrate that the spiral of DnaA protein can reside in locations along the cell axis independent of the MreB helical filament. A similar lack of colocalization was seen in merged images of cells visualized by wide-field fluorescence microscopy (data not shown). Thus, DnaA appears to adopt its helical structure without being directly associated with MreB.
Figure 5.

(A) LBK2 cells (YYH605 mreB-rfpsw yhdE::cat) expressing GFP-tagged DnaA and RFP-tagged MreB were grown in M9 + succinate medium to an OD600 of 0.3 and then imaged sequentially through 0.2 micron z-stack slices for GFP and RFP by confocal fluorescence microscopy. Maximum image projections of GFP-DnaA, RFP-MreB and merged images are shown for two representative cells (a and b). (B) Individual 0.2 micron z-stack slices from cell “a” (above) are shown for GFP-DnaA, RFP-MreB, and merged images.
Discussion
Numerous helical proteins have been the focus in recent literature, the most notable being the cytoskeletal component MreB (van den Ent et al., 2001; Jones et al., 2001; Kruse et al., 2003; Gitai et al., 2005; Shih and Rothfield, 2006; Bendezu et al., 2009). Other proteins that may adopt a helical pattern have been implicated in chromosome and plasmid partitioning (SetB, Par system proteins and SopA and SopB), cell division (MinCD and FtsZ), protein translocation (SecYEG) (Adachi et al., 2006; Espeli et al., 2003; Gerdes et al., 2004; Ramos et al., 2005; Shih et al., 2003; Shiomi et al., 2006; van den Ent et al., 2002), and cell shape (RodZ) (Shiomi et al., 2008; Alyahya et al, 2009; Bendezu et al, 2009). Of these proteins, only SetB (Espeli et al., 2003) and RodZ (Shiomi et al., 2008; Alyahya et al, 2009; Bendezu et al, 2009) have been shown to have a likely direct interaction with MreB. The molecular mechanisms that dictate how these other proteins form cellular helical structures remain unknown. Possibilities include interaction with unknown cytoskeletal proteins, and perhaps for some, localization is guided by the membrane itself.
Continuing studies on both the inner and outer bacterial membrane are changing our perception of the membrane as a static and homogenous structure. In the inner membrane, there is evidence that the acidic phospholipids cardiolipin and phosphatidylglycerol, and the neutral phospholipid, phosphotidylethanolamine, localize in domains, or rafts, within the membrane (Mileykovskaya and Dowhan, 2000; Vanounou et al., 2003). The composition of membrane lipids may also be dynamic, changing in response to the replication status of the cell. Specifically, changes have been seen at certain times in the cell cycle (Joseleau-Petit et al., 1984; Joseleau-Petit et al., 1987), in logarithmically growing versus stationary phase cells (Ichihashi et al., 2003), and also in cells with certain dam, dnaA or seqA mutations (Wegrzyn et al., 1999, Daghfous et al., 2006)
With regard to the membrane, it has been shown that penicillin binding protein 2 (PBP2), which is involved in the synthesis of the peptidoglycan layer, is localized in a helical pattern dependent on MreB in Caulobacter crescentus (Figge et al., 2004). In B. subtilis, PBP2 is colocalized with MreC, though independently of MreB (Scheffers et al., 2004; Divakaruni et al., 2005). Furthermore, the peptidoglycan layer is synthesized in a helical manner (Daniel and Errington, 2003). Thus, there may be an interaction between internal cytoskeletal elements and components of cell membrane structures. Outer membrane proteins, lipids and lipopolysaccharides have also been shown to localize in stable helical ribbons on the lateral sides of the cell (Ghosh and Young, 2005). It remains unknown how this relates to the helical localization of proteins inside the cell.
The ability of acidic phospholipids, especially cardiolipin, to reactivate ADP-DnaA to ATP-DnaA in vitro has been well established (Castuma et al., 1993; Crooke et al., 1992; Sekimizu and Kornberg, 1988; Yung and Kornberg, 1988), and in vivo studies have provided strong evidence that acidic phospholipids are needed for normal DnaA-dependent initiation at oriC (Xia and Dowhan, 1995; Zheng et al., 2001). Earlier immunofluorescence and immunogold cryo-thin section electron microscopy studies revealed that DnaA protein is highly enriched at the cell membrane when compared to the cytosol (Newman and Crooke, 2000). However, during those earlier studies, the fixation process employed disrupted acidic phospholipid-enriched domains (unpublished data) visualized with 10-N-nonyl-acridine orange (NAO) (Mileykovskaya and Dowhan, 2000). Therefore, we created a strain possessing functional DnaA internally tagged with GFP so that we could visualize the cellular location of DnaA protein in living cells with an intact cell membrane.
Using this construct, we observed a helical structure that, while prominent in the nucleoid region, clearly runs along the entire length of the cell. Moreover, this helical structure is found in cells at all stages of growth. A similar cytolocalization pattern was seen with our current immunofluorescence studies. Thus, the helical structure we see with GFP-tagged DnaA in living cells does not seem to be an artifact of the fusion protein.
