

Abstract
The co-occurrence of congenital diaphragmatic hernia (CDH) and cardiovascular malformations (CVMs) has important clinical, genetic, and developmental implications. Previous examinations of this topic often included patients with genetic syndromes. To correct this potential bias, we undertook an extensive review of the literature and obtained new data. The frequency of CVMs associated with isolated CDH was 11–15%. A careful analysis of CVMs indicates that atrial and ventricular septal defects, conotruncal defects, and left ventricular outflow tract obstructive defects were the most common type of CVMs, but proportional to the frequency of occurrence in the general population. The combination of CVM and CDH results in a poorer prognosis than would be expected with either malformation alone. However, the impact on survival from patients with a genetic syndrome has not been consistently evaluated. We encourage researchers to re-analyze existing series and recommend that future studies distinguish isolated CDH from that which is associated with other malformations, especially as part of genetic syndromes. Therapies should be tailored to maximize cardiac output and systemic oxygen delivery rather than systemic oxygen saturation alone. Although there is speculation about the frequency with which isolated left ventricular “hypoplasia” occurs in patients with CDH, we suggest it results from compression of a pre-load deficient left ventricle by the hypertensive right ventricle, and unlike true hypoplasia, is reversible. Irrespective of the type of severity of CVMs in patients with CDH, the degree of pulmonary hypoplasia and pulmonary vascular disease predicts outcome.
Keywords: cardiovascular malformations, congenital diaphragmatic hernia, congenital heart defects, hypoplastic left heart
INTRODUCTION
When a newborn with a congenital diaphragmatic hernia (CDH) is admitted to the neonatal intensive care nursery, the priorities of diagnosis and care are quickly established. “Did the cardiologist see the baby? What did the echocardiogram show?” are the familiar questions which speak volumes about the well-known association of CDH and cardiovascular malformations (CVMs). The neonate with CDH may be cyanotic due to lung hypoplasia, a CVM, reactive pulmonary vascular bed, or a combination. Once the cardiopulmonary status is stabilized, the baby should be examined for the presence of additional malformations. This article discusses both the cardiac and genetic aspects of CDH in association with CVM, and analyzes diagnostic, management, outcome, and developmental perspectives.
CDH and CVMs are both common malformations that impact newborn morbidity and mortality, though CDH is less frequent than CVM. The reported birth prevalence of CDH ranges from 1.4 to 4.5/10,000 births. Dott et al. [2003] discussed possible reasons for this variation including differences in case ascertainment (e.g., clinical series, population-based surveillance programs) and case definition (e.g., the inclusion of eventration). However, the two most recent series, Dott et al. [2003] and Yang et al. [2006], reported a similar prevalence of total CDH in 2.4 and 2.5 per 10,000 births in the Metropolitan Atlanta Congenital Defect Program and the California Birth Defects Monitoring Program, respectively; point estimates for isolated CDH were 1.5 and 1.4 per 10,000. Overall, the occurrence of CDH is less than the prevalence for CVM which has been reported in up to 9/1000 [Botto et al., 2001].
Associated Malformations
Non-cardiac malformations have been reported in approximately 20–60% of patients with a CVM. The wide variation is due to differences in study design, definition of malformations, and patient ascertainment, for example, population-vs. autopsy-based series [Copel et al., 1993; Ferencz et al., 1997; Tennstedt et al., 1999; Bosi et al., 2003; Tegnander et al., 2006]. Data about the association of CVMs with CDH, in particular, are available from two large European population-based studies. Calzolari et al. [2003] reported CDH in 9/1549 (5.8%) of patients who had a CVM. This was similar to the 7.0% (20/3527) frequency noted by Eskedal et al. [2004] in which patients with recognized syndromes were excluded.
Conversely, approximately 30–40% of patients with CDH have an associated malformation. Since some studies use the more general term “anomalies” without providing a full list of case definitions, it is likely that diagnoses which are not true malformations (e.g., Losty et al. [1998] list syndactyly and colonic stenosis) may have been tabulated. Varying prevalence estimates may also be attributed to patient ascertainment from different cohorts including population-based surveys [Dott et al., 2003; Colvin et al., 2005], autopsy examination [Benjamin et al., 1988; Sweed and Puri, 1993], prenatal imaging series [summarized on Table I, Skari et al., 2000] or combined prenatal and postnatal examination [summarized on Table II, Skari et al., 2000; Dillon et al., 2000; Harmath et al., 2006].
TABLE I.
Syndromes and Conditions Frequently* Associated With Congenital Diaphragmatic Hernia: Occurrence of Cardiovascular Malformations
| Syndrome | OMIM # | Locus Gene Inheritance | CVM frequency | Type | References |
|---|---|---|---|---|---|
| Chromosome abnormality | |||||
| Wolf-Hirshhorn syndrome | |||||
| Deletion 4p | #194190 | 4p16 | 30–50% | ASD secundum type | Battaglia et al., 1999 |
| WHCR | PSV | ||||
| WHSCR2 | VSD | ||||
| NA | |||||
| Pallister-Killian syndrome | |||||
| Tetrasomy 12p | #601803 | i(12p) | Low | Asssorted | Schaefer et al., 1997 |
| NA | Doray et al., 2002 | ||||
| NA | |||||
| Trisomy 18 | NA | NA | 95% | Conoventricular VSD | Van Praagh et al., 1989 |
| NA | TOF, DORV | ||||
| NA | Polyvalvar dysplasia | ||||
| Mendelian gene | |||||
| Brachmann-deLange syndrome | #122470 | 5p13.1 | 25% | ASD, VSD | Strauss et al., 2005 |
| NIPBL | PSV | ||||
| AD | |||||
| Meacham syndrome | #608927 | ?11p3 | Low | APVR, ASD, TOF, HLHS | Meacham et al., 1991 |
| ?WT1* | Killeen et al., 2002 | ||||
| AD | |||||
| Simpson-Golabi-Behmel syndrome | #312870 | Xq26 | 26% | No specific type | Lin et al., 1999b |
| GPC3 | |||||
| XLR | |||||
| Presumed mendelian gene | |||||
| Fryns syndrome | %229850 | NA | ~50% | ASD, VSD | Lin et al., 2005 |
| NA | Conotruncal defects | ||||
| AR | |||||
| Thoracoabdominal syndrome | %313850 | Xq25-26.1 | 50% | No specific type | Carmi et al., 1990 |
| NA | Parvari et al., 1996 | ||||
| XLR | |||||
| Teratogens (animal studies) | |||||
| Vitamin A deficiency (rats) | NA | NA | 45% | VSD, defect of aortico-pulmonary septation, aortic anomalies | Wilson and Warkany, 1950 |
| Wilson and Warkany, 1957 | |||||
| Nitrofen (mice, rats) | NA | NA | 60% | Conotruncal/outflow | Migliazza et al., 1999 |
| Truncus, TOF, DORV | Yu et al., 2001 | ||||
| IAA, other arch anomalies | |||||
| Narrow PA outflow | |||||
Syndromes selected in which the association with CDH was reported in at least five patients.
AD, autosomal dominant; AoV, aortic valve; APVR, anomalous pulmonary venous return; AR, autosomal recessive; ASD, atrial septal defect; CHD, congenital heart defect; DORV, double outlet right ventricle; IAA, interrupted aortic arch; NA, not applicable or not available; PA, pulmonary artery; PSV, valvar pulmonic stenosis; PV, pulmonic valve; TOF, tetralogy of Fallot; VSD, ventricular septal defect; XLR, X-linked recessive.
TABLE II.
