

Abstract
The Mo(V) state of the molybdoenzyme sulfite oxidase (SO) is paramagnetic and can be studied by electron paramagnetic resonance (EPR) spectroscopy. Vertebrate SO at pH < 7 and pH > 9 exhibits characteristic EPR spectra that correspond to two structurally different forms of the Mo(V) active center referred to as the low-pH (lpH) and high-pH (hpH) forms, respectively. Both EPR forms have an exchangeable equatorial OH ligand, but its orientation in the two forms is different. It has been hypothesized that the formation of the lpH species is dependent upon the presence of chloride. In this work we have prepared and purified samples of wild type and various mutants of human SO that are depleted in chloride. These samples do not exhibit the typical lpH EPR spectrum at low pH, but rather show spectra that are characteristic of the blocked species that contains an exchangeable equatorial sulfate ligand. Addition of chloride to these samples results in the disappearance of the blocked species and the formation of the lpH species. Similarly, if chloride is added before sulfite, the lpH species is formed instead of the blocked one. Qualitatively similar results were observed for samples of sulfite oxidizing enzymes from other organisms that were previously reported to form a blocked species at low pH. However, the depletion of chloride has no effect upon the formation of the hpH species.
The sulfite-oxidizing enzymes (SOEs), represented by sulfite oxidase (SO) in vertebrates and plants and sulfite dehydrogenase (SDH) in bacteria, catalyze the oxidation of sulfite to sulfate as represented by generic Eq. 1 (1).
| (1) |
In humans SO is essential for normal neonatal neurological development, and inborn deficiencies in SO result in severe physical and neurological disorders and early death (2, 3).
Reaction (1) is catalyzed by the square-pyramidal oxo-molybdenum active center, which has three equatorial sulfur ligands (one from the conserved cysteinyl side chain, and two from the molybdopterin cofactor), one axial oxo ligand, and an exchangeable equatorial oxo ligand in the solvent accessible pocket of the active site (4, 5). During the proposed catalytic cycle (6), sulfite initially reduces Mo(VI) to Mo(IV). Regeneration of the Mo(VI) state involves two one-electron oxidations, as shown in Scheme 1, (only the exchangeable equatorial ligand of the Mo ion is shown). The intermediate Mo(V) state is paramagnetic and has been extensively studied by electron paramagnetic resonance (EPR) spectroscopy (7–14).
Scheme 1.
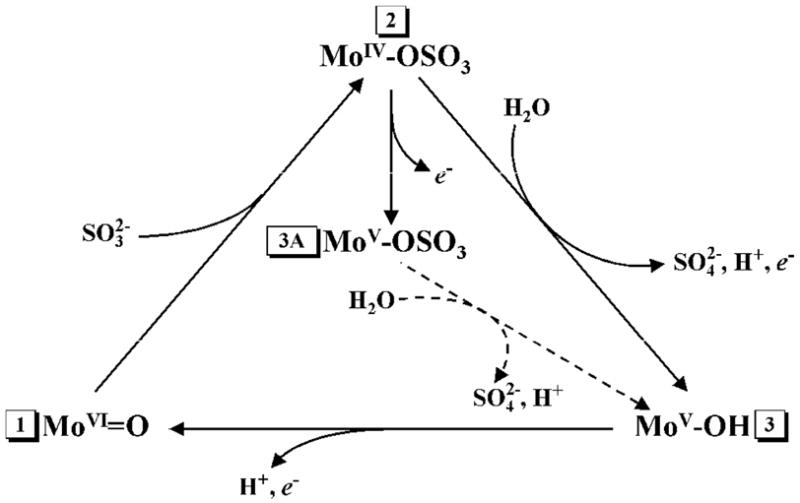
Simplified catalytic cycle of SO and SDH. Stage 1 corresponds to the resting state of the enzyme. The pathway 1→2→3→1 corresponds to the normal catalytic turnover. Depending on the buffer pH, Stage 3 corresponds to the hpH or lpH form of the active center. Stage 3A (typically observed at low pH < 7) corresponds to the blocked form of the active center. In wt vertebrate SO, Stage 3A is generated in chloride-depleted samples at low pH. Addition of chloride results in hydrolysis of sulfate (transition 3A→3).
Unlike X-ray crystallography or extended X-ray absorption fine structure (EXAFS) spectroscopy, EPR can detect protons in the vicinity of a paramagnetic center and is able to unequivocally identify specific nuclei through using substitutions by or permutations of magnetic isotopes (e.g., 16O → 17O, 35Cl → 37Cl, 14N → 15N, etc.). Both, continuous wave (CW) and pulsed EPR spectroscopic approaches have been used to establish the effects of pH, anions in the media, and mutations near the active site on the identity and structure of the exchangeable equatorial ligand of the Mo(V) ion. It was found that in the absence of inhibiting anions (e.g., PO43−, AsO43−), wild type (wt) vertebrate SO can show two distinct types of EPR signals, high-pH (hpH) and low-pH (lpH), corresponding to two different structural forms of the Mo active center. For both of these forms the exchangeable equatorial ligand is OH (see Stage 3 of Scheme 1). However, in some mutant forms of vertebrate SO (Y343F, R160Q hSO), as well as in the R55Q variant of bacterial SDH, and wt plant SO (At-SO from Arabidopsis thaliana), two types of low-pH blocked forms have recently been observed, in which the exchangeable equatorial ligand is sulfate (bound product) rather than OH (Stage 3A of Scheme 1) (15–17). The lethality of the pathogenic R160Q mutation of hSO has been attributed to the occurrence of the blocked form (16), which may represent a catalytically dead end, and inefficient intramolecular electron transfer (18).