With about 1000 molecules per wild-type cell, it is unlikely that DnaA would be able to form a helical filament with the pitch we observed spanning the length of the cell rod. We therefore considered the possibility that DnaA is “decorating” an existing helical structure, with the scaffold protein MreB being a likely candidate. However, DnaA, like some other helical proteins, retains a helical structure independent of intact MreB. Perhaps it is binding to a different membrane helical element or another, yet unknown, scaffold protein. Preliminary data indicates that DnaA helical structures persist in cells with repressed expression of Sec E, suggesting that DnaA can adopt its cellular location independent of the inner membrane protein secretion apparatus (K. L. B. and E. C., unpublished data).
Of further interest, Berger and colleagues have shown that a truncated Aquifex aeolicus ATP-DnaA forms a right-handed helix that may interact with the origin complex. The structure is specific to active ATP-DnaA, not inactive ADP-DnaA (Erzberger et al., 2006). The structure of this helical filament is similar to that of the five AAA+ domains in ORC and therefore may be an important feature in initiator function (Clarey et al., 2006) While we, of course, are not seeing a helix at this molecular resolution by microscopy, the relationship between molecular structure and cellular localization is intriguing. We do not know if helical filaments are required for the cellular localization pattern.
Overall, DnaA’s membrane localization may be needed for the activation of the DnaA into its ATP bound form by acidic phospholipids. While inactive ADP-DnaA can bind to the less stringent early binding DnaA boxes R1, R2, and R4, the ATP-form of DnaA is needed to bind to the lower affinity, more stringent DnaA binding sites, R3, R5, I1, I3, and especially I2, for a functional pre-replication complex to form (Leonard and Grimwade, 2005). Recent work suggests that subtle differences in the ratios of acidic phospholipids to DnaA, as well as macromolecular crowding of DnaA on the membrane, can affect the kinetics of nucleotide release, and thus, the activation of DnaA to its active ATP-bound form (Aranovich et al., 2006). Furthermore, a mutant form of DnaA, DnaA(L366K), which is able to rescue the growth-arrest of acidic phospholipid-deficient cells (Zheng et al., 2001), has recently been shown to have a decreased requirement for cardiolipin for nucleotide release (Aranovich et al., 2007). Perhaps membrane composition and structure affect DnaA localization and activity, thus affecting the ratio of ATP- to ADP-DnaA in the vicinity of the origin, and therefore, the formation of pre-replication complexes.
The helical pattern observed for DnaA was an unexpected result and leaves us with many unanswered questions: 1) With what helical scaffold does DnaA associate? 2) Is the observed cellular location of DnaA required for proper initiation of replication? 3) Does DnaA have a function other than initiation that requires the helical localization? 4) Is the helical structure of DnaA dependent on membrane lipid composition or structure? While the mechanisms and reasons surrounding the findings presented here are unknown, continued advances in microscopy and further research on protein-membrane interactions will hopefully help answer these and other questions related to the chromosomal replication.
Experimental procedures
Transposition Reaction
Transposons EGF-V and TgPT-0 were amplified using primers from the nine base-pair Tn5ME sites flanking the transposons (5′-CTGTCTCTTATACACATCT-3′) from plasmids pBNJ11.7 and pBNJ24.6 (Sheridan et al., 2002). The PCR product was purified and concentrated with PCR Purification Kit (Qiagen) and eluted in sterile distilled water. The transposon (0.2 fmoles) was incubated with 5.0μL of EZ::TN (Epicentre Technologies) transposase (5 μl) in 25% glycerol at 25°C for 30 min. Molar equivalents (0.04 fmoles each) of the transposon and target plasmid, pZL606 (Zheng et al., 2001) were incubated in 10μl of 50 mM Tris-acetate (pH 7.5), 150 mM potassium acetate, 10 mM magnesium acetate and 4 mM spermidine at 37°C for 2 hr reaction. The transposition reaction was stopped by the addition of 1% SDS (1μl) and incubation at 70°C for 10 min. Competent Top 10 F′ E. coli cells (Stratagene) were transformed with 1μl of the transposition reaction and spread onto LB plates that contained ampicillin (100μg/ml) to select for transformation of the original plasmid, kanamycin (50μg/ml) to select for transposition of GFP, and 0.2% arabinose to allow for induction of green fluorescence that could be visualized under a long-wave UV lamp (365nm).
PCR Screening
PCR confirmation for EGFP-V or TgPT-0 insertions within dnaA was performed using a primer spanning the dnaA start codon (5′-CCGGATCCATGTGTCACTTTCGCTTTGGCAG-3′) and a primer complimentary to EGFP 3′ end (5′-ACGTACACGTGTGCTTGTACAGCTCGTCCAT-3′). Plasmid was isolated from candidates that produced clear PCR products within the expected size range (0.8 to 2.2kb). The insertion position was estimated by analyzing the size of PCR product and by restriction digestion of the plasmid with BglII and HindIII (1.4~2.8kb, BglII inside EGFP-V or TgPT-0 and HindIII in the target plasmid beyond the end of dnaA).
Removal of the gene for Kanamycin resistance
Candidate plasmids for carrying GFP insertions were digested with SmaI to remove the kan cassette, thereby creating a sequence encoding a full-length dnaA-gfp-dnaA fusion protein. Following SmaI digestion and ligation, competent cells (Top 10 F′ E. coli) were transformed with the ligation mixture (1μl) and plated on LB agar containing ampicillin. The colonies were replica-plated the following day on LB plates that contained ampicillin and kanamycin to verify loss of kanamycin resistance.
