

Abstract
Lipodystrophic syndromes are characterized by adipose tissue deficiency. Although rare, they are of considerable interest as they, like obesity, typically lead to ectopic lipid accumulation, dyslipidaemia and insulin resistant diabetes. In this paper we describe a female patient with partial lipodystrophy (affecting limb, femorogluteal and subcutaneous abdominal fat), white adipocytes with multiloculated lipid droplets and insulin-resistant diabetes, who was found to be homozygous for a premature truncation mutation in the lipid droplet protein cell death-inducing Dffa-like effector C (CIDEC) (E186X). The truncation disrupts the highly conserved CIDE-C domain and the mutant protein is mistargeted and fails to increase the lipid droplet size in transfected cells. In mice, Cidec deficiency also reduces fat mass and induces the formation of white adipocytes with multilocular lipid droplets, but in contrast to our patient, Cidec null mice are protected against diet-induced obesity and insulin resistance. In addition to describing a novel autosomal recessive form of familial partial lipodystrophy, these observations also suggest that CIDEC is required for unilocular lipid droplet formation and optimal energy storage in human fat.
Keywords: lipodystrophy, insulin resistance, lipid droplet, CIDEC (Fsp27)
INTRODUCTION
White adipose tissue (WAT) is essential for efficient energy (lipid) storage and release (Frayn, 2002). Although all eukaryotic cells can store surplus energy as lipid droplets, white adipocytes are uniquely adapted for the role. Several lipid droplet associated proteins are specifically expressed in white adipocytes, where they appear to be required for efficient lipid storage in and release from lipid droplets (Brasaemle et al, 2008; Traini & Jessup, 2009). Storing triglyceride and cholesterol esters in a single large droplet reduces the lipid droplet surface area available to lipolytic enzymes and optimizes the storage capacity. The importance of fat in human metabolism is highlighted by lipodystrophic syndromes, a heterogeneous cluster of disorders characterized by a lack of WAT (Garg, 2004). Lipodystrophy is distinct from leanness, a state in which ‘empty adipocytes’ can readily adapt to positive energy balance. Instead, in lipodystrophic subjects, positive energy balance leads to ectopic fat deposition in the liver and other organs, insulin resistance and diabetes (Savage et al, 2007).
Lipodystrophies result from either the failure of adipocyte development or premature destruction of adipocytes due to genetic or immunological mechanisms (Garg, 2004). Recent progress in understanding the genetic basis of several forms of familial lipodystrophy has facilitated improved clinical diagnostic workup in patients with lipodystrophy as well as providing novel insights into adipocyte biology. Biallelic loss of function mutations in BSCL2, AGPAT2 or CAV1 account for >90% of all cases of congenital generalized lipodystrophy, whereas heterozygous mutations in LMNA and PPARG account for >50% of all inherited cases of partial lipodystrophy (Garg & Agarwal, 2009). Rare homozygous and compound heterozygous mutations in LMNA and ZMPSTE24 have also been reported and, in one family with partial lipodystrophy, a heterozygous mutation in AKT2 was identified (George et al, 2004).
In this report, we describe a 14-year-old girl with a novel subtype of partial lipodystrophy and ‘ketosis-prone’ insulin resistant diabetes in association with a homozygous nonsense mutation in CIDEC.
CASE HISTORY
A 19-year-old Ecuadorian girl was referred with partial lipodystrophy and insulin resistant diabetes. She first presented at age 14 years with diabetic ketoacidosis (glucose 39.4 mmol/l; pH 7.25; bicarbonate 8.3 mmol/l; strongly positive plasma ketones). Clinically she was noted to have partial lipodystrophy with muscular lower limbs and prominent acanthosis nigricans (Fig 1A). This phenotype was reportedly present from early childhood. Her body mass index (BMI) was 20.8 kg/m2 (BMI Standard deviation score (SDS) +0.52), blood pressure normal (103/64 mm Hg) and she had no signs of virilization or dysmorphic features. Menarche had occurred at age 12 years and menses were regular at presentation, but later became irregular. ICA (islet cell antibodies), anti-GAD (GAD, glutamic acid decarboxylase) and IA2 (protein tyrosine phosphatase-like protein) antibodies were all negative and C-peptide levels were elevated (1274 pmol/l (174–960)). Interestingly, her lipid profile was normal at presentation (total cholesterol 4.3 mmol/l, triglycerides 1.0 mmol/l) but worsening dyslipidaemia developed within 18 months, to the extent that she ultimately had pancreatitis secondary to hypertriglyceridaemia (triglycerides 20.2 mmol/l) and again presented with diabetic ketoacidosis (pH 6.77; strongly positive ketones). Despite high dose insulin therapy (1.6 IU/kg/day), her glycemic control was always poor (HbA1c 11.0–16.3%). She recently developed microalbuminuria and hypertension (blood pressure 153/96 mm Hg).
Figure 1. Identification of a homozygous premature stop E186X CIDEC mutation in a patient with acanthosis nigricans and partial lipodystrophy.