New Patients With Isolated Bochdalek Congenital Diaphragmatic Hernia Associated With CVMs from the Brigham-Women’s Hospital (BWH), Boston, 1972–2002. CVM Frequency Compared to Population-Based Studies
| Type of CVMa | Study |
|||
|---|---|---|---|---|
| BWH |
BWIS, Ferencz et al., 1997 |
Calzolari et al., 2003 |
Comparison of BWH and Calzolari et al. |
|
| Hospital-based surveillance | Population-based surveillance, USA includes syndromes | Population-based surveillance, Italy excluding chromosome and “recognized” syndromes | Chi square analysis, P-value | |
| No. (%) | No. (%) | No. (%) | ||
| Total CDH | 203 | Not applicable | Not applicable | |
| Total CVMs | 23 (11) | 4304 | 1328 | |
| “Early” CVMs (developmental error <4 weeks gestational age) | ||||
| Single ventricle | 1 (4) | 18 (0.4) | 43 (3) DILV | ND |
| Conotruncal CVMs | 5 (22) all types | 670 (16) all types | 205 (15) all types | NS, ≤1 |
| 1TOF | ||||
| 4 DORV | ||||
| 2 with HLHS/MA | ||||
| 1 with AVC | ||||
| Atrioventricular canal defects | 1 canal-type VSD (4) | 320 (7) all types | 24 (2) all types | ND |
| “Later” CVMs (developmental error >4 weeks gestational age) | ||||
| LVOTO CVMs | 3 (13) | 587 (14) all types | 120 (9) all types | NS, ≤1 |
| 2 coarctation | ||||
| 1 possible HLHS | ||||
| RVOTO CVMs | 0 | 427 (10) all types | 97 (7) all types | ND |
| ASD, VSD, ASD+/− VSD | 13 (56) | 1186 (28) | 634 (48) | NS, ≤1 |
| TAPVR/PAPVR | 0 | 68 (2) | 11 (1) TAPVR | ND |
Cardiovascular malformation excludes isolated patent ductus arteriosus, dextrocardia/dextroposition, patent foramen ovale, functional anomalies (valve regurgitation, ventricular hypertrophy); cardiomyopathy, ectopia cordis/Pentalogy of Cantrell, inferior vena cava/pulmonary artery compression, pericardial defect.
ASD, atrial septal defect; AVC, atrioventricular canal; BWIS, Baltimore-Washington Infant Study; CDH, congenital diaphragmatic hernia; CHD, congenital heart defect; DILV, double inlet left ventricle; HLHS, hypoplastic left heart syndrome; LVOTO; left ventricular outflow tract obstruction; MA, mitral atresia; ND, not done if number of cases too small; NS, not significant; RVOTO, right ventricular outflow tract obstruction; TAVPR/PAPVR, total/partial anomalous pulmonary venous return; TOF, tetralogy of Fallot; VSD, ventricular septal defect.
Outcomes of patients with a CVM and extracardiac malformations may be further compromised by low birth weight and prematurity [Gaynor et al., 2002; Karamlou et al., 2005; Andrews et al., 2006; Michielon et al., 2006; Stasik et al., 2006]. As the number of CVMs for which neonatal surgical options increases, prognosis is more often dependent on the presence of extracardiac malformations. Neonates born with both CDH and a CVM have a poorer outcome than those born with either problem alone [Fauza and Wilson, 1994; Cohen et al., 2002]. The combination of a CVM with CDH represents the result of a major abnormality in development, so that superimposed lung hypoplasia, disordered postnatal pulmonary vascular remodeling, abnormal cardiopulmonary physiology, possibly combined with extracardiac, and extra-pulmonary malformations create difficult challenges to the management and survival of many neonates with both CDH and a CVM.
The combination of a CVM with CDH represents the result of a major abnormality in development, so that superimposed lung hypoplasia, disordered postnatal pulmonary vascular remodeling, abnormal cardiopulmonary physiology, possibly combined with extracardiac, and extrapulmonary malformations create difficult challenges to the management and survival of many neonates with both CDH and a CVM.
The presence of multiple congenital malformations can influence decisions made by the parents and caregivers. This applies whether the diagnosis is made prenatally (since pregnancy termination may be a choice for some), postnatally (if transferring a patient for more aggressive surgical management is an option), or postmortem (when knowledge of a syndrome can influence genetic counseling). The importance of recognizing dysmorphic features and associated malformations in CDH patients who may have a genetic syndrome is emphasized (Table II by Dr. Anne Slavotinek, 2007 in the same issue of this Seminar). Syndromes and other multiple malformation associations have tremendous clinical importance in the newborn period so that when a complex, lifesaving cardiac operation is being contemplated, the presence of the CDH and possibly other defects must be considered carefully [Robinson and Newburger, 2003].
CLASSIFICATION AND DEFINITIONS
Before reviewing the clinical management of CVMs, it is helpful to review the role of case classification in the epidemiologic, descriptive, and molecular genetic study of CDH. For families and caregivers, it is practical to view the newborn with a malformation, such as CDH, as having either a solitary malformation, for example, “isolated” CDH, or additional malformations that may or may not constitute a recognizable syndrome (see Table I for list of syndromes in which CDH is frequently associated with a CVM). This distinction has obvious clinical ramifications both in terms of genetic issues, as well as management decisions since patients with more malformations are generally sicker with greater medical needs.
For families and caregivers, it is practical to view the newborn with a malformation, such as CDH, as having either a solitary malformation, for example, “isolated” CDH, or additional malformations that may or may not constitute a recognizable syndrome
Studies of CDH should separate patients with recognizable malformation syndromes from those who have CDH with additional malformations not constituting a recognized syndrome. Analyses that ignore this dichotomy may obscure etiologic heterogeneity [Rasmussen et al., 2003] and provide skewed estimates of the frequency of associated malformations [e.g., Graziano et al., 2005]. For example, many studies have combined bona fide primary cardiovascular “malformations”, which can be viewed as errors of cardiac morphogenesis, together with secondary abnormalities that are due to the positional shifts of the heart in the chest and/or altered hemodynamics, for example pulmonary hypertension.
Distinguishing Form From Function
Narrowly defined, a CVM refers to a structural malformation attributed to an error of cardiac morphogenesis. Unfortunately, the definition of CVM, congenital heart defect, or unspecified heart defect is frequently omitted from articles. For this analysis, we defined CVM in a narrow sense, and excluded heart anomalies that are (a) remnants of fetal hemodynamics which may persist in the presence of pulmonary hypertension (isolated patent ductus arteriosus, PDA; patent foramen ovale, PFO), (b) the result of positional forces (dextrocardia/dextroposition, compression of inferior vena cava or pulmonary artery), (c) functional anomalies (valve regurgitation, ventricular hypertrophy), (d) cardiomyopathy, and e) pericardial defects. We also excluded ectopia cordis/Pentalogy of Cantrell because it represents distinct developmental errors with independent etiologies, management guidelines, and prognosis.
A common challenge is distinguishing patients with classic hypoplastic left heart (HLH) from so-called left heart “hypoplasia” which is not a CVM [Bollmann et al., 1995]. The former is defined as aortic and mitral atresia (or severe stenosis) with left ventricular hypoplasia. The latter refers to a left ventricle which appears small due to compression from an enlarged right ventricle, but structurally normal aortic and mitral valves, and aortic arch with relatively normal volume. Diagnostic studies, especially prenatal ultrasound and fetal echocardiography, may be unable to provide the anatomic details needed to make the distinction, and lack of postnatal confirmation through imaging or autopsy may result in inconclusive reporting. Failure to distinguish between the two entities affects the ascertainment of cases of CVMs which appropriately include HLHS but exclude isolated small left ventricle.
REVIEW OF PATIENTS
New Patients
Table II presents new information on 203 well-studied CDH cases identified through the Brigham-Women’s Malformation Surveillance Program, a hospital-based active surveillance registry. Details of the methods of this program can be found in previous publications [Nelson and Holmes, 1989; Lin et al., 1999a]; of note, individual patients are not examined personally in this program. Using the same dataset as a recent genetic analysis of isolated Bochdalek diaphragmatic hernia [Pober et al., 2005], 11.3% of patients with CDH had a CVM. Table II shows the distribution of CVMs from the Brigham-Women’s Hospital compared to infants in two large studies of CVM prevalence. The Baltimore-Washington Infant Study (BWIS) is a well-reported population-based study of infants ascertained up to age 1 year in the United States [Ferencz et al., 1997]. The Emilia-Romagna Registry of northern Italy is a smaller program which identifies infants <5 days, but allowed exclusion of cases with chromosome abnormalities and syndromes [Calzolari et al., 2003]. Comparisons of CVM type and frequency were made between the 203 CDH cases ascertained from the Brigham-Women’s Hospital and the data from Calzolari et al. [2003], testing for significance performed using a Chi square analysis. Among the CDH cases, there were non-significant increases in atrial and ventricular septal defects (56% vs. 48%), conotruncal CVMs (22% vs. 15%), and left ventricular outflow tract obstructive CVMs (13% vs. 9%) (P-values ≤1.0). In fact, the distribution of CVMs found among cases with CDH mirrored those in the general population.