Temperature Sensitive Rescue Assay
MM294 (dnaA5), CM742 (dnaA46) and CM746 (dnaA204) cells were transformed with pBAD24, pZL606 and dnaA-gfp bearing plasmids and plated onto LB ampicillin plates (100μg/mL) and cultured at 30°C. Transformants were streaked onto ampicillin plates with and without 0.2% arabinose and grown at 42°C. The four plasmids that supported growth of the temperature sensitive dnaA cells at 42°C were pYYH303, pYYH326, pYYH327, pYYH446. pYYH327 showed the strongest effect and was chosen for the subsequent allelic replacements.
Sequencing dnaA-gfp fusion candidates
Insertion sites were determined for pYYH303, pYYH326, pYYH327, and pYYH446 by sequencing from the GFP transposon with a primer complimentary to a region close to the GFP amino-terminus (5′-TGGCCGTTTACGTCGCCGTCCA-3′).
Integration of gfp-tagged dnaA into W1485 and WZ52 and rfp-tagged mreB into YYH605
Gfp-tagged dnaA was subcloned from pYYH327 into pWZC23, a pBluescript derivative containing pdnaA-DnaA(L366K), using EcoRI and BamHI digestion. Ligation products were transformed into DH5α and plated onto LB + ampicillin plates (100μg/ml) at 37°C. The new plasmid, pYYH71, was confirmed with ClaI and BamHI digestion, and fluorescence microscopy revealed that cells harboring the plasmid had green fluorescence after induction with arabinose.
The gfp-tagged dnaA was subcloned from pYYH71 into the pBIP vector (Slater and Maurer 1993) by digestion with ApaI and NotI. pYYH71 gave two bands at the same size, so the whole digestion was purified through a PCR purification Kit (Qiagen) and ligated with a gel-purified pBIP band, also digested with ApaI and NotI, using T4 DNA ligase. Ligations were transformed into DH5α onto LB kanamycin plates (50μg/ml) at 37°C. Clones were screened for green fluorescence under a Nikon Eclipse E600 microscope, then by EcoRI digestion. The resulting plasmid was named pYYH72.
pYYH72 was then used for the integration of dnaA-gfp into W1485 and WZ52 (W1485 background, tna::Tn10) as previously described (Slater and Maurer, 1993). pYYH72 was transformed into the supE strain JM109 on LB agar and colonies were selected by kanamycin resistance. Transformed cells were green under a Nikon Eclipse E600 microscope. JM109/pYYH72 was grown in LB with kanamycin to an OD600~0.2 (~4×108 cell), then ~ 20μl of helper phage f1R189 (~5×1011pfu/ml) was added for an MOI of ~20. The infection was allowed to continue for 7 hrs to convert the independently replicating plasmid into a suicide vector. Phage lysate was collected and used to infect target strains W1485 and WZ52. Infected cells were selected on LB kanamycin plates. The integrant strains are diploid, containing both chromosomal pdnaA-dnaA and suicide vector pdnaA-dnaA-gfp.
To confirm the integration, and to determine which side of pdnaA-dnaA-gfp was involved in the first crossover, 2 pairs of primers were used: p535ApNc and dnaA1120 and dnaAStart and DnDnaA222. p535dnaA (5′-AAACGATGGGCCCCGAAGCCATGGGTG - 3′) is a 5′ primer and 535bp upstream of dnaA and dnaA1120 (5′-CAGGGCGTTGAAGGTGTG-3′) is a 3′ primer at 224aa in dnaA. DnaAStart (5′ CCGAATTCATGTCACTTTCGCTTTGGCAG-3′) is a 5′ primer at the start codon of dnaA and DnDnaA222 (5′-GCGCCGGCCCGGTCGTCGCTCCTGGCTC–3′) is a 3′ primer and 222bp downstream dnaA. For all of the integrations, p535dnaA and dnaA1120 PCR gave 1.2kb (wild-type size) and dnaAStart and DnDnaA222 gave 2.5kb (size with transposon insertion). This means all of the integrations occurred at the longer arm of dnaA (116–467), not the promoter side. The diploid strains were called YYH604 (parental strain W1485) and YYH701 (parental strain WZ52).
To resolve the diploid status a second homologous crossover event which would eliminate the intervening pBIP sequence from the chromosome was needed. YYH604 and YYH701 were grown in LB at 37oC to saturation and the cultures were plated onto salt-free plates LB tetracycline plates (10μg/ml) with 5% sucrose to select for cells that had lost the plasmid, and were thus sucrose resistant. Integrant colonies were found to temperature sensitive at 42°C, which aided in the screening process. Temperature sensitive colonies were further confirmed by PCR. Integrant strains into W1485 and WZ52 were named YYH605 and KB13, respectively.
The rfp-mreB gene (mreB-rfpsw) was moved from strain FB76 (Bendezu et al., 2009) to YYH605 by P1 transduction. The initial screening of candidates was for growth on LB + chloramphenicol (25 μg/ml) medium at 30°C, with chloramphenicol resistance arising from the closely linked yhdE::cat locus. Cells from chloramphenicol-resistant colonies were cultured in M9 + succinate medium at 30°C and screened by confocal fluorescence microscopy for red-fluorescent MreB filaments.