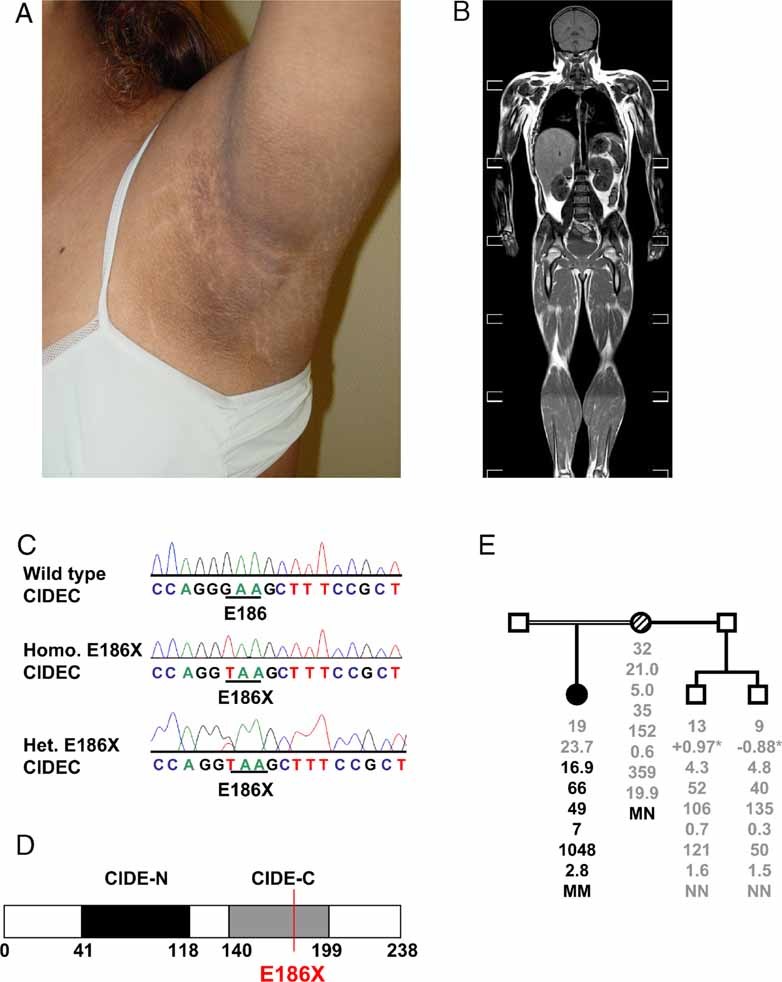
- Photograph demonstrating axillary acanthosis nigricans in the proband.
- T1 weighted magnetic resonance image of the proband indicating the paucity of leg-, forearm-, femorogluteal- and subcutaneous abdominal fat. Axilliary fat and neck fat are preserved. The liver is significantly enlarged due to severe steatosis. She also manifests prominent muscle bulk.
- Wild type CIDEC, E186X CIDEC homozygous (Homo; proband) and heterozygous (Het; proband's mother) mutant sequence traces. Homozygous transversion of guanine to thymine at nucleotide position 556 in exon 6 of CIDEC, results in the substitution of a premature stop codon (TAA) for glutamine (GAA) at codon 186.
- Schematic representation of CIDEC showing the position of the E186X premature stop mutation (red line), resulting in loss of a significant portion of the CIDE-C domain (grey) of CIDEC. The CIDE-N domain is highlighted in black and amino acid numbers are indicated below.
- Proband's family pedigree (squares represent male and circles female family members). Below each symbol, age (years) is given, followed by the BMI (kg/m2), fasting glucose (mmol/l), insulin (pmol/l), HOMA (homeostasis model assessment value; %), triglycerides (mmol/l), free fatty acids (µmol/l), leptin (µg/l) and genotype, with N denoting the normal (wild type) allele and M the mutant allele.
*BMI for children is given as BMI SDS.
Filled symbol represents the affected proband and hatched symbol her heterozygous mother.
Magnetic resonance imaging (MRI) of her fat distribution confirmed the virtual absence of lower limb and femorogluteal fat pads and the preservation of visceral, neck and axilliary fat (Fig 1B and Fig 1 of Supporting Information). Her total fat mass was 20% by DXA (dual energy X-ray absorptiometry), which is 1 SD below the age-matched mean. Plasma leptin (2.8 µg/l; BMI- and gender-matched reference range 2.4–24.4 µg/l) and adiponectin levels (1.7 mg/l; BMI- and gender-matched reference range 2.6–12.6 mg/l) were correspondingly low. She was also noted to have striking hepatomegaly and hepatic steatosis on MRI (Fig 1B).
RESULTS
Identification of a homozygous nonsense CIDEC mutation
The proband was wild type for all coding exons and splice junctions of LMNA, PPARG, ZMPSTE24 and AKT2, so we went on to sequence additional candidate genes for lipodystrophy. We identified a homozygous transversion of guanine to thymine at cDNA nucleotide position 556 in exon 6 of CIDEC, resulting in the substitution of a premature stop codon (TAA) for a glutamine codon (GAA) at codon 186 (ca 556G → T, p.Glu186* (E186X); Fig 1C). The mutation is predicted to truncate the protein at amino acid 186, resulting in loss of a significant portion of the CIDE-C domain of CIDEC (Fig 1D). It was absent in 120 ethnically matched control alleles. The proband's unaffected mother was heterozygous for the CIDEC E186X variant. Her BMI, fat distribution and biochemical parameters were all entirely normal (Fig 1E). Two maternal half brothers were wild type for this variant and manifested normal fat distribution and biochemistry. The proband's father was not available for genetic testing but single nucleotide polymorphism (SNP) genotyping analysis of the proband revealed a total genomic homozygosity value of 13.5% which is consistent with parental consanguinity (A-M Patch, unpublished observations; Woods et al, 2006). The mutation containing the homozygous segment was found to be 19.03-Mb long, spanning 3p26.3–3p24.3 delimited by the SNPs rs163577–rs17070741.