Previous pH titrations of vertebrate SO have shown that the equilibrium between the lpH and hpH forms depends on the amount of Cl− added to the buffer in the form of NaCl (12). From these experiments it was hypothesized that Cl− is an integral part of the lpH form, likely to be coordinated to the Mo ion. Attempts to detect the hyperfine interaction (hfi) with this chloride were later undertaken by Doonan et al. using CW EPR and 35Cl or 37Cl-enriched NaCl (19). While some differences between the 37Cl- and 35Cl-enriched samples were observed, they were at the limit of the experimental accuracy. The weakness of the experimental CW EPR evidence for chloride interaction was, however, alleviated by the fact that when fluoride, bromide or iodide were used instead of chloride, clearly observable EPR splittings due to the halogen nucleus hfi were detected (12, 19). Based on these data, the chloride ligand was suggested to be weakly coordinated at the axial position of the Mo(V) center trans to the axial oxo ligand (19).
More conclusive and detailed information about the interaction between Mo and Cl in lpH SO was obtained recently using pulsed EPR and DFT calculations (17). The electron spin echo envelope modulation (ESEEM) caused by 35Cl and 37Cl was unequivocally detected, and the hfi and nuclear quadrupole interaction (nqi) parameters of the nearby chlorine nucleus were determined. DFT calculations performed for various structural models for incorporation of Cl− into the active site established that Cl− does not coordinate in the axial position, but rather is hydrogen bonded to the equatorial OH ligand, to the hydrogen atoms of the surrounding OH groups, and to amino acid residues of the binding site. Interestingly, even a sample of hSO purified elsewhere and prepared without added chloride showed the CW EPR spectrum characteristic of the lpH form (12, 19), as well as a 35,37Cl ESEEM (17). Thus, it was hypothesized that Cl− may be essential for the formation of the lpH form, and that wt vertebrate SO has a high affinity for Cl− at low pH (17).
Here we report that an SOE apparently lacking chloride near the Mo active site can be prepared when the enzyme is purified using an initial desalting column about twice as large as that employed in earlier preparations (20, 21). The wt hSO purified in this manner does not exhibit the typical lpH species at pH ~6, but shows instead formation of the blocked species, as confirmed by detecting characteristic 33S ESEEM in a sample prepared with 33S-labeled sulfite. A similar result was also obtained for various mutated hSO enzymes. Addition of chloride to these samples converted their EPR spectra to the typical lpH form (transition between Stages 3A and 3 in Scheme 1) with the exception of the lethal R160Q mutant of hSO. Similarly, adding chloride before the reduction with sulfite resulted in formation of the lpH species instead of the blocked one.
MATERIALS AND METHODS
Recombinant wt hSO and mutant forms of hSO were expressed and purified as previously described (20, 21). The chloride content of the enzyme samples was analyzed using QuantiChrom™ Chloride assay kit (DICL-250) from BioAssay Systems. Steady-state kinetic measurements were performed using the sulfite/cytochrome c assay reaction for SO and monitoring the reduction of cytochrome c at 550 nm. Concentrations of chloride and sulfite were varied and plots of l/rate vs. l/substrate were made for a series of chloride concentrations. The binding constant for chloride was determined from the x intercept of a replot of the slopes of the lines (l/rate vs. l/substrate) vs. chloride concentration (see Supporting Information).
The EPR samples at pH 5.8 were prepared in 100 mM Bis-Tris buffer. The samples at pH 7 and 9 were prepared in 100 mM Bis-Tris propane buffer. The enzyme was reduced with a 20-fold excess of sodium sulfite under argon and immediately frozen in liquid nitrogen. The same buffer system and procedure were used for reduction with 33S-labeled sulfite, prepared as previously described (15). After the blocked form was detected by EPR measurements of the samples at low pH, the samples were thawed at 0°C, 10 or 100 mM NaCl was added, the samples were frozen again, and the EPR measurements were repeated. In a parallel set of experiments, 100 mM NaCl was added to chloride-depleted enzymes prior to adding Na(SO3)2.
The X-band (~9 GHz) CW EPR experiments were performed on a Bruker ESP300 spectrometer at 77 K. The ESEEM measurements were done on a homebuilt Ka-band (26–40 GHz) pulsed EPR spectrometer (22). The measurement temperature was about 21 K.
The concentrations of Mo(V) in the SO enzyme samples were determined by comparing the double integrals of the X-band CW EPR spectra recorded at 77 K with that of the 5 mM Cu(NO3)2 in glassy water/glycerol solution. For double intergation, only the well-defined EPR features of the active centers containing non-magnetic (I = 0, ~ 75% natural abundance) molybdenum isotopes were used. The broad and rather featureless EPR lines of the Mo(V) centers containing magnetic molybdenum isotopes (95Mo, 97Mo, both I = 5/2, total natural abundance ~ 25%) were suppressed by the baseline subtraction preceding the double integration. Therefore, to account for the magnetic isotopes and obtain the total concentration of the Mo(V) active centers, the estimated concentration of the centers with non- magnetic isotopes was multiplied by 4/3. Typical total concentrations of Mo(V) centers relative to the enzyme concentrations were about 40% – 50%. Thawing the samples to add NaCl (overall handling time at 0 °C about 2 min) resulted in a decrease of the Mo(V) concentration to about 30% – 40%.
RESULTS AND DISCUSSION
Numerous previous EPR experiments on wt vertebrate SO at low pH (< 7) performed by us and other authors (7–10, 12, 17, 19) always showed formation of the lpH EPR-active Mo(V) species with characteristic hyperfine coupling from an exchangeable proton. Recently, we began preparing and purifying recombinant wt hSO in our own laboratory and were initially surprised to find that the EPR signal we systematically obtained at low pH (typically, 5.8) was similar to that characteristic of the blocked form of SO (trace 1 in Figure 1) with coordinated sulfate (product). Similar to the blocked forms observed earlier in wt At-SO (15) and Y343F hSO (16), this EPR signal is characterized by the principal g-values (g1, g2, g3) ≈ (1.999, 1.972, 1.963) and does not show any hyperfine splittings because the exchangeable equatorial ligand, sulfate, does not have any magnetic nuclei (the natural abundance of 16O is 99.757%, and that of 32S+34S is 99.2%).