Western Blotting
Cells were grown to an OD600nm of approximately 0.5 and total cell protein from 1ml of culture was precipitated with ice-cold TCA (10%), washed with chilled acetone (67%) resuspended in SDS-PAGE loading buffer, resolved on a 12% SDS-PAGE gel and transferred to nitrocellulose paper (Whatman). Samples of purified, non-tagged DnaA protein (0.5 to 4 ng) were included as a standard curve. Blots were probed with DnaA antisera (1:1000), and goat anti-rabbit IgG alkaline phosphatase (Amersham Biosciences) as a secondary antibody. Signal was detected by ECL detection agent (Amersham Biosciences) on a Storm Imager.
Flow Cytometry
Cells were grown either in M9 minimal media (Miller, 1972) containing 0.2% succinate, 40μg/ml thymine and 1μg/ml vitamin B1 or in LB at 25°C to an optical density of 0.15 at 450nm or 600nm for cells grown in M9 + succinate or LB medium, respectively. They were treated with 300μg/ml rifampicin and 10μg/ml cephalexin for approximately four doubling times. Cells were fixed in 70% ethanol and flow cytometry was performed as described (Torheim et al., 2000). Integral numbers of chromosomes per cell represent numbers of origins per cell at the time of drug addition. DNA content and cell mass prior to drug addition was also monitored and a lowered DNA concentration confirmed in cells said to underinitiate.
GFP-DnaA and RFP-MreB Microscopy
YYH605 and LBK2 cells were grown in M9 minimal medium (Miller, 1972) containing 0.2% succinate, 40μg/ml thymine and 1μg/ml vitamin B1 (M9 + succinate medium_ at approximately 30°C. Cells were harvested by centrifugation in early log phase (an OD600nm of approximately 0.15 to 0.4), and resuspended in M9 medium with 1% agarose. Samples (5 μl) were put onto glass slides and quickly covered with a glass coverslip. Cells were viewed with either an Olympus BX61 or an Olympus IX-70 inverted microscope with an Olympus Fluoview- FV300 or FV500 Series Confocal Laser Scanning System using UPlanApo X 100 objectives with numerical apertures of 1.3 and 1.35, respectively. Cells were imaged in 0.2 micron z-stacks with excitation at 488nm and an EGFP a BA 505–525 bandpass filter for DnaA, and 543nm excitation and a Texas Red BA 610IF bandpass filter for RFP. Cells containing both GFP-tagged DnaA and RFP-tagged MreB were imaged sequentially for GFP and then RFP at each z-level. Field images were acquired with a 0.18μm pixel resolution.
To view filamentous cells, cultures were treated with 10μg/ml of cephalexin for approximately three to four doubling times before imaging. The membrane dye N-(3-triethylammoniumpropyl)-4-(6-(4-(diethylamino)phenyl)hexatrienyl)pyridinium dibromide (FM4-64) was added to a final concentration of 30μM approximately 30 minutes before imaging, and 1 μg/ml 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) and 200μg/ml chloramphenical were added 10 minutes before imaging. Cells were harvested and mounted on glass slides as described above. DAPI and Texas Red filters (BA 430–460 and BA 610IF bandpass filters, respectively) were used to visualize these dyes. DnaA images were deconvolved using Metamorph software, which uses nearest neighbor analysis and DeltaVision software, which uses iterative deconvolution algorithims based on the specific point-spread function (PSF) of the objective used. Images were prepared for publication using Adobe Photoshop version 6.0.
Immunofluorescence
YYH605 and W3110 cells were grown in LB at 37°C to an OD600nm of approximately 0.5 to 0.6. One milliliter of cells was spun down and resuspended in an equal volume of freshly made fixative (30mM Na2HPO4/NaH2PO4 pH 7.4, 2.5% paraformaldehyde, 0.04% gluteraldehyde) and incubated at room temperature for 10 min, then on ice for 50 minutes. Cells were washed three times with PBS, then resuspended in 50μl GTE buffer (50mM glucose, 20mM TrisCl pH 7.5, 10mM EDTA) with 4μg/ml lysozyme. Cells (10μl) were immediately put onto poly-L-lysine coated 15-well slides and lysis was allowed to continue for 1 minute. The liquid was removed by needle aspiration and the wells were washed twice with 10μl PBS and allowed to air dry for 10–15 minutes. The wells were then re-hydrated with PBS (10μl) for four minutes. Affinity-purified DnaA antibody or leader peptidase antiserum, diluted 1:10 or 1:100, respectively in PBS (10μl total) was added to the wells, and the slides were incubated overnight at 4°C.
The next day the primary antibody was removed by needle aspiration and the wells were washed ten times with 10μl PBS. Ten microliters of goat anti-rabbit FITC or TRITC conjugated secondary antibody (Sigma) diluted 1:100 in PBS was added to each well. Slides were incubated for two hours in the dark at room temperature. Wells were washed again 10 times with 10μl PBS. Five microliters of SlowFade equilibrium buffer (Molecular Probes) was added to each well for five minutes. The excess liquid was removed by needle aspiration and slides were allowed to air dry for 10–15 minutes. One microliter SlowFade (Molecular Probes) was added to each well, a coverslip was added and sealed. Slides were stored at −20°C for up to three hours and visualized on an Olympus BX61 microscope with an Olympus Fluoview- FV500 Series Confocal Laser Scanning System using an UPlanApo X 100 objective with a numerical aperture of 1.35. Cells were imaged in 0.2μm stacks with a FITC BA 505IF filter or a Texas Red BA 610IF filter. Field images were acquired with a pixel resolution of 0.18μm. Final images were deconvolved with Metamorph nearest neighbor software version 6.0 and prepared for publication by using Adobe Photoshop version 6.0.