Adipose tissue histology
Since CIDEC (mouse orthologue is known as Fsp27) is a lipid droplet protein implicated in the formation of white adipocytes with unilocular lipid droplets in mice (Nishino et al, 2008; Toh et al, 2008), the proband went on to have a subcutaneous fat biopsy. The most striking finding in the tissue sample was the presence of many adipocytes with multiple small lipid droplets, each of which was strongly immunoreactive to perilipin (so called ‘multilocular adipocytes’), rather than the normal single large droplet (Fig 2A). The number of lipid droplets per cell varied inversely with cell size (Fig 2B). Unilocular adipocytes were similar in size to adipocytes from a lean control (Fig 2 of Supporting Information). Immunohistochemistry for UCP1 (uncoupling protein 1) was negative (data not shown), suggesting that these were white-, not brown adipocytes, but intense focal cytochrome c immunoreactivity in some cells with multilocular lipid droplets suggests that mitochondrial mass is increased in at least some of the proband's adipocytes (Fig 2C). Electron microscopy (EM) confirmed the multilocular nature of lipid droplets in many white adipocytes (Fig 2D) and the apparent focal increase in mitochondrial density (Fig 2D). There was no evidence of excess crown-like structures or inflammation in the adipose tissue.
Figure 2. Adipose tissue histology from the E186X CIDEC proband.
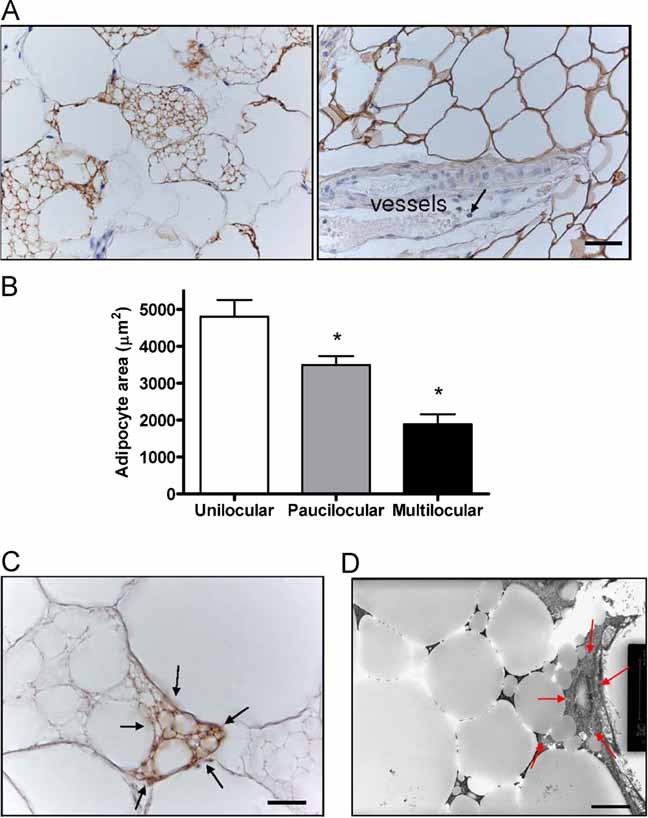
- Light microscopy of a subcutaneous adipose tissue biopsy taken from the lateral chest wall (axillary region) of the CIDEC E186X proband showing several perilipin positive multilocular adipocytes (left). Perilipin staining in control adipose tissue is seen in the right hand panel. Note that vessels and granulocytes (arrow) inside the vessels are negative to perilipin. Mean unilocular adipocyte cross sectional area is 4799.8 ± 451.04 µm2 corresponding to a mean adipocyte weight of 0.22 ± 0.00 µg lipid per cell in the patient's sample. In comparison, the mean adipocyte area in dorsal subcutaneous adipose tissue from a BMI-matched person was 5379.84 ± 702.07 µm2 and the mean adipocyte weight from the same control was 0.27 ± 0.01 µg lipid per cell. Results are given as mean ± SE.
- Light microscopy revealed a mixed cell population containing unilocular (UL), multilocular (ML) and intermediate paucilocular (PL) adipocytes in the proband's biopsy. Mean ± SE cell size was estimated by analysing 21 cells of each type. *p < 0.05 compared with unilocular adipocytes.
- Immunoreactivity for cytochrome c, a mitochondrial marker, is increased focally (arrows) in some multilocular adipocytes from the E186X CIDEC proband, but not in unilocular adipocytes. The ‘dot-like’ immunoreactivity corresponds to mitochondria.
-
Electron micrograph (EM) showing multilocular lipid droplets in adipose tissue from the proband with focally increased mitochondria (red arrows point to the area containing mitochondria).Scale bar length: panel A = 36.23 µm; C = 14.32 µm; D = 5 µm.
Although the ‘multilocular adipocytes’ seen in this patient are not typical brown adipocytes, we wondered if these cellular changes might alter the resting metabolic rate (RMR). The proband's RMR was significantly increased when compared to healthy controls. The elevation in RMR remained after correction for lean body mass (Fig 3A of Supporting Information) and for fat and lean mass (Fig 3B of Supporting Information). Thyroid function tests and plasma metanephrine concentrations were normal (data not shown).