Figure 1.
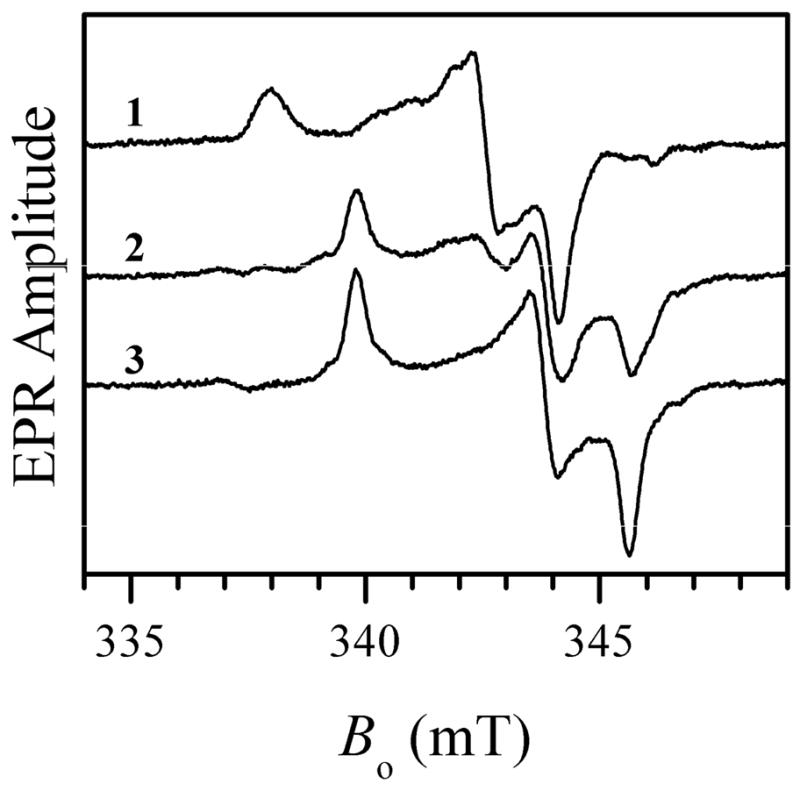
Traces 1, 2, and 3, X-band CW EPR spectra of wt hSO at pH 5.8, 7.0 and 9.0, respectively. No NaCl was added to the samples. Experimental conditions: microwave (mw) frequency, 9.455 GHz; mw power, 2 mW; magnetic field modulation amplitude, 0.1 mT; temperature, 77 K, enzyme concentrations, 430, 570, and 500 μM for traces 1, 2, and 3, respectively. Estimated relative concentrations of Mo(V) centers are 51%, 47% and 48% for traces 1, 2, and 3, respectively.
In order to verify that the EPR signal we detected for wt hSO indeed belongs to the blocked form, the enzyme was reduced with 33S-enriched sulfite, and Ka-band ESEEM experiments were performed similar to those described elsewhere (15, 16). The ESEEM spectra (see Figure 2) confirm that the observed EPR signal belongs to the blocked form of SO. The 33S hfi constant (aiso ≈ 2.6 MHz) and quadrupole coupling constant (e2Qq/h ≈ 36 MHz) estimated from hypefine sublevel correlation (HYSCORE) spectra (see Supporting Information) are similar to those found for the blocked species in other SOEs (15, 16). The samples prepared at high pH (~ 9.0) show the formation of the usual hpH species (trace 3 in Figure 1) characterized by the principal g-values (g1, g2, g3) ≈ (1.987, 1.964, 1.953) (10). At intermediate pH (e.g., pH 7) a mixture of the blocked and hpH forms was observed (trace 2 in Figure 1). The typical lpH form was not generated at any pH within the studied range from 5.8 to 9.
Figure 2.
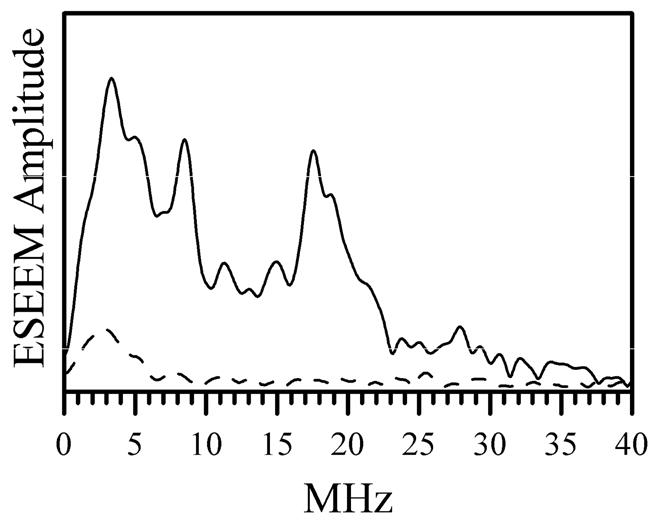
Amplitude Fourier transformation spectra of normalized two-pulse ESEEM spectra of 33S-labeled (i.e., reduced with 33S-enriched sulfite, solid trace) and unlabeled (dashed trace) chloride-depleted wt hSO at pH 5.8 obtained at gY EPR turning point. Experimental conditions: mw frequency, 29.51 GHz; Bo = 1074.1 mT; mw pulses, 9 and 15 ns; temperature, 21 K, enzyme concentrations, 600 μM for trace 1 and 430 μM for trace 2. Since the only difference between the samples was in the enrichment with magnetic isotope of sulfur (33S, natural abundance is 0.76%), the lines observed in the spectrum shown by the black trace are attributed to 33S.