Literature
Table III summarizes 30 years of articles which report the type and frequency of CDH associated with a CVM (rounded figures have been used throughout). The data are presented as: the reported frequency in the publication; the frequency after our additional analyses subjecting the cohort to more rigorous CVM definition (see Table III footnote listing exclusions); and the frequency after excluding patients with recognized chromosome and Mendelian syndromes. Several papers contain analyses devoted specifically to CDH and CVMs [Greenwood et al., 1976; Migliazza et al., 1999; Cohen et al., 2002; Graziano et al., 2005], whereas others are general reviews of CDH and associated anomalies [Cunniff et al., 1990; Fauza and Wilson, 1994; Bollmann et al., 1995; Enns and Cox, 1998; Losty et al., 1998]. A few papers focus on prenatal detection [Allan et al., 1996; Dillon et al., 2000; Witters et al., 2001] and several articles are population-based surveys of CDH [Martinez-Frias et al., 1996; Dott et al., 2003; Stege et al., 2003; Tonks et al., 2004; Colvin et al., 2005]. No single paper classified the cases from both the CDH and CVM point of view. Because of dramatically different methodologies, comparisons between studies have been made cautiously, and do not lend themselves to rigorous meta-analysis, statistical analysis, or simple generalizations. The reported “raw” frequency figures for the co-occurrence of CDH and CVM ranged from 9–42%. The number of patients with recognized syndromes was not always available, but after excluding them in a few reports, a refined estimate is 12–15%, similar to the 11.3% frequency in the Brigham-Women’s Hospital cohort of new patients (Table II).
TABLE III.
Summary of the Literature Describing Congenital Diaphragmatic Hernia Associated With Cardiovascular Malformations*,**
| Author | Year | Type of study | CDH patients | CVMs among all CDH as published | CVMs among all CDH, revised | CVMs among nonsyndromic patientsa,b | Comments |
|---|---|---|---|---|---|---|---|
| Greenwood et al. | 1976 | Cardiology center Hospital medical records | Forty-eight Total Eleven with CVM |
11/48 (23%) | 6/48 (12%) excluding non-CVM | 6/43 (14%) 2/6 (33%) TOF 2/6 (33%) ASD, VSD |
Seminal article included one pericardial defect, one “compressive” IVC, two ectopia cordis Individual patients NS |
| Benjamin et al. | 1988 | Hospital records | One hundred and eight Seventy-three Died Autopsy 60/73 |
25/60 (42%) At autopsy 7/24 (28%) Conotruncal 3/25 (12%) Coarctation ASDs, VSD, NS Other artery and venous anomaly |
12% Conotruncal | Sixty-five isolated CDH. Could not calculate frequency of CVMs |
Detailed autopsy series NS how many had CVMs |
| Cuniff et al. | 1990 | Genetics center Hospital records |
One hundred and two Total Ninety-two Bochdalek or Morgagni Ten eventration |
18/102 (18%) | 15/102 (15%) | Could not calculate individual CVMs | Emphasis on associated malformations and patterns Included pulmonary artery hypoplasia/sequestration and pericardial defect |
| Sweed and Puri | 1993 | Hospital records Autopsy |
One hundred and sixteen Total | 16/40 (40%) | VSD 5/16 (31%) TOF, TGA 6/16 (38%) HLHS 2/16 (12%) |
16/33 (48%) Could not calculate individual CVMs |
Specified syndromes However, reported total cardiac anomalies instead of patients |
| Fauza and Wilson | 1994 | Surgical team Hospital record |
One hundred and sixty-six Total | “Anomaly” in 63% of patients At least 37 (22%) |
Syndromes NS Could not be calculated |
Data reported as total Cardiac anomalies rather than CHD per patient |
|
| Bollman et al. | 1995 | Prenatal diagnosis unit records | Forty-four Total Type NS |
17/44 (39%) | Could not be calculated | Could not be calculated | Analysis of associated malformations and chromosome defects Data reported as cardiac defects, not patients |
| Allan et al. | 1996 | Two prenatal diagnostic units. | Ninety-three Total Type NS |
14/93 (15%) 4/14 (29%) VSD 3/14 (31%) TOF 3/14 (31%) LVOTO 1/14 COA, 2/14 HLHS 1/14 VSD + PSV |
Could not be calculated | Could not be calculated | Expert fetal echocardiography diagnosis, but not all confirmed postnatally |
| Martinez-Frias et al. | 1996 | Population-based survey | Two hundred and ninety-seven Total Type NS |
29/297 (10%) | Could not be calculated | Extensive epidemiologic analyses | |
| Enns et al. | 1998 | Genetic and surgical centers Hospital records |
Sixty Total 16 with genetics evaluation | NS | 6/9 Excluded 1 PDA, 5/8 (62%) 2/5 (40%) conotruncal |
Excellent genetic analysis Difficult to analyze CVM patients |
|
| Losty et al. | 1998 | Surgical centers Autopsy records |
Three hundred and one Total: Two hundred and sixty left CDH Forty-one right CDH Two hundred and one (58) Isolated: 177/260 (68) Left CDH 24/41 (89) Right CDH |
28/301 (9%) Total 22/260 (8%) Left CDH 4/41 (10%) Right CDH |
27/97 (28%) Left CDH: 7/22 (32%) LVOTO 5/22 (23%) Conotruncal 5/22 (23%) ASD, VSD |
Could not be calculated | Emphasis on CDH and sidedness Only three patients with syndromes. Individual patients not listed |
| Migliazza et al. | 1999 | Surgery center | One hundred and thirty-six total | 33/136 (24%) | 25/128 (20%) 7/25 (28%) conotruncal 6/25 (24%) ASD, VSD 5/25 (20%) hypoplastic heart 2/25 (8%) COA+VSD 5/25 (20%)other | Could not be calculated | Discusses experimental work with nitrofen in mice, and human data |
| Dillon et al. | 2000 | Population-based Emphasis on prenatal. diagnosis | Two hundred and one total One hundred and eighty-seven CDH without eventration 139/201 (69%) Isolated 62/201 (31%) Multiple |
NS | 18/187 (10%) CDH only |
12/37 (32%) 2/12 (17%) conotruncal 3/12 (25%) LVOTO 4/12 (33%) Septal defects |
Individual patient data with CVM and syndromes. Ectopia/Pentalogy of Cantrell excluded |
| Witters et al. | 2001 | Genetics center Emphasis on prenatal diagnosis |
Forty-two total Left CDH | 6/42 (14%) | 4/32 (12%) 4/4 “mild VSD” |
Unusual CVM distribution, that is, four mild VSDs. | |
| Cohen et al. | 2002 | Fetal diagnosis and surgical center | One seventy-one total No eventration Only CDH |
31/171 (18%) | 30/171 (18%) 7/30 (23%) Conotruncal 8/30 (27%) Isolated arch obstruction, “HLHS” 11/30 (37%) VSD, One with arch obstruction |
Main cohort reported with”lethal anomalies excluded” | |
| Stege et al. | 2003 | Population-based Emphasis on outcome and mortality | One eighty-five total Types NS | 31/185 (17%) | 31/156 (20%) | Could not be calculated | No details about CVM, just the summary figure. |
| Dott et al. | 2003 | Population-based study | Two hundred and forty-nine total Including TOPs One hundered twenty-nine left Forty-three right Four bilateral |
37/249 (15%) | 37/157 (24%) Anomalies, not classified defects |
Entire case classified, but heart reported as anomalies, not classified as defect per patient | |
| Tonks et al. | 2004 | Population-based study Emphasis on survival, outcome | One sixty-one total One forty-five CDH Sixteen eventration |
25/161 (16%) | 14/129 (11%) | Could not be calculated | Includes all births, that is, LBs, stillbirths and TOPs. Table 6 presents data clearly Lacks details about CVM types. |
| Colvin et al. | 2005 | Population-based study Emphasis on survival, outcome | One hundred sixteen total All CDH |
18/116 (16%) | 18/99 (19%) | Could not be calculated | Includes all births, that is, LBs, stillbirths and TOPs |
| Graziano et al. | 2005 | 82 cardiology and surgical centers. Emphasis on prognosis. | Two thousand six hundred thirty-six total enrolled | 385/2636 (15%) Exclude isolated ASD, other minor CHDs: 280/2636 (11%) 118/280 (42%) septal 80/280 (29%) LVOTO 47/280 (17%) conotruncal |
No data about syndromes | ||
| Harmath et al. | 2006 | Prenatal diagnosis center | One hundred total pre- and postnatal diagnosis | 31/71 (44%) 5/31(16%) “complex” 5/31 (16%) “conotruncal” 5/31 (22%) right heart 9/31 (29%) “minor anomalies” |
28/71 (39%) excluding “minor” anomalies; 3/28 conotruncal (11%) | Syndrome data cannot be integrated into CVM calculations | Complex cohort analysis: Prenatal and postnatal data; 1990–97 versus 1998–2005 Increased CVMs in second half of study period. COA reported as conotruncal. |
Figures rounded. Selected figures listed based on both published and revised data. For example, the column “CVMs among all CDH, revised” listed the results of the analysis of specific CVM types, beyond information about overall CVM frequency discussed in the text of the article. It also included the analysis after excluding certain cardiac defects which did not meet our definition ofe CVM. The column “CVMs among nonsyndromic patients” reported CVM frequency data after removing patients with multiple defects or syndromes.