Cellular characterization of the E186X CIDEC mutant
In order to examine the function of the mutant CIDEC protein, we electroporated COS cells and 3T3L1 pre-adipocytes with N-terminal green fluorescent protein-tagged (GFP-tagged) human CIDEC vectors. Although lipid droplet size was significantly increased in COS cells over expressing wild type CIDEC compared to untransfected cells (Fig 3A), lipid droplet size was not altered in cells over expressing the mutant protein at a similar level (Fig 3A). Similar effects were observed in 3T3L1 pre-adipocytes (Fig 3B). Formal morphometric analysis confirmed these visual observations in both cell types (Fig 3C) and suggested that the mutant protein is biologically inert, at least in terms of the protein's actions on lipid droplet size in cultured cells. Overlay of the GFP and oil-red-O stained images shows some co-localization of wild type CIDEC and lipid droplets whereas this is not seen in cells expressing the truncated CIDEC protein (Fig 3A,B). The fact that most of the WT CIDEC is not localized around lipid droplets in these cells could be due to the need for other differentiation-dependent proteins present in adipocytes to facilitate CIDEC localization to lipid droplets. Thus we also assessed localization of the wild type and E186X CIDEC mutant in transfected adipocytes (six days after initiating differentiation). Whereas most of the wild type CIDEC is localized around lipid droplets in these transfected adipocytes, the CIDEC truncated mutant is not (Fig 4), suggesting that the C-terminal part of CIDEC is required for lipid droplet localization.
Figure 3. Cellular analysis of the impact of wild type and E186X truncated CIDEC expression on lipid droplet size.
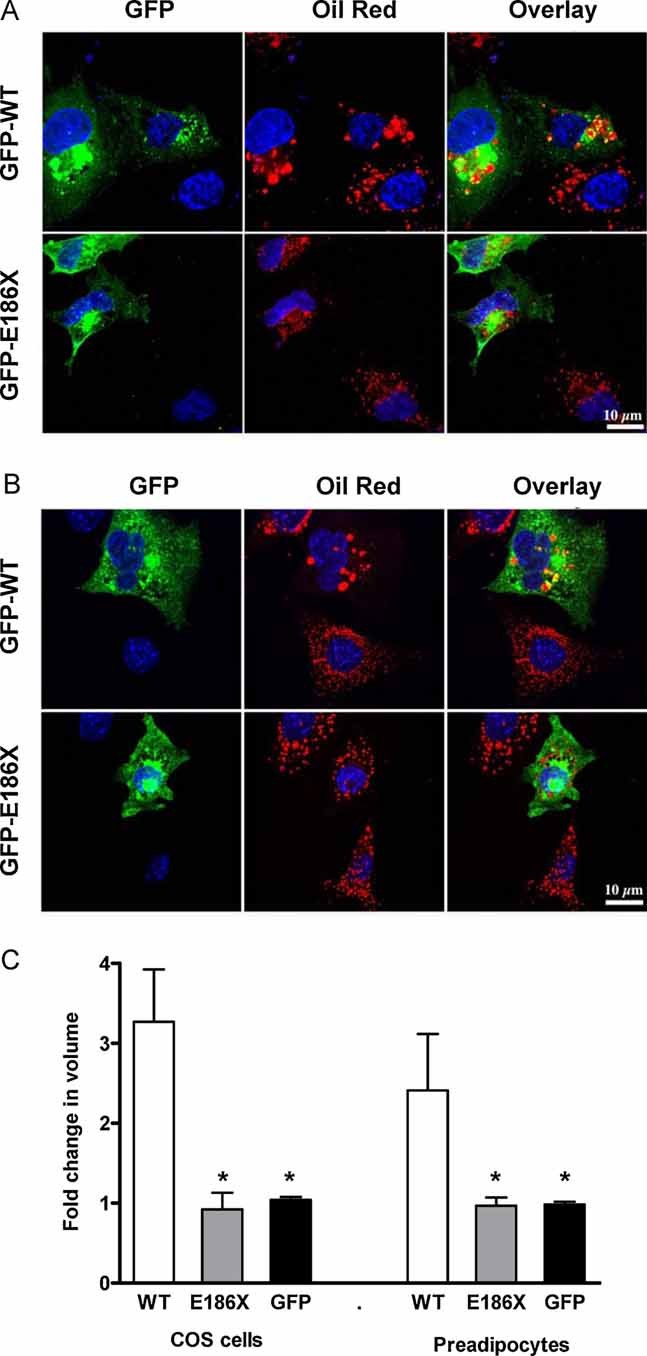
- COS cells transfected for 24 h.
- 3T3L1 pre-adipocytes transfected for 32 h with human wild type GFP-CIDEC (WT, top row) and E186X truncated GFP-CIDEC (E186X, bottom row) and cultured in a medium containing 20 µM OA/BSA (oleic acid conjugated to bovine serum albumin). Middle panel in the top row of each panel shows increased oil red staining of neutral lipids in cells expressing human wild type CIDEC. Overlay of the GFP and oil red stains indicate localization of some wild type CIDEC to lipid droplets, whereas there is no overlap of E186X GFP-CIDEC and oil red staining. Expression of GFP alone has no effect on lipid droplet size in COS and 3T3lL1 adipocytes (Fig 4 of Supporting Information). Scale bar: 10 µm.
- Quantitative morphometric analysis of lipid droplets in COS cells and 3T3-L1 pre-adipocytes transfected with human wild type (WT) GFP-CIDEC or E186X truncated GFP-CIDEC (E186X) or GFP alone (GFP) for 24 h. The values represent a fold change (mean ± SE) of lipid droplet volume of transfected cells as compared to untransfected cells in the same field. *p < 0.01 compared to WT.
Figure 4. Mislocalization of E186X CIDEC in adipocytes.