Careful examination of the enzyme purification and sample preparation procedures has revealed that the only difference between the published procedure (20, 21) and our current procedure is in the larger size of the G25 desalting column (500 mL vs. 250 mL bed volume) used in our initial chromatographic purification step. Taking into account the recent suggestion that Cl− may be indispensable for the formation of the lpH species (17), we hypothesized that our EPR results can be explained if we assume that our purification procedure sufficiently reduces the Cl− concentration to preclude formation of the usual lpH form of SO. Indeed, the measurement of the chloride content of our enzyme samples prepared for EPR studies at pH 5.8 yielded the ratio [Cl−]/[SO] to be about 20. For comparison, the chloride concentration in samples of hSO purified elsewhere with the smaller desalting column was significantly larger, [Cl−]/[SO] ≈ 30–40.
To test if this low chloride concentration was responsible for the formation of the blocked species instead of the lpH form, Cl− was added to the low-pH sample of wt hSO that was purified in our laboratory as described above. Adding 10 mM NaCl resulted in formation of a substantial amount of the lpH species (characterized by the principal g-values (g1, g2, g3) ≈ (2.004, 1.973, 1.966) and showing the hyperfine splittings (A1, A2, A3) ≈ (0.8, 0.8, 1.3) mT at the EPR turning points caused by the hfi with the OH ligand proton (10), and a significant decrease in the amount of the blocked form (compare traces 1 and 2 in Figure 3). An EPR sample with 100 mM NaCl gave entirely the typical lpH form (trace 3 in Figure 3). When 100 mM NaCl was added to an EPR sample before the reduction by sulfite, the same end result was obtained: the usual lpH species was generated instead of the blocked one.
Figure 3.
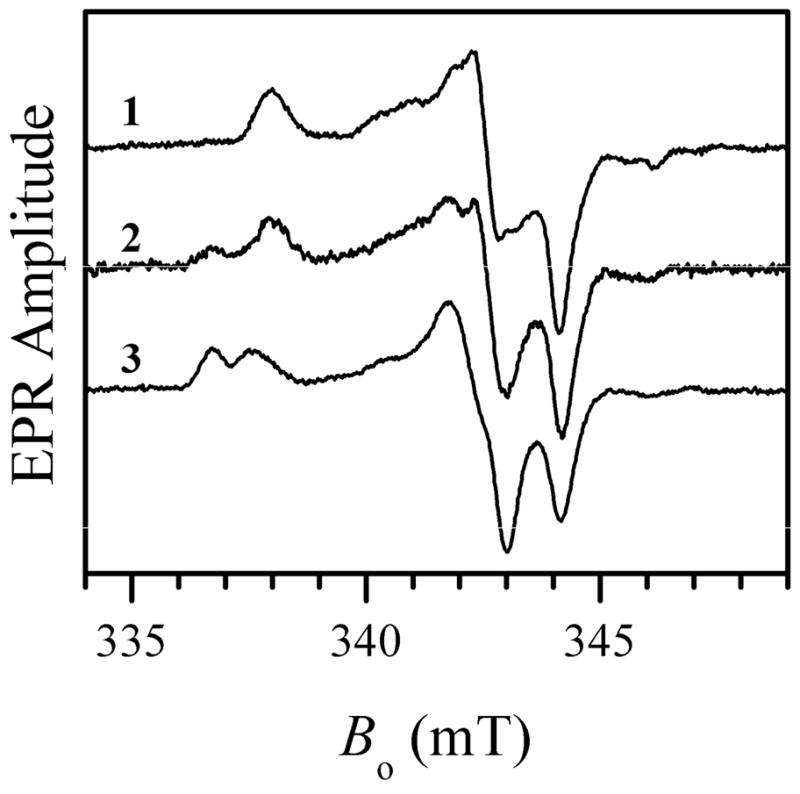
X-band CW EPR spectra of wt hSO at pH 5.8 without adding NaCl (trace 1, same as trace 1 in Figure 1), with 10 mM NaCl (trace 2), and with 100 mM NaCl (trace 3). Experimental conditions: mw frequency, 9.455 GHz; mw power, 2 mW; magnetic field modulation amplitude, 0.1 mT; temperature, 77 K, enzyme concentrations, 430 μM for traces 1 and 3, and 270 μM for trace 2. Estimated relative concentrations of Mo(V) centers are 51%, 30%, and 40% for traces 1, 2, and 3, respectively.
The effect of added Cl− on various preparations of SO and SDH that exhibited the blocked form was also studied for the purpose of comparison and generalization. With one exception, all of the hSO mutants purified in our lab (see Supporting Information) that exhibited the blocked form when prepared at low pH without added chloride, were completely converted to the typical lpH form by adding 100 mM NaCl (as exemplified in Figure 3 for wt hSO). The exception is the pathogenic R160Q mutant (16), which was not affected by added Cl− (up to 300 mM NaCl) and remained in the blocked form. On the other hand, for the analogous mutation (R55Q) of bacterial SDH the blocked form was converted to the lpH form by adding 100 mM NaCl (see Figure 4) (23). For At-SO and Y343F hSO at pH 6 adding 100 mM NaCl resulted in formation of measurable amounts of the typical lpH species, although the EPR spectra were still dominated by the blocked form (as an example, see Figure 5 for the results obtained for At-SO).
Figure 4.