Supplementary Material
Acknowledgments
We would like to acknowledge the Lombardi Cancer Center Microscopy and Imaging Shared Resource, which is supported in part by a grant from the National Cancer Institute (P30CA051008) and the Biochemistry and Molecular & Cellular Biology Confocal Microscopy shared facility for equipment use and technical assistance. We would also like to thank Drs. James McNally and Tatiana Karpova of the CCR Fluorescence Imaging Facility at the NCI (Bethesda, MD) for technical assistance with deconvolution methods and use of Deltavision software. We would like to thank the laboratory of Dr. Thomas E. Hughes of Yale University for sending us the EGF-V and TgPT-0 transposons and for technical suggestions for the transposition reaction, and the laboratory of Dr. Piet de Boer of Case Western Reserve University for the MreB-RFP containing strain FB76. This work was supported in part by a grant from NIH (GM49700) to E.C. K.B. was supported in part by a T32 Predoctoral Training Grant in Tumor Biology (CA009686) and S.F. was supported by the Norwegian Cancer Society and the University of Oslo, EMBIO.
References
- Abe Y, Jo T, Matsuda Y, Matsunaga C, Katayama T, Ueda T. Structure and function of DnaA N-terminal domains: specific sites and mechanisms in inter-DnaA interaction and in DnaB helicase loading on oriC. J Biol Chem. 2007;282:17816–17827. doi: 10.1074/jbc.M701841200. [DOI] [PubMed] [Google Scholar]
- Adachi S, Hori K, Hiraga S. Subcellular positioning of F plasmid mediated by dynamic localization of SopA and SopB. J Mol Biol. 2006;356:850–863. doi: 10.1016/j.jmb.2005.11.088. [DOI] [PubMed] [Google Scholar]
- Alyahya SA, Alexander R, Costa T, Henriques AO, Emonet T, Jacobs-Wagner C. RodZ, a component of the bacterial core morphogenic apparatus. Proc Natl Acad Sci USA. 2009;106:1239–1244. doi: 10.1073/pnas.0810794106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranovich A, Gdalevsky GY, Cohen-Luria R, Fishov I, Parola AH. Membrane-catalyzed nucleotide exchange on DnaA. Effect of surface molecular crowding. J Biol Chem. 2006;281:12526–12534. doi: 10.1074/jbc.M510266200. [DOI] [PubMed] [Google Scholar]
- Aranovich A, Parola AH, Fishov I. The reactivation of DnaAL366K requires less acidic phospholipids supporting their role in the initiation of chromosome replication in Escherichia coli. FEBS letters. 2007;581:4439–4442. doi: 10.1016/j.febslet.2007.08.019. [DOI] [PubMed] [Google Scholar]
- Bendezu FO, Hale CA, Bernhardt TG, de Boer PAJ. RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E. Coli. EMBO J. 2009;28:193–204. doi: 10.1038/emboj.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal P, Segura A, Ramos JL. Compensatory role of the cis-trans-isomerase and cardiolipin synthase in the membrane fluidity of Pseudomonas putida DOT-T1E. Environ Microbiol. 2007;9:1658–1664. doi: 10.1111/j.1462-2920.2007.01283.x. [DOI] [PubMed] [Google Scholar]
- Castuma CE, Crooke E, Kornberg A. Fluid membranes with acidic domains activate DnaA, the initiator protein of replication in Escherichia coli. J Biol Chem. 1993;268:24665–24668. [PubMed] [Google Scholar]
- Clarey MG, Erzberger JP, Grob P, Leschziner AE, Berger JM, Nogales E, Botchan M. Nucleotide-dependent conformational changes in the DnaA-like core of the origin recognition complex. Nat Struct Mol Biol. 2006;13:684–690. doi: 10.1038/nsmb1121. [DOI] [PubMed] [Google Scholar]
- Crooke E, Castuma CE, Kornberg A. The chromosome origin of Escherichia coli stabilizes DnaA protein during rejuvenation by phospholipids. J Biol Chem. 1992;267:16779–16782. [PubMed] [Google Scholar]
- Daghfous D, Chatti A, Marzouk B, Landoulsi A. Phospholipid changes in seqA and dam mutants of Escherichia coli. C R Biol. 2006;329:271–276. doi: 10.1016/j.crvi.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Daniel RA, Errington J. Control of cell morphogenesis in bacteria: two distinct ways to make a rod-shaped cell. Cell. 2003;113:767–776. doi: 10.1016/s0092-8674(03)00421-5. [DOI] [PubMed] [Google Scholar]
- Divakaruni AV, Loo RR, Xie Y, Loo JA, Gober JW. The cell-shape protein MreC interacts with extracytoplasmic proteins including cell wall assembly complexes in Caulobacter crescentus. Proc Natl Acad Sci USA. 2005;102:18602–18607. doi: 10.1073/pnas.0507937102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzberger JP, Mott ML, Berger JM. Structural basis for ATP-dependent DnaA assembly and replication-origin remodeling. Nat Struct Mol Biol. 2006;13:676–683. doi: 10.1038/nsmb1115. [DOI] [PubMed] [Google Scholar]