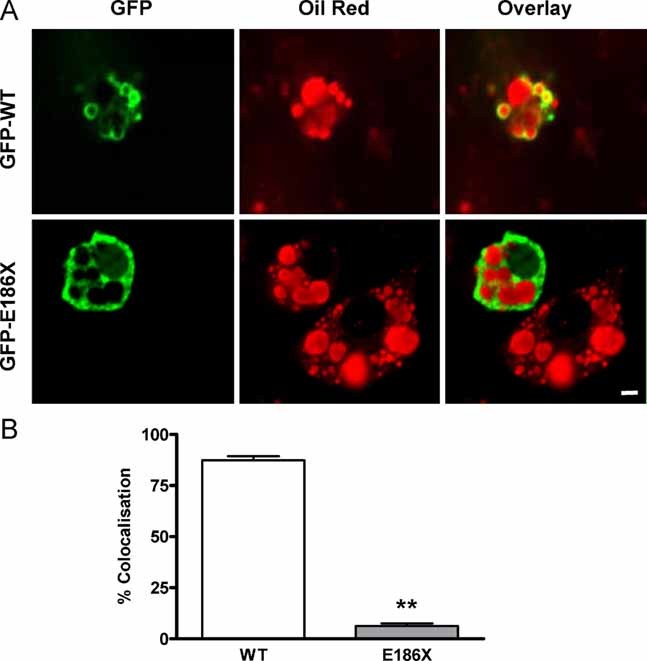
- Day six differentiated 3T3L1 adipocytes transfected with human wild type GFP-CIDEC (WT, top row) and E186X truncated GFP-CIDEC (E186X, bottom row). Overlay (right panels) of the GFP (left panels) and oil red stains (middle panels) suggest that most WT CIDEC surrounds lipid droplets whereas the mutant protein is almost entirely cytosolic. Scale bar: 10 µm.
- Co-localization analysis of GFP florescence with oil red-labelled lipid droplets in each Z-section of a transfected adipocyte. The data are from 8–10 adipocytes in each condition from three independent experiments. **p < 0.0001 compared to WT.
DISCUSSION
In a patient with unexplained partial lipodystrophy, we identified a homozygous premature stop mutation in CIDEC. CIDEC/Fsp27 is a lipid droplet protein most highly expressed in adipocytes where its expression is induced during adipocyte differentiation (Keller et al, 2008; Puri et al, 2007). Fsp27 knockdown in 3T3L1 pre-adipocytes reduces triglyceride accumulation and lipid droplet size, and increases lipid droplet number and lipolysis (Keller et al, 2008; Nishino et al, 2008). In keeping with these data, Fsp27 knockout (KO) mice manifest reduced adipose tissue mass and white adipocytes with multilocular lipid droplets (Nishino et al, 2008; Toh et al, 2008). CIDEC is expressed in human WAT where its mRNA level is inversely correlated with fat mass, reduced by a very low calorie diet (Magnusson et al, 2008), and lower in adipose tissue from obese insulin resistant compared to equally obese insulin sensitive subjects (Puri et al, 2008). The latter observation is particularly interesting as it suggests that reduced CIDEC expression may contribute to the pathogenesis of obesity-induced insulin resistance.
The CIDEC E186X mutation disrupts the highly conserved CIDE-C domain which is known to be required for the protein's function (Chen et al, 2000; Keller et al, 2008). Expressing the N-terminal half of Fsp27 in fibroblasts had no effect on lipid droplet formation, whereas expression of the C-terminal CIDE-C domain was sufficient to stimulate lipid accumulation (Keller et al, 2008). The homologous CIDE-C domain of Cideb is required for phospholipid membrane targeting and appears to be equally critical for its biological activity (Ye et al, 2009). When expressed in COS cells or 3T3L1 pre-adipocytes, the mutant CIDEC protein is mistargeted and has similar effects on lipid droplet morphology to those of an empty vector, whereas expression of wild type CIDEC significantly enhanced the volume of cellular lipid droplets. The truncated CIDEC protein is also mistargeted in differentiated 3T3L1 adipocytes where the bulk of wild type CIDEC localizes around lipid droplets. The most striking finding in our patient was the presence of many white adipocytes with multilocular lipid droplets and focal increases in mitochondrial mass. These observations are consistent with what was seen in Fsp27 knockdown cells and in Fsp27 null mice (Nishino et al, 2008; Toh et al, 2008) but in contrast to the KO mouse phenotype, these changes were associated with an adverse lipodystrophic, rather than a healthy lean, phenotype. The presence of severe dyslipidaemia, fatty liver, low leptin and adiponectin levels, coupled with insulin resistance and diabetes in the CIDEC E186X proband is typical of human lipodystrophy (Garg, 2004). Discrepancies between mouse and human phenotypes have been reported previously: PPARG P467L and other ligand binding domain variants consistently cause lipodystrophy and severe insulin resistance in humans (Semple et al, 2006), whereas mice with the equivalent Pparg P465L mutation are neither lipodystrophic nor insulin resistant (Gray et al, 2006). It is currently too early to say if this discrepancy indicates the existence of species-specific differences in the global metabolic response to perturbations of adipocyte biology.
In Fsp27 KO mice, all white adipocytes appeared to have multilocular lipid droplets (Nishino et al, 2008; Toh et al, 2008), whereas we observed adipocytes with unilocular and paucilocular lipid droplets in addition to the multilocular cells (Fig 2A). Until more is known about the function of CIDEC, we can only speculate about the molecular origins of this finding. Although our transfection studies suggest that the truncated protein fails to increase lipid droplet size, it remains possible that it does retain some in vivo activity (for instance, the CIDE-N domain remains intact). The progressive increase in the size of adipocytes with fewer and ultimately a single lipid droplet compared to multilocular cells (Fig 2B) also suggests that lipid droplets may eventually fuse to generate a single large droplet despite ‘CIDEC deficiency’. CIDEA, another member of the CIDE family of proteins, is predominantly expressed in brown, rather than white, adipose tissue in mice, but is expressed in white adipocytes in humans (Puri et al, 2008). It is therefore also possible that CIDEA may compensate, at least in part, for the loss of CIDEC function. We have sequenced the coding regions of CIDEA in our patient and her family. She was heterozygous for an arginine to tryptophan substitution at codon 5 (R5W) (data not shown). This variant was also present in one of her half-brothers, who was phenotypically normal, and in 16.7% of 108 South American control alleles. It is located in an N-terminal lead sequence before the CIDE-N domain and hence is not expected to alter the protein structure. These observations suggest that the CIDEA variant is a common polymorphism and is unlikely to be pathogenic but we cannot exclude the possibility that it might contribute to the proband's extreme phenotype.