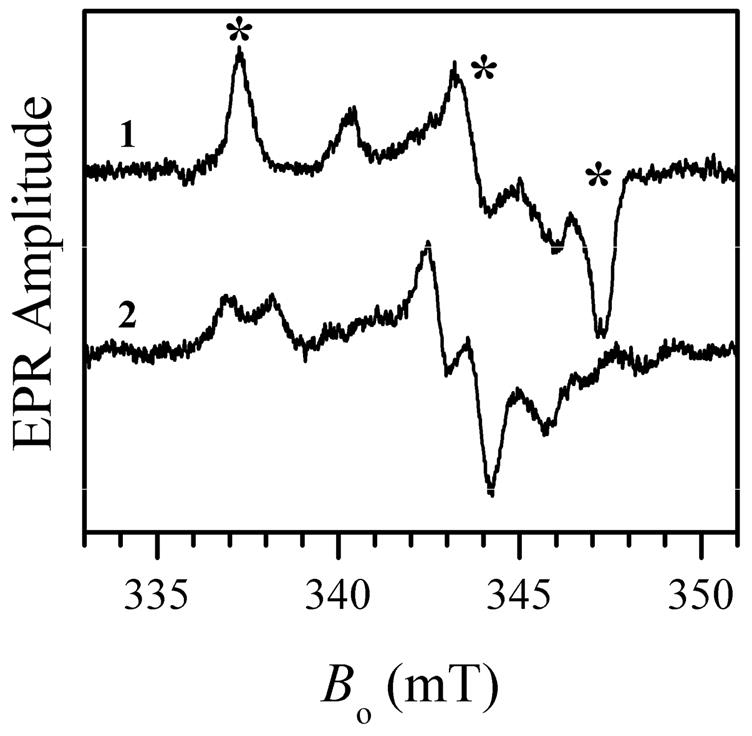
X-band CW EPR spectra of R55Q SDH at pH 4.9 without added NaCl (trace 1) and with 100 mM NaCl (trace 2). Experimental conditions: mw frequency, 9.470 GHz; mw power, 2 mW; magnetic field modulation amplitude, 0.1 mT; temperature, 77 K, enzyme concentration, 300 μM. The asterisks over trace 1 show the EPR turning points of the blocked species. The minor features in trace 1 belong to the hpH form. Estimated relative concentrations of Mo(V) centers are 42% and 30% for traces 1 and 2, respectively.
Figure 5.
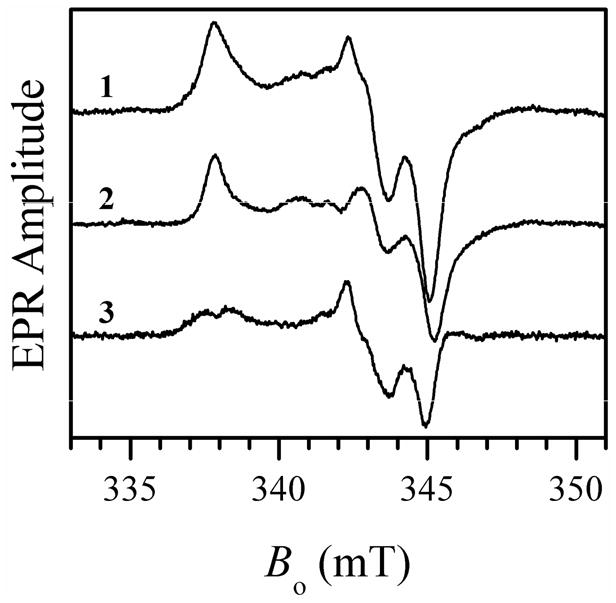
X-band CW EPR spectra of wt At-SO at pH 6.0 with added 100 mM NaCl (trace 1) and without NaCl (trace 2). Trace 3 represents the difference between traces 1 and 2 showing the contribution of the lpH SO spectrum obtained after addition of chloride. Experimental conditions: mw frequency, 9.483 GHz; mw power, 2 mW; magnetic field modulation amplitude, 0.1 mT; temperature, 77 K, enzyme concentration, 460 μM. Estimated relative concentrations of Mo(V) centers are 35% and 42% for traces 1 and 2, respectively.
The formation of the blocked form of hSO in chloride-depleted samples is consistent with the earlier proposal that multiple mechanisms may be possible for SO (24). The blocked form results from one-electron oxidation of the molybdenum center prior to product release to give a Mo(V)-OSO3 moiety (2 → 3a of Scheme 1). However, the weakly coordinating sulfate ion is readily replaced in the presence of other anions at low pH. Addition of chloride gives the well-known lpH form (3 in Scheme 1); other halogens (F−, Br−, I−) produce the same effect (12, 19). For those anions, however, the EPR splittings due to the halogen nucleus hfi are directly observed by CW EPR because the hfi constants of 19F, 79Br and 127I per unit unpaired electron spin density are several fold greater than that of 35,37Cl (25). Distinct EPR forms with coordinated phosphate (11, 26) and arsenate (9) have also been observed at low pH. It has also been suggested that the blocked form actually contains bound sulfite, the reactant, which is present in large excess in the EPR samples (27, 28).
Recently we used 35,37Cl ESEEM spectroscopy and DFT calculations to obtain information about the chloride binding site in the vicinity of the molybdenum active center (17). In contrast to earlier suggestions (19) that Cl− may be coordinated in the axial position to Mo(V), trans to the oxo ligand, our data have shown that in lpH SOEs Cl− is most likely hydrogen-bonded to the proton of the equatorial OH ligand as shown in Figure 6a. Other details of the Cl− binding site are less definitive, although it is clear the Cl− is stabilized in place by several hydrogen bonds, and the overall environment is fairly spherical. This is suggested by a relatively weak nuclear quadrupole interaction of this chloride ion. Based on X-ray structures (PDB 2A99 (4)), one of the amino acid residues involved in the Cl− binding site in chicken SO (cSO) was thought to be W204, which corresponds to W226 in hSO. However, mutations of W226 residue to Ala or Phe did not affect the formation of the typical lpH form with added 100 mM NaCl at low pH. Therefore, the role of W226 in stabilization of Cl− in the enzyme in solution is questionable.
Figure 6.

Putative structures of the substrate-accessible side of the Mo active center in (a) lpH, (b) blocked, and (c) hpH forms
One possible qualitative explanation of the chloride effect on hydrolysis of sulfate from the Mo active center is that Cl− competes with one of the distal oxygens of SO42− for the same binding site (see Figure 6b), so that when Cl− displaces this oxygen, SO42− is hydrolyzed more easily. This model is indirectly supported by the fact that neither the blocked species detected in this work nor those studied earlier (15, 16), exhibit 35,37Cl ESEEM spectra.