Cardiovascular “malformation” excludes isolated patent ductus arteriosus, dextrocardia/dextroposition, patent foramen ovale, functional anomalies (valve regurgitation, ventricular hypertrophy); cardiomyopathy, ectopia cordis/Pentalogy of Cantrell, inferior vena cava/pulmonary artery compression, pericardial defect.
Syndromes include chromosome and mendelian gene conditions. Nonsyndromic patients defined as isolated CDH, and CDH with associated malformations not known to be a syndrome.
Refer to Table II for comparison frequencies for the general population from the Baltimore-Washington Infant Study (syndromes included) [Ferencz et al., 1997] and the Emilia-Romagna Registry (syndromes excluded) [Calzolari et al., 2003].
CDH, congenital diaphragmatic hernia; COA, coarctation; CVM, cardiovascular malformation; HLHS, hypoplastic left heart syndrome; LB, liveborn; LVOTO, left ventricular outflow tract obstruction; NS, not specified or not stated; TOF, tetralogy of Fallot; TOP, termination of pregnancy; VSD, ventricular septal defect.
A wide spectrum of CVMs associated with CDH has been reported in the literature. As noted previously in the discussion under “New Patients”, two population-based comparison groups were used because of heterogeneity of the data. We analyzed each study for the type of CVM that was proportionally more common within the study. When sufficient data allowed exclusion of syndromic cases, we re-analyzed to determine if the most common defect persisted. Comparisons were made (without using tests of significance) to the BWIS (if the reported data included syndromes), or the data from Calzolari et al. [2003] (when syndromes could be excluded).
“Septal defects”, was often broadly used and typically included secundum-type or unspecified atrial septal defects, and isolated ventricular septal defects, usually membranous and muscular types. This group was reported as the most common type of CDH-associated CVM by Graziano et al. [2005] (42% compared to the BWIS general population septal defect prevalence of 28%). Further analysis by excluding syndromes was not possible. Excluding syndrome cases from the data published by Dillon et al. [2000] failed to show an increase (33% compared to 48% in Calzolari et al., 2003]. It is likely that the overall frequency of ventricular septal defects would be erroneously increased if their occurrence as essential components of other CVMs, such as ventricular septal defect in tetralogy of Fallot, truncus arteriosus and type B interrupted aortic arch were included.
It has been suggested that conotruncal CVMs are associated with CDH more frequently than would be expected by chance. Based on the “raw” published figures, conotruncal CVMs were proportionally more common in one autopsy series [28%, Benjamin et al., 1988], in data from prenatal ultrasonographic diagnostic centers [31% tetralogy of Fallot, Allan et al., 1996], and a large surgery center [28%, Migliazza et al., 1999]. However, they were not increased in a large surgical center collaboration [17%, Graziano et al., 2005] or a prenatal diagnostic center review [16%, Harmath et al., 2006] when compared to the approximate 16% conotruncal frequency in the BWIS general population [Ferencz et al., 1997]. By revising the published data (e.g., by excluding cases with syndromes, eventration, or CVMs that did not meet our definitions, etc.), we observed that conotruncal CVMs were proportionally increased in a few studies [33% tetralogy of Fallot in Greenwood et al., 1976; 40% in Enns and Cox, 1998; 23% in both Losty et al., 1998 and Cohen et al., 2002], but not in the studies of Dillon et al. [2000] and Harmath et al. [2006] after re-analysis of their data (17% and 11%, respectively). Thus, an increase frequency of conotruncal CVMs in association with CDH is not consistent in the literature. However, it is also likely that hemodynamically insignificant aortic arch malformations such as aberrant origin of the subclavian and right aortic arch are underreported (which we believe also represent neural crest derived CVMs) [Hutson and Kirby, 2003].
The spectrum of left ventricular outflow tract obstruction, notably aortic and mitral atresia (classic HLH) has been described often in infants with CDH [Greenwood et al., 1976; Sweed and Puri, 1993; Fauza et al., 1994; Ryan et al., 1994; Pfleghaar et al., 1995; Witters et al., 2001; Cohen et al. 2002; Graziano et al., 2005]. Viewing published data, these defects also appear to show an increased frequency in infants with CDH compared to the BWIS frequency of approximately 14%. In the “raw” figures of Allan et al., 1996 (31%); Graziano et al., 2005 (29%); and Harmath et al., 2006 (22%), an increase was observed for left-sided defects reported as a group, as well as for coarctation [12% vs. 4.6%, Benjamin et al., 1988] and HLH [12% vs. 3.8%, Sweed et al., 1993]. Revision of published data by excluding cases with syndromes reinforced this apparent increase in left-sided obstruction [32%, Losty et al., 1998; 25%, Dillon et al., 2000; 27%, Cohen et al., 2002, compared to 9.0% in Calzolari et al., 2003]. However, it is not clear whether patients diagnosed with HLH in some series had a bona fide CVM or a small left ventricle because of altered hemodynamics (see below).
Other CVMs did not seem to be more commonly associated with CDH compared to the general population. It will require additional research to determine whether those that were infrequently reported (e.g., heterotaxy, atrioventricular canal defects) represent significant negative associations or proportionally low frequency defects.
When a syndrome with known association with CDH has a co-existing CVM (Table I), it is the experience of one of the authors (I.A.) that the CVM is usually of the same class typically associated with the syndrome. For example, patients with trisomy 18 and CDH usually have one of the conotruncal CVMs, such as conoventricular ventricular septal defect, tetralogy of Fallot, or double outlet right ventricle [Van Praagh et al., 1989; Musewe et al., 1990], which is the most common CVM type found among all infants with trisomy 18. Turner syndrome has been reported in patients with CDH in whom the cardiac status was not specified [Table III in Tibboel and Gaag, 1996; Table II, Dillon et al., 2000]; however, an infant with Turner syndrome and CDH accompanied by coarctation of the aorta and bicuspid aortic valve has also been reported [Adatia 2004].