The perception that converting white adipocytes into thermogenic brown adipocytes could cause weight loss and improve insulin sensitivity has prompted successful attempts to do this in rodents. These efforts included both genetic (Tsukiyama-Kohara et al, 2001) and pharmacological approaches such as chronic treatment with a selective β 3-adrenergic agonist (Himms-Hagen et al, 2000). In all cases, UCP1 expression and mitochondrial mass were increased in multilocular adipocytes. Although the limited amount of available tissue meant that we could not formally assess mitochondrial mass or function in WAT from the CIDEC E186X proband, cytochrome c immunoreactivity and electron micrographs suggested that mitochondrial mass was increased. The absence of UCP1 immunostaining suggests that these cells are not brown adipocytes. Nevertheless, are these multilocular adipocytes likely to account for the increased RMR observed in our patient? It is currently not possible to accurately measure the tissue specific contributions to the total energy expenditure in humans, but the mass specific contribution of fat to energy expenditure is roughly 17 kJ/kg/day, about one sixth of that of lean mass (Nelson et al, 1992). To account for a 1.3 kJ/min elevation in resting energy expenditure relative to healthy volunteers, fat associated energy expenditure would need to be double that seen in healthy fat metabolism. Changes in oxygen consumption of this magnitude were recorded in adipocytes from the Fsp27 KO mouse (Nishino et al, 2008).
Limitations of our study include the lack of additional affected family members or other kindreds with loss of function variants in CIDEC. The fact that we did not identify additional pathogenic CIDEC mutations in 168 patients with unexplained lipodystrophy suggests that this is likely to be a rare cause of partial lipodystrophy, much like AKT2 (see Supporting Information). The proband's mother is heterozygous for the CIDEC variant but is clinically and biochemically normal, which is consistent with the normal phenotype of Fsp27 heterozygous KO mice (Nishino et al, 2008). The proband's presentation with diabetic ketoacidosis despite manifesting acanthosis nigricans and high plasma C-peptide levels is intriguing and suggests that lipolytic regulation may be impaired. The presence of high fasting plasma free fatty acid levels (1048 µmol/l (280–920)) and elevated urine glycerol levels (713 µmol/mol creatinine (17–183)) is in keeping with this possibility. Plasma free fatty acids were normal in Fsp27 KO mice, although one study suggested that elevated intracellular fatty acid levels may be responsible for inducing mitochondrial biogenesis and fat oxidation (Nishino et al, 2008; Puri & Czech, 2008; Toh et al, 2008).
In summary, we report a novel autosomal recessive cause of partial lipodystrophy. The clinical phenotype is similar to that caused by mutations in LMNA, PPARG and AKT2, all of which manifest striking lower limb and femorogluteal lipodystrophy, fatty liver, dyslipidaemia and insulin-resistance. Together with observations made in cultured cells and Fsp27 KO mice, our observations also suggest that CIDEC is required for optimal energy storage in ‘unilocular adipocytes’ in humans, although we recognize that this statement is tempered by the fact that it is currently based on observations in a single small kindred.
METHODS
Genetic and phenotypic studies
The study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the UK National Health Service Research Ethics Committee. Each participant, or a parent in the case of minors, provided written informed consent; minors provided oral consent.
Genomic DNA was isolated from peripheral-blood leukocytes. After excluding mutations in the coding regions and splice junctions of LMNA, PPARG, ZMPSTE24 and AKT2, the coding regions and splice junctions of CIDEC were amplified by PCR and sequenced (primer sequences available upon request).
Insulin sensitivity was calculated using HOMA %S (homeostasis model assessment) (Levy et al, 1998). Body composition was measured by whole-body DXA and fat distribution was assessed using a 1.5 T MR scanner to acquire T1-weighted water suppressed images. RMR was measured using a ventilated canopy indirect calorimeter after an overnight fasting. Subcutaneous fat was studied using light microscopy, immunohistochemistry, and EM (see Supporting Information for details).
Cellular functional analysis
3T3-L1 pre-adipocytes and COS cells were cultured and differentiated in standard conditions (Puri et al, 2008). For cells incubated with fatty acids, 20 µM oleic acid/BSA mixture was added to the medium 8 h after transfection. CIDEC plasmid DNA was procured from Open Biosystems. For full length CIDEC, PCR was performed by using a 5′-linker with a BglII restriction site and a 3′-linker containing an EcoRI site. For truncated CIDEC (E186X), PCR was performed by using the same 5′-linker and a 3′-linker containing a HindIII site. Following a restriction digest, purified PCR fragments were cloned into pEGFPC1 vector (Clontech, USA). GFP-CIDEC cDNA (5 µg) was transfected into COS cells using the Lipofectamine Plus Reagent (GIBCO Life Technologies, Rockville, Maryland). Pre-adipocytes and adipocytes were electroporated with 6 µg cDNA (200,000 cells/200 µl PBS) in a 0.4 cm cuvette at 180 V and 950 µF with a time constant of 25 ms on a Bio-Rad Gene Pulser II system. After transfection, the cells were cultured for 24–48 h before fixing and staining with oil red. Oil-red-O staining and morphometric analysis of lipid droplet size using confocal microscopy were undertaken as described previously (Puri et al, 2008). Measurements of the total volume of lipid droplets per cell (10–25 cells in each case) were based on at least three independent experiments. Association of full length or truncated CIDEC with lipid droplets in adipocytes was assessed using Metamorph software by performing a co-localization analysis of GFP florescence with Oil-red-O labelled lipid droplets in each Z-section of a transfected cell; these results were then averaged to calculate the overall co-localization in a given cell. The data are from 8–10 adipocytes in each condition from three independent experiments.