At high pH, the hydrolysis of sulfate and the complete catalytic turnover occur without the presence of halogens, with the Mo(V) hpH species being formed at Stage 3 of Scheme 1. The results of recent 1H and 17O ESEEM measurements suggest that in this case the exchangeable equatorial OH ligand is hydrogen-bonded to a second-sphere OH− (29, 30). We hypothesize, therefore, that at high pH the OH− anion plays the same role in facilitating the hydrolysis of sulfate as the halogen anions do at low pH. The provisional structure for an hpH SOE based on this hypothesis is shown in Figure 6c.
It would be tempting to interpret the EPR results described above as suggesting that chloride plays a physiological role in SO by facilitating the hydrolysis of the product from the molybdenum active center. The EPR experiments, however, do not represent a sufficient ground for such interpretation because the processes in the EPR samples of SO are limited by a single electron transfer event, and the catalytic cycle is incomplete. In order to assess the effects of chloride ion concentration on the overall catalytic cycle, we have carried out steady-state activity measurements on wt hSO at pH 5.8 and 8.0 (Table 1). At low pH (5.8), high concentrations of chloride greatly decrease the catalytic activity of recombinant hSO and exhibit “mixed” type inhibition. This result is in agreement with previous studies of the effect of chloride on the activity of native chicken SO (31). In addition, the presence of high concentrations of chloride (and other small anions) have an inhibitory effect on the rate constant for intramolecular electron transfer (IET) between the Mo(VI)/Fe(II) and Mo(V)/Fe(III) forms of SO (32). It was proposed that small anions which can fit into the Mo active site will weaken the electrostatic attraction between the Mo and heme domains and disfavor IET. The dissociation constant for Cl− from hSO, measured under steady-state conditions using the sulfite/cytochrome c assay reaction at pH 5.8, was 13 ± 3 mM (Table 1). From this result it follows that the chloride concentration in the EPR sample of trace 1 of Figure 1 (~5 mM) is insufficient to produce the lpH EPR signal. Hence, we observe the blocked form. The subsequent addition of chloride results in sufficient population of Cl in the active site to observe the lpH signal (traces 2 and 3 of Figure 1).
Table 1.
Steady-state data for wt-hSO with various amounts of NaCl added
| pH | Amount of added chloride, mM | kcat, s−1 | KM (sulfite), μM | Cl− dissociation constant, mM |
|---|---|---|---|---|
| 5.8 | 0 | 23 ± 2 | 2 ± 1 | 13 ± 3 |
| 2.5 | 26 ± 1 | 4 ± 1 | ||
| 5 | 28 ± 3 | 6 ± 1 | ||
| 15 | 15 ± 1 | 6 ± 1 | ||
| 30 | 17 ± 1 | 5 ± 1 | ||
| 100 | 4 ± 1 | 4 ± 1 | ||
| 8.0 | 0 | 27 ± 2 | 9 ± 3 | 14 ± 3 |
| 2.5 | 25 ± 1 | 9 ± 2 | ||
| 5 | 25 ± 1 | 7 ± 1 | ||
| 10 | 18 ± 1 | 8 ± 1 | ||
| 30 | 15 ± 1 | 8 ± 1 | ||
| 100 | 19 ± 1 | 12 ± 1 |
At high pH (8.0) the activity decreases by ~30% as the chloride concentration is increased to 100 mM, substantially less than the seven-fold decrease at pH 5.8. The steady-state results at high pH are consistent with the fact that these samples do not exhibit 35,37Cl ESEEM spectra (15, 16) and give a typical hpH EPR spectrum (29, 30).
The EPR results indicate that reduction of hSO with sulfite at low chloride concentrations and at low pH involves the one-electron oxidation of the MoIV-OSO3 center to MoV-OSO3 (pathway 2 → 3A in Scheme 1) to give the blocked form. However, the predominant Mo(V) species observed by EPR in the reductive half-reaction may not necessarily be the most catalytically efficient one. The steady-state experiments on SOEs involve a second one-electron oxidation to regenerate the MoVI form of the enzyme, and the possibility of multiple kinetic pathways for SOEs has been discussed previously (24). If the steady-state kinetics experiments at low chloride and low pH presented here for hSO also involve the blocked Mo(V) form (3A of Scheme 1), then a second one-electron oxidation to Mo(VI) could generate a catalytically competent species that could be hydrolyzed to product. Consequently, increasing the chloride concentration can have two competing effects. It can facilitate the dissociation of the product/substrate from Mo(V) state (Scheme 1, pathway 3A → 3) to give the typical lpH EPR form, and the amount of this form that is observed depends upon the SOE and the mutation. However, large concentrations of chloride simultaneously decrease the IET rates (32) and have an inhibitory effect on the catalytic activity (Table 1). These observations are consistent with a previous extensive microcoulometric study of the effects of anions and pH on the potentials of SO (33).
CONCLUSIONS
In contrast to all of the earlier experimental observations (7–10, 12, 17, 19), in this work we detected, for the first time, the formation of the blocked Mo(V) species in wt hSO at low pH (pH < 7). The formation of the blocked species was traced to the depletion of chloride from the purified enzyme by employing a large (compared to those used in other laboratories) desalting column. This observation supports the formation of Mo-OSO3 as the first step of the catalytic cycle of SO (Scheme 1). Adding NaCl to the wt hSO samples where the blocked species was formed results in hydrolysis of the product, sulfate, from the Mo active center and the formation of the usual lpH species. Similar experiments on several mutant forms of hSO have shown that the formation of the lpH species upon addition of Cl− is mutation-dependent, and the observation of an irreversible blocked species for the R160Q mutant of hSO supports the earlier hypothesis for the lethality of this mutation (16). Steady-state assays as a function of pH and chloride concentration show that kcat for wt hSO decreases with increasing chloride concentration, but this effect is much smaller at high pH. The combined EPR and kinetic data presented here are a further indication that the results of specific active site mutations on the chemical and spectroscopic properties of SOEs reflect a subtle interplay of inner- and outer-sphere effects among anions in the media, nearby amino acid side chains, and solvent molecules.