THE LEFT VENTRICLE IN CDH
In general, a slender compressed left ventricular cavity is a common feature of congenital and acquired heart defects associated with pulmonary hypertensive vascular disease, deficient left ventricular pre-load, or preferential right ventricular volume loading. The enlarged right ventricle compresses the left ventricle which may be interpreted as left ventricular “hypoplasia”. The distorted left ventricular geometry resulting from right ventricular compression does not predict the true or potential left ventricular volume [Phoon and Silverman, 1997]. Examples of other conditions with apparent left ventricular “hypoplasia” but normal aortic and mitral valves and normal ventricular volumes include total anomalous pulmonary venous drainage, atrioventricular canal defect, arteriovenous malformations, and almost any cause of severe pulmonary arterial hypertension (Fig. 1A, B—congenital diaphragmatic hernia; Fig. 2A, B—total anomalous pulmonary venous drainage; Fig. 3A, B—cerebral arteriovenous malformation) [Graham et al., 1972; Bove et al., 1975; Nakazawa et al., 1977; Phoon and Silverman, 1997]. Indeed some patients with total anomalous pulmonary venous drainage and right dominant atrioventricular canal defects appear to have apex-forming right ventricles with left ventricular volumes that are less than 15 ml/m2. However, if potential volumes are calculated (by taking into account the degree of left ventricular compression so elegantly demonstrated by Phoon et al.) these correlate well with postoperative volumes which are within the normal range [Phoon and Silverman, 1997], and autopsy studies of neonates with total anomalous pulmonary venous drainage demonstrate normal left ventricular volume [Bove et al., 1975; Rosenquist et al., 1985]. After surgical correction of the total anomalous pulmonary venous drainage and resolution of the pulmonary hypertension, the left ventricle resumes a normal shape, size and contour, and develops normally. [Graham et al., 1972; Nakazawa et al., 1977; Phoon and Silverman, 1997]. Similar changes in left ventricular morphology occur in other lesions that result in abnormal disproportion between the left and right ventricle size [Phoon and Silverman, 1997; Van Son et al., 1997].
Figure 1–3.
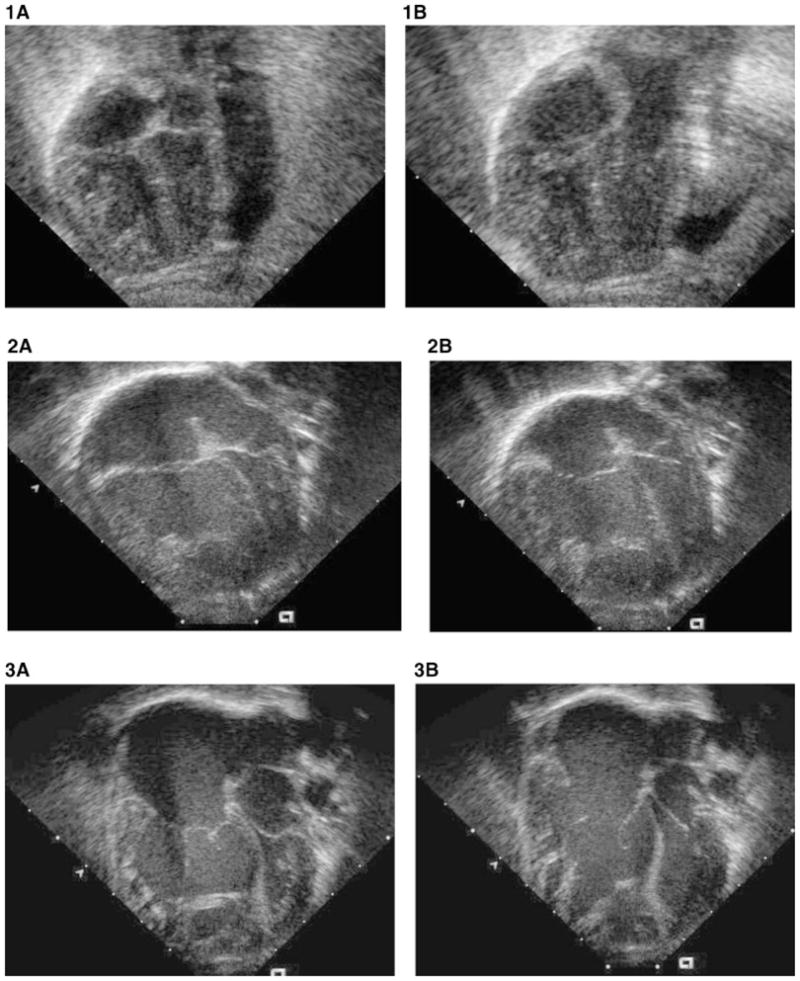
This series of four-chamber two-dimensional echocardiographic images compares the disproportion between right ventricle and left ventricle in congenital diaphragmatic hernia (Fig. 1A, B); total anomalous pulmonary venous drainage (Fig. 2A, B); and cerebral arteriovenous malformation (Fig. 3A, B) in systole (Fig. 1,2,3A) and diastole (Fig. 1,2,3B). The enlarged apex-forming right ventricle compresses the left ventricle. The left ventricle in these conditions differs from true HLHS because the aortic and mitral valves are morphologically normal, and when pulmonary hypertension resolves, the left ventricle assumes a normal shape and volume.
Similarly, in CDH, it seems likely that apparent left ventricular “hypoplasia”, especially in the presence of a normal aortic and mitral valve, is due to compression of an under-filled pre-load deficient left ventricle by the hypertensive right ventricle rather than true left ventricular hypoplasia. Indeed, left ventricular mass, left ventricular size, aortic size, left ventricle to right ventricle ratio, and aortic to pulmonary artery discrepancy do not predict postnatal outcome in patients with CDH [Suda et al., 2000; Sokol et al., 2002, 2006].
In patients with CDH, it seems likely that severe lung hypoplasia and elevated pulmonary vascular resistance, which cause marked right ventricular enlargement and the apparent left heart “hypoplasia” are the real modifiers of outcome. Indeed, indicators of lung hypoplasia such as branch pulmonary artery size, lack of branch pulmonary artery growth in utero, and postnatal lung area to head circumference ratio may be better predictors of mortality and morbidity than left ventricular volume [Cohen et al., 2002; Sokol et al., 2002, 2006]. Thus, the morphology of the left ventricle in CDH is more likely to be related to the state of the pulmonary vascular bed.
In patients with CDH it seems likely that severe lung hypoplasia and elevated pulmonary vascular resistance, which cause marked right ventricular enlargement and the apparent left heart “hypoplasia” are the real modifiers of outcome.
EXPERIMENTAL MODELS OF CDH
Cardiac defects similar to human CVMs have been reported in experimental studies following exposure to the teratogenic herbicide, nitrofen, which induces CDH in rat fetuses [Momma et al., 1992; Losty et al., 1998; Migliazza et al., 1999]. These animal studies, similar to the human data mentioned above, indicate that ventricular septal defect, especially with an outflow tract anomaly (conotruncal CVM) is the most frequent CVM associated with CDH. The predominance of conotruncal CVMs in rat pups with CDH exposed to nitrofen [Migliazza et al., 1999] is compelling. This argues against the simplistic notion that alterations in fetal blood flow related to cardiac compression by the hernia are responsible solely for CVMs. There was no increase in the relative proportion of bona fide HLHS in humans, though approximately 4% of nitrofen-exposed rat fetuses with CDH had “aortic valve dysplasia” [Migliazza et al., 1999].
INSIGHTS INTO CAUSATION
If a specific cardiac phenotype had been dramatically over-represented in cases with isolated (nonsyndromic) CDH, such an association could be a major clue to errant developmental pathways for CDH. As noted above, several studies have suggested that there is an increased risk of conotruncal CVMs, but the strength of this association is modest [Greenwood et al., 1976; Allan et al., 1996; Enns and Cox, 1998; Migliazza et al., 1999; Cohen et al., 2002; our series]. The lack of a robust association in human studies does not preclude the existence of common developmental genes, or weaken the hypothesis that CDH has genetic determinants.