The paper explained
PROBLEM
Lipodystrophy is a rare disease characterized by a partial or complete lack of adipose tissue. Paradoxically, both ‘too much’ (obesity) and ‘too little’ (lipodystrophy) fat lead to adipose tissue dysfunction, ectopic lipid accumulation, insulin resistance and diabetes.
Within the last decade, investigators have identified mutations in seven different genes in patients with lipodystrophy. In this study, we focused on a novel subtype of partial lipodystrophy.
RESULTS
We report the identification of a homozygous nonsense mutation in CIDEC in a patient with partial lipodystrophy, fatty liver, severe insulin resistance, dyslipidaemia and diabetes. Histological studies of residual adipose tissue from the patient revealed white adipocytes with multiloculated lipid droplets and excess mitochondria. Functional studies of the mutant protein indicated that it fails to increase the lipid droplet size and, contrary to the wild type protein, it does not localize around lipid droplets in transfected cells.
IMPACT
The results add the CIDEC gene to the list of genes whose mutation is associated with lipodystrophy. CIDEC is a recently identified lipid droplet protein whose function remains incompletely understood. CIDEC (Fsp27) null mice manifest low fat mass and multilocular adipocytes with excess mitochondria, but in contrast to our patient, these mice are insulin sensitive. Together, these studies suggest that CIDEC is required for unilocular lipid droplet formation and optimal lipid storage in white adipose tissue. The differences between the human and mouse phenotypes raise intriguing questions about interspecies differences in adipose tissue metabolism.
Statistical analysis
Quantitative data are presented as mean ± SE. Two-tailed Student's t-tests or one-way ANOVA with post hoc Bonferroni analyses were performed on data at a minimum p < 0.05 threshold.
Author contributions
Oscar Rubio Cabezas, Jesu´s Argente and David Savage conducted the clinical investigations. Vishwajeet Puri, aided by Anil Chawla, performed the functional studies which he planned together with Michael Czech, Stephen O'Rahilly and David Savage. Incoronata Murano, Jeremy Skepper and Saverio Cinti performed the histological studies. Vladimir Saudek provided bioinformatic analyses on all the mutations identified. Oscar Rubio Cabezas, Robert Semple, Satya Dash, Caroline Hyden, William Bottomley, Ann-Marie Patch, Ines Barroso, Stephen O'Rahilly and David Savage contributed to the genetic studies. Philippa Raymond-Barker, Peter Murgatroyd and David Savage analysed the metabolic rate data and provided reference data from a control sample. Robert Semple, Corinne Vigouroux, Jocelyne Magré, V. Krishna Chatterjee, Sara Suliman, LD Screening Consortium, Anil Agarwal, Abhimanyu Garg, Stephen O'Rahilly and David Savage contributed DNA samples from patients with unexplained lipodystrophy. Oscar Rubio Cabezas, Vishwajeet Puri, Jesu´s Argente, Stephen O'Rahilly and David Savage wrote the paper, which was then reviewed by all the authors.
Acknowledgments
We thank the participants. We thank Margaret Blount for assistance with histological analysis. O.R.C. is supported by an ‘Ayuda para contratos post-Formación Sanitaria Especializada’ from the ‘Instituto de Salud Carlos III’ (FIS CM06/00013), Spain. This work was also supported by grants from the Wellcome Trust (R.K.S., V.K.K.C., S.O'R., D.B.S., W.B., I.B. (WT Grant 077016/Z/05/Z)), CIBER Fisiopatología de la Obesidad y Nutrición (CIBEROBN) from the ‘Instituto de Salud Carlos III’ and Fundación Endocrinología y Nutrición, Madrid, Spain (J.A.), GlaxoSmithKline (D.B.S.), the U.K. NIHR Cambridge Biomedical Research Centre, National Institutes of Health grants DK30898, DK60837 (to M.P.C.) and DK54387 (to A.G.), the Biomedical Imaging Core Facility of the University of Massachusetts Diabetes and Endocrinology Center (National Institutes of Health grant DK32520) (M.P.C.), Diabetes UK (S.S.), the Italian Ministry of University (FIRB RBIN047PZY_000 Internazionalizzazione, to S.C.) and Cofin. PRIN bando 2007 (S.C.), Aide aux Jeunes Diabétiques (AJD), Association de Langue Française pour l'Etude du Diabète et des Maladies Métaboliques (ALFEDIAM) (J.M.) and from the French National Institute for Health and Medical Research (INSERM) (J.M., C.V.).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
For more information
Online Mendelian Inheritance in Man:
CIDEC:
http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=612120
David Savage Laboratory:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Brasaemle DL, Subramanian V, Garcia A, Marcinkiewicz A, Rothenberg A. Perilipin A and the control of triacylglycerol metabolism. Mol Cell Biochem. 2009;326:15–21. doi: 10.1007/s11010-008-9998-8. [DOI] [PubMed] [Google Scholar]