Supplementary Material
Acknowledgments
This research was supported by NIH Grant GM-037773 (to JHE) and Ruth L. Kirchstein-NIH Fellowship 1F32GM082136-01 (to KJW)). The construction of the pulsed EPR spectrometers was supported by grants from the NSF (DBI-0139459, DBI-9604939, BIR-9224431) and the NIH (S10RR020959) for development of the pulsed EPR facility.
We are indebted to Professor K. V. Rajagopalan for providing the pTG918 plasmid containing the hSO gene for preparing recombinant human sulfite oxidase and the protocols for purifying the enzyme. We are grateful to Professor F. Ann Walker for the use of equipment. We thank Dr. Eric L. Klein for preparing the 33S-labeled sulfite and Professor G. Tollin and Dr. A. Raitsimring for helpful discussions. Samples of At-SO and SDH were from previous collaborations (ref. (15) and (24), respectively).
Abbreviations
- SO
sulfite oxidase
- SOEs
sulfite oxidizing enzymes
- SDH
sulfite dehydrogenase
- EPR
electron paramagnetic resonance
- lpH
low-pH
- hpH
high-pH
- DFT
density functional theory
- ESEEM
electron spin echo envelope modulation
- HYSCORE
hypefine sublevel correlation
- hfi
hyperfine interaction
- wt
wild type
Footnotes
SUPPORTING INFORMATION: List of mutants, HYSCORE spectra of wt hSO enriched with 33S-enriched sulfite at pH 5.8, and Kinetic analyses plots for determination of binding constant for Cl−. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rajagopalan KV. In: Molybdenum and Molybdenum Containing Enzymes. Coughlan M, editor. Pergamon Press; New York: 1980. pp. 243–272. [Google Scholar]
- 2.Johnson JL, Rajagopalan KV, Renier WO, Van der Burgt I, Ruitenbeek W. Isolated Sulfite Oxidase Deficiency: Mutation Analysis and DNA-Based Prenatal Diagnosis. Prenat Diagn. 2002;22:433–436. doi: 10.1002/pd.335. [DOI] [PubMed] [Google Scholar]
- 3.Dublin AB, Hald JK, Wootton-Gorges SL. Isolated Sulfite Oxidase Deficiency: MR Imaging Features. Am J Neuroradiol. 2002;23:484–485. [PMC free article] [PubMed] [Google Scholar]
- 4.Karakas E, Wilson HL, Graf TN, Xiang S, Jaramillo-Buswuets S, Rajagopalan KV, Kisker C. Structural Insights into Sulfite Oxidase Deficiency. J Biol Chem. 2005;280:33506–33515. doi: 10.1074/jbc.M505035200. [DOI] [PubMed] [Google Scholar]
- 5.Kisker C, Schindelin H, Pacheco A, Wehbi W, Garrett RM, Rajagopalan KV, Enemark JH, Rees DC. Molecular Basis of Sulfite Oxidase Deficiency from the Structure of Sulfite Oxidase. Cell. 1997;91:973–983. doi: 10.1016/s0092-8674(00)80488-2. [DOI] [PubMed] [Google Scholar]
- 6.Hille R. The Mononuclear Molybdenum Enzymes. Chem Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 7.Cohen HJ, Fridovich I, Rajagopalan KV. Hepatic Sulfite Oxidase- A Functional Role for Molybdenum. J Biol Chem. 1971;246:374–382. [PubMed] [Google Scholar]
- 8.Kessler DL, Rajagopalan KV. Purification and Properties of Sulfite Oxidase from Chicken Liver. J Biol Chem. 1972;247:6566–6573. [PubMed] [Google Scholar]
- 9.George GN, Garrett RM, Graf T, Prince RC, Rajagopalan KV. Interaction of Arsenate with the Molybdenum Site of Sulfite Oxidase. J Am Chem Soc. 1998;120:4522–4523. [Google Scholar]
- 10.Lamy MT, Gutteridge S, Bray RC. Electron-Paramagnetic-Resonance Parameters of Molybdenum(V) in Sulphite Oxidase from Chicken Liver. Biochem J. 1980;185:397–403. doi: 10.1042/bj1850397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutteridge S, Lamy MT, Bray RC. The Nature of the Phosphate Inhibitor Complex of Sulphite Oxidase from Electron-Paramagnetic-Resonance Studies using Oxygen-17. Biochem J. 1980;191:285–288. doi: 10.1042/bj1910285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bray RC, Gutteridge MT, Lamy MT, Wilkinson T. Equilibria Amongst Different Molybdenum(V)-Containing Species from Sulphite Oxidase. Evidence for a Halide Ligand of Molybdenum in the Low-pH Species. Biochem J. 1983;211:227–236. doi: 10.1042/bj2110227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enemark JH, Astashkin AV, Raitsimring AM. High Resolution EPR Spectroscopy of Mo-Enzymes. Sulfite Oxidases: Structural and Functional Implications, in Biological Magnetic Resonance. In: Hanson GR, Berliner LJ, editors. Metals in Biology: Applications of High Resolution EPR to Metalloenzymes. Vol. 29. Springer; 2010. pp. 121–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enemark JH, Astashkin AV, Raitsimring AM. Investigation of the Coordination Structures of the Molybdenum(V) Sites of Sulfite Oxidizing Enzymes by Pulsed EPR Spectroscopy. Dalton Trans. 2006:3501–3514. doi: 10.1039/b602919a. [DOI] [PubMed] [Google Scholar]