MANAGEMENT GUIDELINES
Because the neonate with both CDH and CVM may have additional major malformations and minor anomalies, a thorough evaluation should be undertaken. In addition to multi-organ imaging, a genetic consultant may be useful to guide the diagnostic evaluation (e.g., chromosome testing vs. more sophisticated comparative genomic hybridization), especially when dysmorphic facial features are observed. If a syndrome is identified prenatally the information from the prenatal diagnostic team should be available to the postnatal specialists. A multidisciplinary approach is essential.
When discussing outcome and prognosis, the family should be counseled about the institution-specific experience in the management of CDH with CVM. Inferences should not be drawn from the outcome of either lesion in isolation, and data from the literature should be applied with caution. The family should be informed that survival is diminished even for relatively simple CVMs in the presence of CDH, since the combination of CVM and CDH results in poorer survival and increased complexity of management when compared to either defect in isolation. This is best illustrated by isolated ventricular septal defect which is a well-tolerated lesion, with less than 1% mortality for surgical correction and good long-term outcome. However, VSD with CDH has been reported to carry a 60% mortality (though inclusion of cases with syndromes and chromosome abnormalities could contribute to this 60-fold risk increase) [Cohen et al., 2002; Graziano et al. 2005]. During a 23-year period at the Hospital for Sick Children in Toronto, the survival for patients with CDH combined with any CVM was 45%, for a hemodynamically significant lesion 25%, and for a lesion requiring surgical intervention only 15% [Adatia, 2004]. In general, interventions to stabilize and permit the pulmonary vascular resistance to decrease and subsequently repair the CDH take priority over cardiac surgical intervention. Cardiac diagnoses that would be exceptions include obstructed total anomalous pulmonary venous drainage and duct-dependent CVMs unresponsive to PGE-1 infusion. Duct-dependent lesions that usually respond to PGE-1 infusion include pulmonary atresia/critical pulmonic stenosis, interrupted aortic arch, severe coarctation, severe aortic stenosis, and HLHS.
Baseline Testing
All patients with CDH should undergo a high quality diagnostic echocardiogram with Doppler and color flow mapping as soon as the clinical condition permits. Even patients who have undergone a prenatal examination should have a postnatal study since functional and anatomic changes occurring with birth mandate a full postnatal study. Certain defects, such as coarctation of the aorta, are difficult to diagnose prenatally. Patients with isolated CDH may share many similarities in the prenatal echocardiogram to patients with isolated coarctation including left ventricular to right ventricular disproportion, transverse arch and isthmus hypoplasia, and a reversal of the interatrial shunt [Allan et al., 1996]. Although CDH and coarctation can occur together, coarctation with CDH tends to be overdiagnosed prenatally. Postnatally, it may be difficult to differentiate a coarctation of the aorta from isthmal hypoplasia with certainty, especially in the presence of CDH because patients with pulmonary hypertension and right to left ductal flow may have isthmal hypoplasia. However, resolution of the pulmonary hypertension and ductal closure increases flow across the arch and isthmus resulting in normal development. In contrast, coarctation of the aorta is a true malformation in which there is a posterior shelf due to abnormal insertion of ductal tissue in the back wall of the aorta that constricts progressively to create an aortic obstruction with ductal closure [Kappetein et al., 1991]. It may be necessary to permit the ductus to close before an aortic coarctation can be ruled in or out with certainty especially in patients with CDH. Patients with CDH and blood pressure gradients from arm to leg after ductal closure should be followed closely for the later development of coarctation.
Significance of the Foramen Ovale and Ductus Arteriosus
Persistence of the foramen ovale and ductus arteriosus are a normal part of early postnatal adaptation and are not considered CVMs. However, their persistence has a profound, and often, misunderstood influence on the outcome and management of the patient with CDH. An atrial communication that permits intracardiac right to left shunting of blood has an important survival benefit in patients with right ventricular dysfunction. Early observations in patients with primary pulmonary arterial hypertension demonstrated prolonged survival in patients with patent foramen ovale [Rozkovec et al., 1986]. The concept that systemic arterial desaturation is well tolerated if cardiac output is adequate or improved is well accepted in the management of congenital heart disease. This physiologic principle lies behind the rationale for fenestration of the atrial septum to alleviate postoperative right ventricular dysfunction and in symptomatic patients with pulmonary hypertension [Kerstein et al., 1995; Sandoval et al., 1998; Micheletti et al., 2006]. Thus, cyanosis in CDH maybe beneficial when due to right to left atrial shunting. Indeed, any neonate with CDH who develops increasing or fluctuating cyanosis should undergo an echocardiogram to detect atrial shunting, using saline contrast if necessary.
Management should aim to maximize oxygen delivery and cardiac output indicated by a narrow arteriovenous oxygen saturation difference or low serum lactate rather than an increase in ventilator support or inspired oxygen concentration in response to a decreased systemic oxygen saturation. Iatrogenic pulmonary damage has a profound effect on outcome in infants with CDH and permissive ventilation strategies improve short and long term outcome [Bohn, 2002]. Unnecessary barotraumas may result if management is dictated by arterial oxygen saturation alone.
In contrast to an atrial level shunt which maintains systemic output after the onset of right ventricular dysfunction or failure, a patent ductus arteriosus will decompress the pulmonary circulation and relieve right ventricular after-load whenever the pulmonary vascular resistance exceeds systemic vascular resistance. This may prevent right ventricular failure. Likewise, closure of a PDA may precipitate right ventricular failure if the pulmonary vascular resistance remains elevated. PGE-1 can reopen a closed ductus in patients with CDH and right ventricular failure [Bohn, 2002; Buss et al., 2006]. The presence of an oximetric saturation difference between upper and lower limbs can be regarded as physiologically beneficial rather than an indication for escalation of therapy. Paradoxically, when the upper and lower limb saturation difference decreases or disappears with ductal closure in the presence of a severely elevated pulmonary vascular resistance, the patient is most at risk for acute right ventricular failure. However, once the pulmonary vascular resistance has decreased, the presence of a PDA shunting purely from left to right poses a hemodynamic burden and the PGE-1 infusion should be discontinued. If the ductus fails to close spontaneously at this stage it should be obliterated surgically or by using transcatheter techniques. Patients with a hypoplastic pulmonary vascular bed do not tolerate even small left to right shunts and until the PDA is closed, medical management is difficult and prolonged.
However, patients with right or left ventricular outflow tract obstruction will require support with PGE-1 infusion to maintain ductal patency until a management plan can be executed. The combination of transposition of the great arteries with CDH and poor mixing may require a balloon atrial septostomy.
Cardiac Surgery
Cardiac surgical intervention in a patient with a co-existing CDH and CVM is usually delayed until the pulmonary vascular resistance has decreased to acceptable levels. Cardiac catheterization can be extremely useful even for the evaluation of CVMs that would not routinely undergo cardiac catheterization and angiography prior to repair. Since CVM outcome is exquisitely dependent on pulmonary vascular resistance, hemodynamic measurements in response to inhaled nitric oxide may be needed. However, management decisions should take into account the uneven distribution of pulmonary blood flow in patients with all but mild diaphragmatic hernias. Patients with CDH with hypoplastic pulmonary vascular beds do not tolerate the increased flow associated with an intracardiac shunt. A decline in pulmonary vascular resistance may cause symptoms out of proportion to the shunt. These patients may benefit from surgical correction of even anatomically small lesions.
Patients with CDH and a functional single ventricle, such as tricuspid atresia or mitral atresia, have a poor prognosis and present unique challenges [Gaynor et al., 2002; Stasik et al., 2006]. Creating a stable source of pulmonary blood flow is essential to neonatal palliation. Sufficient pulmonary blood flow to provide adequate systemic oxygen saturation is important for somatic and pulmonary vascular growth and development. However, the surgically created or modified source of pulmonary blood flow (e.g., modified Blalock-Taussig shunt or right ventricle to pulmonary artery shunt as part of the modified Norwood procedure or pulmonary artery banding) should be restrictive enough to permit remodeling of the pulmonary vasculature. The pulmonary vascular resistance should decrease to levels that permit subsequent palliation. Understandably, performing a single ventricle palliation in a patient with an elevated or markedly fluctuating pulmonary vascular resistance represents significant short and long term management challenges in an already challenging group of patients. However, one of the authors (I.A.) has cared for a patient with mitral atresia and double outlet right ventricle who survived atrial septectomy and pulmonary artery banding, and had sufficient reduction of pulmonary vascular resistance at 4 months of age to permit a second palliative procedure with bidirectional cavopulmonary anastomosis [Adatia, 2004]. Thus, each case must be assessed individually.