- Chen Z, Guo K, Toh SY, Zhou Z, Li P. Mitochondria localization and dimerization are required for CIDE-B to induce apoptosis. J Biol Chem. 2000;275:22619–22622. doi: 10.1074/jbc.C000207200. [DOI] [PubMed] [Google Scholar]
- Frayn KN. Adipose tissue as a buffer for daily lipid flux. Diabetologia. 2002;45:1201–1210. doi: 10.1007/s00125-002-0873-y. [DOI] [PubMed] [Google Scholar]
- Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350:1220–1234. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- Garg A, Agarwal AK. Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta. 2009;1791:507–513. doi: 10.1016/j.bbalip.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SL, Dalla Nora E, Backlund EC, Manieri M, Virtue S, Noland RC, O'Rahilly S, Cortright RN, Cinti S, Cannon B, et al. Decreased brown adipocyte recruitment and thermogenic capacity in mice with impaired peroxisome proliferator-activated receptor (P465L PPARgamma) function. Endocrinology. 2006;147:5708–5714. doi: 10.1210/en.2006-0684. [DOI] [PubMed] [Google Scholar]
- Himms-Hagen J, Melnyk A, Zingaretti MC, Ceresi E, Barbatelli G, Cinti S. Multilocular fat cells in WAT of CL-316243-treated rats derive directly from white adipocytes. Am J Physiol Cell Physiol. 2000;279:C670–C681. doi: 10.1152/ajpcell.2000.279.3.C670. [DOI] [PubMed] [Google Scholar]
- Keller P, Petrie JT, De Rose P, Gerin I, Wright WS, Chiang SH, Nielsen AR, Fischer CP, Pedersen BK, MacDougald OA. Fat-specific protein 27 regulates storage of triacylglycerol. J Biol Chem. 2008;283:14355–14365. doi: 10.1074/jbc.M708323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JC, Matthews DR, Hermans MP. Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care. 1998;21:2191–2192. doi: 10.2337/diacare.21.12.2191. [DOI] [PubMed] [Google Scholar]
- Magnusson B, Gummesson A, Glad CA, Goedecke JH, Jernas M, Lystig TC, Carlsson B, Fagerberg B, Carlsson LM, Svensson PA. Celldeath-inducing DFF45-like effector C is reduced by caloric restriction and regulates adipocyte lipid metabolism. Metabolism. 2008;57:1307–1313. doi: 10.1016/j.metabol.2008.04.028. [DOI] [PubMed] [Google Scholar]
- Nelson KM, Weinsier RL, Long CL, Schutz Y. Prediction of restingenergy expenditure from fat-free mass and fat mass. Am J Clin Nutr. 1992;56:848–856. doi: 10.1093/ajcn/56.5.848. [DOI] [PubMed] [Google Scholar]
- Nishino N, Tamori Y, Tateya S, Kawaguchi T, Shibakusa T, Mizunoya W, Inoue K, Kitazawa R, Kitazawa S, Matsuki Y, et al. FSP27 contributes to efficient energy storage in murine white adipocytes by promotingthe formation of unilocular lipid droplets. J Clin Invest. 2008;118:2808–2821. doi: 10.1172/JCI34090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri V, Czech MP. Lipid droplets: FSP27 knockout enhances their sizzle. J Clin Invest. 2008;118:2693–2696. doi: 10.1172/JCI36554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri V, Konda S, Ranjit S, Aouadi M, Chawla A, Chouinard M, Chakladar A, Czech MP. Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J Biol Chem. 2007;282:34213–34218. doi: 10.1074/jbc.M707404200. [DOI] [PubMed] [Google Scholar]
- Puri V, Ranjit S, Konda S, Nicoloro SM, Straubhaar J, Chawla A, Chouinard M, Lin C, Burkart A, Corvera S, et al. Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc Natl Acad Sci USA. 2008;105:7833–7838. doi: 10.1073/pnas.0802063105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87:507–520. doi: 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple RK, Chatterjee VK, O'Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest. 2006;116:581–589. doi: 10.1172/JCI28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh SY, Gong J, Du G, Li JZ, Yang S, Ye J, Yao H, Zhang Y, Xue B, Li Q, et al. Up-regulation of mitochondrial activity and acquirement of brown adipose tissue-like property in the white adipose tissue of fsp27 deficient mice. PLoS ONE. 2008;3:e2890. doi: 10.1371/journal.pone.0002890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traini M, Jessup W. Lipid droplets and adipose metabolism: a novel role for FSP27/CIDEC. Curr Opin Lipidol. 2009;20:147–149. doi: 10.1097/MOL.0b013e32832956c7. [DOI] [PubMed] [Google Scholar]
- Tsukiyama-Kohara K, Poulin F, Kohara M, DeMaria CT, Cheng A, Wu Z, Gingras AC, Katsume A, Elchebly M, Spiegelman BM, et al. Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nat Med. 2001;7:1128–1132. doi: 10.1038/nm1001-1128. [DOI] [PubMed] [Google Scholar]
- Woods CG, Cox J, Springell K, Hampshire DJ, Mohamed MD, McKibbin M, Stern R, Raymond FL, Sandford R, Malik Sharif S, et al. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am J Hum Genet. 2006;78:889–896. doi: 10.1086/503875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Li JZ, Liu Y, Li X, Yang T, Ma X, Li Q, Yao Z, Li P. Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab. 2009;9:177–190. doi: 10.1016/j.cmet.2008.12.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.