- 15.Astashkin AV, Johnson-Winters K, Klein EL, Byrne RS, Hille R, Raitsimring AM, Enemark JH. Direct Demonstration of the Presence of Coordinated Sulfate in the Reaction Pathway of Arabidopsis thaliana Sulfite Oxidase using 33S Labeling and ESEEM Spectroscopy. J Am Chem Soc. 2007;129:14800–14810. doi: 10.1021/ja0704885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Astashkin AV, Johnson-Winters K, Klein EL, Feng C, Wilson HL, Rajagopalan KV, Raitsimring AM, Enemark JH. Structural Studies of the Molybdenum Center of the Pathogenic R160Q Mutant of Human Sulfite Oxidase by Pulsed EPR Spectroscopy and 17O and 33S Labeling. J Am Chem Soc. 2008;130:8471–8480. doi: 10.1021/ja801406f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein EL, Astashkin AV, Ganyushin D, Riplinger C, Johnson-Winters K, Neese F, Enemark JH. Direct Detection and Characterization of Chloride in the Active Site of the Low-pH Form of Sulfite Oxidase using Electron Spin Echo Envelope Modulation Spectroscopy, Isotopic Labeling, and Density Functional Theory Calculations. Inorg Chem. 2009;48:4743–4752. doi: 10.1021/ic801787s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng C, Wilson HL, Hurley JK, Hazzard JT, Tollin G, Rajagopalan KV, Enemark JH. Essential Role of Conserved Arginine 160 in Intramolecular Electron Transfer in Human Sulfite Oxidase. Biochemistry. 2003;42:12235–12242. doi: 10.1021/bi0350194. [DOI] [PubMed] [Google Scholar]
- 19.Doonan CJ, Wilson HL, Rajagopalan KV, Garrett RM, Bennett B, Prince RC, George GN. Modified Active Site Coordination in a Clinical Mutant of Sulfite Oxidase. J Am Chem Soc. 2008;130:6298–6298. doi: 10.1021/ja071402a. [DOI] [PubMed] [Google Scholar]
- 20.Temple CA, Graf TN, Rajagopalan KV. Optimization of Expression of Human Sulfite Oxidase and its Molybdenum Domain. Arch Biochem Biophys. 2000;383:281–287. doi: 10.1006/abbi.2000.2089. [DOI] [PubMed] [Google Scholar]
- 21.George GN, Garrett RM, Prince RC, Rajagopalan KV. The Molybdenum Site of Sulfite Oxidase: A Comparison of Wild-Type and the Cysteine 207 to Serine Mutant using X-Ray Absorption Spectroscopy. J Am Chem Soc. 1996;118:8588–8592. [Google Scholar]
- 22.Astashkin AV, Enemark JH, Raitsimring A. 26.5–40 GHz K-a-Band Pulsed EPR Spectrometer. Concepts in Magnetic Resonance Part B-Magnetic Resonance Engineering. 2006;29B:125–136. [Google Scholar]
- 23.Rapson TD, Astashkin AV, Johnson-Winters K, Bernhardt KV, Kappler U, Raitsimring AM, Enemark JH. Pulsed EPR Investigations of the Mo(V) Centers of the R55Q and R55M Variants of Sulfite Dehydrogenase from Starkeya novella. J Biol Inorg Chem. 2010;15:505–514. doi: 10.1007/s00775-009-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emesh S, Rapson TD, Rajapakshe A, Kappler U, Bernhardt PV, Tollin G, Enemark JH. Intramolecular Electron Transfer in Sulfite-Oxidizing Enzymes: Elucidating the Role of a Conserved Active Site Arginine. Biochemistry. 2009;48:2156–2163. doi: 10.1021/bi801553q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morton JR, Preston KF. Atomic Parameters for Paramagnetic Resonance Data. J Magn Reson. 1978;30:577–582. [Google Scholar]
- 26.Pacheco A, Basu P, Borbat P, Raitsimring AM, Enemark JH. Multifrequency ESEEM Spectroscopy of Sulfite Oxidase in Phosphate Buffer: Direct Evidence for Coordinated Phosphate. Inorg Chem. 1996;35:7001–7008. doi: 10.1021/ic9607806. [DOI] [PubMed] [Google Scholar]
- 27.Bray RC, Lamy MT, Gutteridge SWT. Evidence from Electron-Paramagnetic-Resonance Spectroscopy for a Complex of Sulfite Ions with the Molybdenum Centre of Sulfite Oxidase. Biochem J. 1982;201:241–243. doi: 10.1042/bj2010241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Enemark JH, Raitsimring AM, Astashkin AV, Klein EL. Implications for the Mechanism of Sulfite Oxidizing Enzymes from Pulsed EPR Spectroscopy and DFT Calculations for “Difficult” Nuclei. Faraday Discuss. (148) doi: 10.1039/c004404k. Submitted 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Astashkin AV, Mader ML, Enemark JH, Pacheco A, Raitsimring AM. Direct Detection of the Proton-Containing Group Coordinated to Mo(V) in the High-pH Form of Chicken Liver Sulfite Oxidase by Refocused Primary ESEEM Spectroscopy: Structural and Mechanistic Implications. J Am Chem Soc. 2000;122:5294–5302. [Google Scholar]
- 30.Astashkin AV, Klein EL, Ganyushin D, Johnson-Winters K, Neese F, Kappler U, Enemark JH. Exchangeable Oxygens in the Vicinity of the Molybdenum Center of the High-pH Form of Sulfite Oxidase and Sulfite Dehydrogenase. Phys Chem Chem Phys. 2009;11:6733–6742. doi: 10.1039/b907029j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan EP, Hazzard JT, Tollin G, Enemark JH. Electron Transfer in Sulfite Oxidase: Effects of pH and Anions on Transient Kinetics. Biochemistry. 1993;32:12465–12470. doi: 10.1021/bi00097a026. [DOI] [PubMed] [Google Scholar]
- 32.Pacheco A, Hazzard JT, Tollin G, Enemark JH. The pH Dependence of Intramolecular Electron Transfer Rates in Sulfite Oxidase at High and Low Anion Concentrations. J Biol Inorg Chem. 1999;4:390–401. doi: 10.1007/s007750050325. [DOI] [PubMed] [Google Scholar]
- 33.Spence JT, Kipke CA, Enemark JH, Sunde RH. Stoichiometry of Electron Uptake and the Effect of Anions and pH on the Molybdenum and Heme Reduction Potentials of Sulfite Oxidase. Inorg Chem. 1991;30:3011–3015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.