Late Cardiac Followup
All patients with CDH who survive the neonatal period, with or without a CVM, often have complex medical and social needs and are served best by frequent evaluations in a multidisciplinary clinic that should include a pediatric cardiologist. Defects such as coarctation of the aorta, mitral stenosis, subaortic stenosis, and cor triatriatum may progress. Late intervention may be required for atrial septal defect and persistent ductus arteriosus. In addition, a few patients will have ongoing problematic pulmonary vascular disease and require therapy from cardiologists skilled in the drug therapy of pulmonary artery hypertension.
All patients with CDH who survive the neonatal period, with or without a CVM, often have complex medical and social needs and are served best by frequent evaluations in a multidisciplinary clinic that should include a pediatric cardiologist.
SUMMARY
The association of CDH with CVMs is important because of the clinical, genetic, and developmental implications. A careful evaluation for other congenital anomalies and underlying syndromes should be undertaken. When CVMs are present, the acute management, estimations of prognosis, and genetic counseling are clearly influenced.
An extensive reviewof the literature (Table III) and new data (Table II) show that CVMs occur in 11–15% of CDH without a recognizable genetic syndrome. Septal defects were the most common type of CVM in many series, but in a proportion similar to the general population. Conotruncal CVMs were inconsistently more common in patients with CDH, but this potential association merits further attention in light of its existence in animal studies. Left ventricular outflow tract obstructive defects also may be more common, but a rigorous definition of this group is needed in future studies. Available data suggests that the combined presence of CVM and CDH result in a poorer prognosis than would be expected with either malformation in isolation. However, the impact on these survival data from patients with a malformation syndrome or other genetic etiology has not been consistently evaluated, and we encourage researchers to re-analyze existing series and ensure that future studies distinguish CDH which is isolated from that which is associated with other malformations, especially as part of genetic syndromes. Therapies should be tailored to maximize cardiac output and systemic oxygen delivery rather than systemic oxygen saturation alone. When the pulmonary vascular resistance is greater than the systemic vascular resistance the ductus arteriosus unloads, the right ventricle and ductal closure may precipitate acute right ventricular failure. Thus, persistence of the atrial foramen ovale and the ductus arteriosus may be helpful to maintain cardiac output at the expense of cyanosis. Reopening the ductus with PGE-1 infusion may salvage the CDH patient with suprasystemic pulmonary artery hypertension and right ventricular failure Although there is speculation about the frequency of isolated left ventricular “hypoplasia”, we suggest that in most cases it results from compression of a pre-load deficient left ventricle by the hypertensive right ventricle, and unlike true hypoplasia is reversible. Irrespective of the type of severity of CVMs in patients with CDH, the degree of pulmonary hypoplasia and pulmonary vascular disease predicts outcome.
Survival for patients with isolated CDH has improved dramatically [Bohn, 2002], even with differing approaches to treatment [Azarow et al., 1997; Wilson et al., 1997]. Current management strategies permit survival of more severely affected cases, but in contrast to patients from earlier cohorts [Trachsel et al., 2006], serious late morbidity is recognized increasingly [Chiu et al., 2006]. The occurrence of CDH with a CVM adversely affects both early mortality and later morbidity, so must be taken into account when planning the medical and/or surgical management of a neonate who has both CDH and a CVM, and when discussing prognosis and treatment options with the family.
Acknowledgments
Grant sponsor: Massachusetts Department of Public Health, Massachusetts Centers for Birth Defects Research and Prevention; Grant number: (#U50/CCU 1132247-03) (A.E.L.); Grant sponsor: NICHD; Grant number: RO1 HD55150-01 (B. R. P.).
We thank Dr. Lewis B. Holmes and Marie-Noel Westgate, MEd for access to, and assistance in reviewing the Brigham-Women’s Hospital Malformation Surveillance Program patients. Meaghan Muir provided research assistance.
Biographies
Angela E. Lin, M.D. is an Associate Clinical Professor of Pediatrics at Harvard Medical School, a member of the Genetics Unit, Department of Pediatrics, at the MassGeneral Hospital for Children, and consultant to the Massachusetts Department of Public Health, Massachusetts Centers for Birth Defects Research and Prevention in Boston, Massachusetts. Having trained in pediatric cardiology and medical genetics, Dr. Lin’s interests include the epidemiology and delineation of cardiovascular malformations in genetic disorders
Barbara R. Pober, M.D. is an Associate Professor of Pediatrics at Harvard Medical School and member of the Department of Surgery Children’s Hospital of Boston, Genetics Unit, and the Department of Pediatrics at the MassGeneral Hospital for Children in Boston, Massachusetts. Dr. Pober’s interests include the genetic delineation of congenital diaphragmatic hernia, as well as a longstanding interest in Williams syndrome
Ian Adatia, M.B.ChB., M.R.C.P., F.R.C.P. is a Professor of University of San Francisco, Department of Pediatrics, Division of Cardiology and Critical Care Medicine, University of San Francisco Children’s Hospital, California. Dr. Adatia’s interests include pulmonary vascular disease and cardiac intensive care, including the management and outcome of infants with congenital diaphragmatic hernia
References
- Adatia I. Congenital diaphragmatic hernia and congenital heart disease. Congenital Diaphragmatic Hernia Working Study Group; Houston: 2004. Feb 19–20, [Google Scholar]
- Allan LD, Irish MS, Glick PL. The fetal heart in diaphragmatic hernia. Clin Perinatol. 1996;23:795–812. [PubMed] [Google Scholar]
- Andrews RE, Simpson JM, Sharland GK, Sullivan ID, Yates RW. Outcome after preterm delivery of infants antenatally diagnosed with congenital heart disease. J Pediatr. 2006;148:213–216. doi: 10.1016/j.jpeds.2005.10.034. [DOI] [PubMed] [Google Scholar]
- Azarow K, Messineo A, Pearl R, Filler R, Barker G, Bohn D. Congenital diaphragmatic hernia—A tale of two cities: The Toronto experience. J Pediatr Surg. 1997;32:395–400. doi: 10.1016/s0022-3468(97)90589-3. [DOI] [PubMed] [Google Scholar]
- Battaglia A, Carey JC, Cederholm P, Viskochil D, Brothman AR, Galasso C. Natural history of Wolf-Hirschhorn syndrome: Experience with 15 cases. Pediatrics. 1999;103:830–836. doi: 10.1542/peds.103.4.830. [DOI] [PubMed] [Google Scholar]
- Benjamin DR, Juul S, Siebert JR. Congenital posterolateral diaphragmatic hernia: Associated malformations. J Pediatr Surg. 1988;23:899–903. doi: 10.1016/s0022-3468(88)80380-4. [DOI] [PubMed] [Google Scholar]