- Erzberger JP, Pirruccello MM, Berger JM. The structure of bacterial DnaA: implications for general mechanisms underlying DNA replication initiation. EMBO J. 2002;21:4763–4773. doi: 10.1093/emboj/cdf496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espeli O, Nurse P, Levine C, Lee C, Marians KJ. SetB: an integral membrane protein that affects chromosome segregation in Escherichia coli. Mol Microbiol. 2003;50:495–509. doi: 10.1046/j.1365-2958.2003.03736.x. [DOI] [PubMed] [Google Scholar]
- Figge RM, Divakaruni AV, Gober JW. MreB, the cell shape-determining bacterial actin homologue, co-ordinates cell wall morphogenesis in Caulobacter crescentus. Mol Microbiol. 2004;51:1321–1332. doi: 10.1111/j.1365-2958.2003.03936.x. [DOI] [PubMed] [Google Scholar]
- Fujikawa N, Kurumizaka H, Nureki O, Terada T, Shirouzu M, Katayama T, Yokoyama S. Structural basis of replication origin recognition by the DnaA protein. Nucleic Acids Res. 2003;31:2077–2086. doi: 10.1093/nar/gkg309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner J, Crooke E. Membrane regulation of the chromosomal replication activity of E. coli DnaA requires a discrete site on the protein. EMBO J. 1996;15:3477–3485. [PMC free article] [PubMed] [Google Scholar]
- Garner J, Durrer P, Kitchen J, Brunner J, Crooke E. Membrane-mediated release of nucleotide from an initiator of chromosomal replication, Escherichia coli DnaA, occurs with insertion of a distinct region of the protein into the lipid bilayer. J Biol Chem. 1998;273:5167–5173. doi: 10.1074/jbc.273.9.5167. [DOI] [PubMed] [Google Scholar]
- Gerdes K, Moller-Jensen J, Ebersbach G, Kruse T, Nordstrom K. Bacterial mitotic machineries. Cell. 2004;116:359–366. doi: 10.1016/s0092-8674(04)00116-3. [DOI] [PubMed] [Google Scholar]
- Ghosh AS, Young KD. Helical disposition of proteins and lipopolysaccharide in the outer membrane of Escherichia coli. J Bacteriol. 2005;187:1913–1922. doi: 10.1128/JB.187.6.1913-1922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitai Z, Dye NA, Reisenauer A, Wachi M, Shapiro L. MreB actin-mediated segregation of a specific region of a bacterial chromosome. Cell. 2005;120:329–341. doi: 10.1016/j.cell.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Gordon GS, Sitnikov D, Webb CD, Teleman A, Straight A, Losick R, Murray AW, Wright A. Chromosome and low copy plasmid segregation in E. coli: visual evidence for distinct mechanisms. Cell. 1997;90:1113–1121. doi: 10.1016/s0092-8674(00)80377-3. [DOI] [PubMed] [Google Scholar]
- Grimwade JE, Torgue JJ, McGarry KC, Rozgaja T, Enloe ST, Leonard AC. Mutational analysis reveals. Escherichia coli oriC interacts with both DnaA-ATP and DnaA-ADP during pre-RC assembly. Mol Microbiol. 2007;66:428–439. doi: 10.1111/j.1365-2958.2007.05930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen FG, Koefoed S, Atlung T. Cloning and nucleotide sequence determination of twelve mutant dnaA genes of Escherichia coli. Mol Gen Genet. 1992;234:14–21. doi: 10.1007/BF00272340. [DOI] [PubMed] [Google Scholar]
- Heacock PN, Dowhan W. Alteration of the phospholipid composition of Escherichia coli through genetic manipulation. J Biol Chem. 1989;264:14972–14977. [PubMed] [Google Scholar]
- Ichihashi N, Kurokawa K, Matsuo M, Kaito C, Sekimizu K. Inhibitory effects of basic or neutral phospholipid on acidic phospholipid-mediated dissociation of adenine nucleotide bound to DnaA protein, the initiator of chromosomal DNA replication. J Biol Chem. 2003;278:28778–28786. doi: 10.1074/jbc.M212202200. [DOI] [PubMed] [Google Scholar]
- Iwai N, Nagai K, Wachi M. Novel S-benzylisothiourea compound that induces spherical cells in Escherichia coli probably by acting on a rod-shape-determining proteins other than penicillin-binding protein 2. Biosci Biotechnol Biochem. 2002;66:2658–2662. doi: 10.1271/bbb.66.2658. [DOI] [PubMed] [Google Scholar]
- Jones LJ, Carballido-Lopez R, Errington J. Control of cell shape in bacteria: helical actin-like filaments in Bacillus subtilis. Cell. 2001;104:913–922. doi: 10.1016/s0092-8674(01)00287-2. [DOI] [PubMed] [Google Scholar]
- Joseleau-Petit D, Kepes F, Kepes A. Cyclic changes of the rate of phospholipid synthesis during synchronous growth of Escherichia coli. European journal of biochemistry/FEBS. 1984;139:605–611. doi: 10.1111/j.1432-1033.1984.tb08047.x. [DOI] [PubMed] [Google Scholar]
- Joseleau-Petit D, Kepes F, Peutat L, D’Ari R, Kepes A. DNA replication initiation, doubling of rate of phospholipid synthesis, and cell division in Escherichia coli. J Bacteriol. 1987;169:3701–3706. doi: 10.1128/jb.169.8.3701-3706.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaguni JM. DnaA: controlling the initiation of bacterial DNA replication and more. Annu Rev Microbiol. 2006;60:351– 375. doi: 10.1146/annurev.micro.60.080805.142111. [DOI] [PubMed] [Google Scholar]
- Kawai F, Shoda M, Harashima R, Sadaie Y, Hara H, Matsumoto K. Cardiolipin domains in Bacillus subtilis marburg membranes. J Bacteriol. 2004;186:1475–1483. doi: 10.1128/JB.186.5.1475-1483.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyamura K, Fujikawa N, Ishida T, Ozaki S, Su’etsugu M, Fujimitsu K, Kagawa W, Yokoyama S, Kurumizaka H, Katayama T. The interaction of DiaA and DnaA regulates the replication cycle in E. coli by directly promoting ATP DnaA-specific initiation complexes. Genes Dev. 2007;21:2083–2099. doi: 10.1101/gad.1561207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen JL, Li Z, Crooke E. Electrostatic interactions during acidic phospholipid reactivation of DnaA protein, the Escherichia coli initiator of chromosomal replication. Biochemistry. 1999;38:6213–6221. doi: 10.1021/bi982733q. [DOI] [PubMed] [Google Scholar]
- Kruse T, Moller-Jensen J, Lobner-Olesen A, Gerdes K. Dysfunctional MreB inhibits chromosome segregation in Escherichia coli. EMBO J. 2003;22:5283–5292. doi: 10.1093/emboj/cdg504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, Ito Y, Sekimizu K, Tagaya M, Katayama T. DnaA protein Lys-415 is close to the ATP-binding site: ATP-pyridoxal affinity labeling. Biochem Biophys Res Comm. 2001;288:1141–1148. doi: 10.1006/bbrc.2001.5898. [DOI] [PubMed] [Google Scholar]
- Lau IF, Filipe SR, Soballe B, Okstad OA, Barre FX, Sherratt DJ. Spatial and temporal organization of replicating Escherichia coli chromosomes. Mol Microbiol. 2003;49:731–743. doi: 10.1046/j.1365-2958.2003.03640.x. [DOI] [PubMed] [Google Scholar]
- Leonard AC, Grimwade JE. Building a bacterial orisome: emergence of new regulatory features for replication origin unwinding. Mol Microbiol. 2005;55:978–985. doi: 10.1111/j.1365-2958.2004.04467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Kitchen JL, Boeneman K, Anand P, Crooke E. Restoration of growth to acidic phospholipid-deficient cells by DnaAL366K is independent of its capacity for nucleotide binding and exchange and requires DnaA. J Biol Chem. 2005;280:9796–9801. doi: 10.1074/jbc.M413923200. [DOI] [PubMed] [Google Scholar]
- Marszalek J, Zhang W, Hupp TR, Margulies C, Carr KM, Cherry S, Kaguni JM. Domains of DnaA protein involved in interaction with DnaB protein, and in unwinding the Escherichia coli chromosomal origin. J Biol Chem. 1996;271:18535–18542. doi: 10.1074/jbc.271.31.18535. [DOI] [PubMed] [Google Scholar]
- Messer W, Blaesing F, Majka J, Nardmann J, Schaper S, Schmidt A, Seitz H, Speck C, Tungler D, Wegrzyn G, Weigel C, Welzeck M, Zakrzewska-Czerwinska J. Functional domains of DnaA proteins. Biochimie. 1999;81:819–825. doi: 10.1016/s0300-9084(99)00215-1. [DOI] [PubMed] [Google Scholar]
- Messer W, Blaesing F, Jakimowicz D, Krause M, Majka J, Nardmann J, Schaper S, Steitz H, Speck C, Weigel C, Wegrzyn G, Welzeck M, Zakrzewska-Czerwinska J. Bacterial replication initiator DnaA. Rules for DnaA binding and and roles of DnaA in origin unwinding and helicase loading. Biochimie. 2001;83:5–12. doi: 10.1016/s0300-9084(00)01216-5. [DOI] [PubMed] [Google Scholar]
- Mileykovskaya E, Dowhan W. Visualization of phospholipid domains in Escherichia coli by using the cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J Bacteriol. 2000;182:1172–1175. doi: 10.1128/jb.182.4.1172-1175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott ML, Berger JM. DNA replication initiation: mechanisms and regulation in bacteria. Nat Rev Microbiol. 2007;5:343–354. doi: 10.1038/nrmicro1640. [DOI] [PubMed] [Google Scholar]
- Newman G, Crooke E. DnaA, the initiator of Escherichia coli chromosomal replication, is located at the cell membrane. J Bacteriol. 2000;182:2604–2610. doi: 10.1128/jb.182.9.2604-2610.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki S, Kawakami H, Nakamura K, Fujikawa N, Kagawa W, Park SY, Yokoyama S, Kurumizaka H, Katayama T. A common mechanism for the ATP-DnaA-dependent formation of open complexes at the replication origin. J Biol Chem. 2008;283:8351–8362. doi: 10.1074/jbc.M708684200. [DOI] [PubMed] [Google Scholar]
- Ramos A, Letek M, Campelo AB, Vaquera J, Mateos LM, Gil JA. Altered morphology produced by ftsZ expression in Corynebacterium glutamicum ATCC 13869. Microbiology. 2005;151:2563–2572. doi: 10.1099/mic.0.28036-0. [DOI] [PubMed] [Google Scholar]
- Roth A, Messer W. The DNA binding domain of the initiator protein DnaA. EMBO J. 1995;14:2106–11. doi: 10.1002/j.1460-2075.1995.tb07202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper S, Messer W. Prediction of the structure of the replication initiator protein DnaA. Proteins. 1997;28:1–9. [PubMed] [Google Scholar]
- Scheffers DJ, Jones LJ, Errington J. Several distinct localization patterns for penicillin-binding proteins in Bacillus subtilis. Mol Microbiol. 2004;51:749–764. doi: 10.1046/j.1365-2958.2003.03854.x. [DOI] [PubMed] [Google Scholar]
- Sekimizu K, Kornberg A. Cardiolipin activation of dnaA protein, the initiation protein of replication in Escherichia coli. J Biol Chem. 1988;263:7131–7135. [PubMed] [Google Scholar]
- Sheridan DL, Berlot CH, Robert A, Inglis FM, Jakobsdottir KB, Howe JR, Hughes TE. A new way to rapidly create functional, fluorescent fusion proteins: random insertion of GFP with an in vitro transposition reaction. BMC Neurosci. 2002;3:7. doi: 10.1186/1471-2202-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih YL, Le T, Rothfield L. Division site selection in Escherichia coli involves dynamic redistribution of Min proteins within coiled structures that extend between the two cell poles. Proc Natl Acad Sci USA. 2003;100:7865–7870. doi: 10.1073/pnas.1232225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih YL, Rothfield L. The bacterial cytoskeleton. Microbiol Mol Biol Rev. 2006;70:729–754. doi: 10.1128/MMBR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi D, Yoshimoto M, Homma M, Kawagishi I. Helical distribution of the bacterial chemoreceptor via colocalization with the Sec protein translocation machinery. Mol Microbiol. 2006;60:894–906. doi: 10.1111/j.1365-2958.2006.05145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi D, Sakai M, Niki H. Determination of bacterial rod shape by a novel cytoskeletal membrane protein. EMBO J. 2008;27:3081–3091. doi: 10.1038/emboj.2008.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater S, Maurer R. Simple phagemid-based system for generating allele replacements in Escherichia coli. J Bacteriol. 1993;175:4260–4262. doi: 10.1128/jb.175.13.4260-4262.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MD, Kaguni JM. Novel alleles of the Escherichia coli dnaA gene are defective in replication of pSC101 but not of oriC. J Bacteriol. 1995;177:6657–6665. doi: 10.1128/jb.177.22.6657-6665.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torheim NK, Boye E, Lobner-Olesen A, Stokke T, Skarstad K. The Escherichia coli SeqA protein destabilizes mutant DnaA204 protein. Mol Microbiol. 2000;37:629–638. doi: 10.1046/j.1365-2958.2000.02031.x. [DOI] [PubMed] [Google Scholar]
- van den Ent F, Amos LA, Lowe J. Prokaryotic origin of the actin cytoskeleton. Nature. 2001;413:39–44. doi: 10.1038/35092500. [DOI] [PubMed] [Google Scholar]
- van den Ent F, Moller-Jensen J, Amos LA, Gerdes K, Lowe J. F-actin-like filaments formed by plasmid segregation protein ParM. EMBO J. 2002;21:6935–6943. doi: 10.1093/emboj/cdf672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanounou S, Parola AH, Fishov I. Phosphatidylethanolamine and phosphatidylglycerol are segregated into different domains in bacterial membrane. A study with pyrene-labelled phospholipids. Mol Microbiol. 2003;49:1067–1079. doi: 10.1046/j.1365-2958.2003.03614.x. [DOI] [PubMed] [Google Scholar]
- Wegrzyn A, Wrobel B, Wegrzyn G. Altered biological properties of cell membranes in Escherichia coli dnaA and seqA mutants. Mol Gen Genet. 1999;261:762–769. doi: 10.1007/s004380050019. [DOI] [PubMed] [Google Scholar]
- Weigel C, Schmidt A, Seitz H, Tungler D, Welzeck M, Messer W. The N-terminus promotes oligomerization of the Escherichia coli initiator protein DnaA. Mol Microbiol. 1999;34:53–66. doi: 10.1046/j.1365-2958.1999.01568.x. [DOI] [PubMed] [Google Scholar]
- Woldringh CL, Nanninga N. Structural and physical aspects of bacterial chromosome segregation. J Struct Biol. 2006;156:273–283. doi: 10.1016/j.jsb.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Xia W, Dowhan W. In vivo evidence for the involvement of anionic phospholipids in initiation of DNA replication in Escherichia coli. Proc Natl Acad Sci USA. 1995;92:783–787. doi: 10.1073/pnas.92.3.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung BY, Kornberg A. Membrane attachment activates dnaA protein, the initiation protein of chromosome replication in Escherichia coli. Proc Natl Acad Sci USA. 1988;85:7202–7205. doi: 10.1073/pnas.85.19.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Li Z, Skarstad K, Crooke E. Mutations in DnaA protein suppress the growth arrest of acidic phospholipid-deficient Escherichia coli cells. EMBO J. 2001;20:1164–1172. doi: 10.1093/emboj/20.5.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.