- Bohn D. Congenital diaphragmatic hernia. Am J Respir Crit Care Med. 2002;166:911–915. doi: 10.1164/rccm.200204-304CC. [DOI] [PubMed] [Google Scholar]
- Bollmann RK, Kalache K, Mau H, Chaoui R, Tennstedt C. Associated malformations and chromosomal defects in congenital diaphragmatic hernia. Fetal Diagn Ther. 1995;10:52–59. doi: 10.1159/000264193. [DOI] [PubMed] [Google Scholar]
- Bosi G, Garani G, Scoranno M, Calzolari E the IMER Working Party. Temporal variability in birth prevalence of congenital heart defects as recorded by a general birth defects registry. J Pediatr. 2003;142:690–698. doi: 10.1067/mpd.2003.243. [DOI] [PubMed] [Google Scholar]
- Botto LD, Correa A, Erickson JD. Racial and temporal variations in the prevalence of heart defects. Pediatrics. 2001;107:E32. doi: 10.1542/peds.107.3.e32. [DOI] [PubMed] [Google Scholar]
- Bove KE, Geiser EA, Meyer RA. The left ventricle in anomalous pulmonary venous return. Morphometric analysis of 36 fatal cases in infancy. Arch Pathol. 1975;99:522–528. [PubMed] [Google Scholar]
- Buss M, Williams G, Dilley A, Jones O. Prevention of heart failure in the management of congenital diaphragmatic hernia by maintaining ductal patency. A case report. J Pediatr Surg. 2006;41:e9–11. doi: 10.1016/j.jpedsurg.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Calzolari E, Garani G, Cocchi G, Magnani C, Rivieri F, Neville A, Astolfi G, Baroncini A, Garavelli L, Gualandi F, Scorrano M, Bosi G IMERWorking Group. Congenital heart defects: 15 years of experience of the Emilia-Romagna Registry (Italy) Eur J Epidemiol. 2003;18:773–780. doi: 10.1023/a:1025312603880. [DOI] [PubMed] [Google Scholar]
- Carmi R, Barbash A, Mares AJ. The thoracoabdominal syndrome (TAS): A new X-linked dominant disorder. Am J Med Genet. 1990;36:109–114. doi: 10.1002/ajmg.1320360122. [DOI] [PubMed] [Google Scholar]
- Chiu PP, Sauer C, Mihailovic A, Adatia I, Bohn D, Coates AL, Langer JC. The price of success in the management of congenital diaphragmatic hernia: is improved survival accompanied by an increase in long-term morbidity? J Pediatr Surg. 2006;41:888–892. doi: 10.1016/j.jpedsurg.2006.01.026. [DOI] [PubMed] [Google Scholar]
- Cohen MS, Rychik J, Bush DM, Tian ZT, Howell LJ, Adzick NS, Flake AW, Johnson MP, Spray TL, Crombleholme TM. Influence of congenital heart disease on survival in children with congenital diaphragmatic hernia. J Pediatr. 2002;141:25–30. doi: 10.1067/mpd.2002.125004. [DOI] [PubMed] [Google Scholar]
- Colvin J, Bower C, Dickinson JE, Sokol J. Outcomes of congenital diaphragmatic hernia: A population-based study in Western Australia. Pediatrics. 2005;116:e356–363. doi: 10.1542/peds.2004-2845. [DOI] [PubMed] [Google Scholar]
- Copel JA, Pilu G, Kleinman CS. Extracardiac anomalies and congenital heart disease. Semin Perinatol. 1993;17:89–105. [PubMed] [Google Scholar]
- Cunniff C, Jones KL, Jones MC. Patterns of malformation in children with congenital diaphragmatic defects. J Pediatr. 1990;116:258–261. doi: 10.1016/s0022-3476(05)82884-7. [DOI] [PubMed] [Google Scholar]
- Dillon E, Renwick M, Wright C. Congenital diaphragmatic herniation: Antenatal detection and outcome. Br J Radiol. 2000;73:360–365. doi: 10.1259/bjr.73.868.10844860. [DOI] [PubMed] [Google Scholar]
- Doray B, Girard-Lemaire F, Gasser B, Baldauf JJ, de Geeter B, Spizzo M, Zeidan C, Flori E. Pallister-Killian syndrome: difficulties of prenatal diagnosis. Prenat Diagn. 2002;22:470–477. doi: 10.1002/pd.342. [DOI] [PubMed] [Google Scholar]
- Dott MM, Wong LYC, Rasmussen S. Population-based study of congenital diaphragmatic hernia: risk factors and survival in Metropolitan Atlanta, 1968–1999. Birth Defects Res Part A Clin Mol Teratol. 2003;68:261–267. doi: 10.1002/bdra.10039. [DOI] [PubMed] [Google Scholar]
- Enns GM, Cox VA. Congenital diaphragmatic defects and associated syndromes, malformations, and chromosome anomalies: A retrospective study of 60 patients and literature review. Am J Med Genet. 1998;79:215–225. [PubMed] [Google Scholar]
- Eskedal L, Hagemo P, Eskild A, Aamodt G, Seiler KS, Thaulow E. A population-based study of extra-cardiac cardiac anomalies in children with congenital cardiac malformations. Cardiol Young. 2004;14:600–607. doi: 10.1017/S1047951104006043. [DOI] [PubMed] [Google Scholar]
- Fauza DO, Wilson JM. Congenital diaphragmatic hernia and associated anomalies: Their incidence, identification, and impact on prognosis. J Pediatr Surg. 1994;29:1113–1117. doi: 10.1016/0022-3468(94)90290-9. [DOI] [PubMed] [Google Scholar]
- Ferencz C, Loffredo CA, Correa-Villaseñor A, Wilson PD. Genetic and Environmental Risk Factors of Major Congenital heart defects: The Baltimore-Washington Infant Study: 1981–1989. Armonk, NY: Futura Publishing Company, Inc; 1997. [Google Scholar]
- Gaynor JW, Mahle WT, Cohen MI, Ittenbach RF, DeCampli WM, Steven JM, Nicolson SC, Spray TL. Risk factors for mortality after the Norwood procedure. Eur J Cardiothorac Surg. 2002;22:82–89. doi: 10.1016/s1010-7940(02)00198-7. [DOI] [PubMed] [Google Scholar]
- Graham TP, Jr, Jarmakani JM, Canent RV., Jr Left heart volume characteristics with a right ventricular volume overload. Total anomalous pulmonary venous connection and large atrial septal defect. Circulation. 1972;45:389–396. doi: 10.1161/01.cir.45.2.389. [DOI] [PubMed] [Google Scholar]
- Graziano JN For the Congenital Diaphragmatic Hernia Study Group. Cardiac anomalies in patients with congenital diaphragmatic hernia and their prognosis: A report from the Congenital Diaphragmatic Hernia Group. J Pediatr Surg. 2005;40:1045–1050. doi: 10.1016/j.jpedsurg.2005.03.025. [DOI] [PubMed] [Google Scholar]
- Greenwood RD, Rosenthal A, Nadas AS. Cardiovascular abnormalities associated with congenital diaphragmatic hernia. Pediatrics. 1976;57:92–97. [PubMed] [Google Scholar]
- Harmath A, Hajdu J, Csaba A, Hauzman E, Pete B, Gorbe E, Beke A, Papp Z. Associated malformations in congenital diaphragmatic hernia cases in the last 15 years in a tertiary referral institute. Am J Med Genet Part A. 2006;140A:2290–2304. doi: 10.1002/ajmg.a.31470. [DOI] [PubMed] [Google Scholar]
- Hutson MR, Kirby ML. Neural crest and cardiovascular development: A 20 year perspective. Birth Defects Res Part C Embryo Today. 2003;69:2–13. doi: 10.1002/bdrc.10002. [DOI] [PubMed] [Google Scholar]
- Kappetein AP, Gittenberger-de Groot AC, Zwinderman AH, Rohmer J, Poelmann RE, Huysmans HA. The neural crest as a possible pathogenetic factor in coarctation of the aorta and bicuspid aortic valve. J Thorac Cardiovasc Surg. 1991;102:830–836. [PubMed] [Google Scholar]
- Karamlou T, Ashburn DA, Caldarone CA, Blackstone EH, Jonas RA, Jacobs ML, Williams MG, Ungerleider RM, McCrindle BW. Matching procedure to morphology improves outcomes in neonates with tricuspid atresia. J Thorac Cardiovasc Surg. 2005;130:1503–1510. doi: 10.1016/j.jtcvs.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Kerstein D, Levy P, Hsu D, Hordorf A, Gersony W, Barst R. Blade balloon atrial septostomy in patients with with severe primary pulmonary hypertension. Circulation. 1995;91:2028–2035. doi: 10.1161/01.cir.91.7.2028. [DOI] [PubMed] [Google Scholar]
- Killeen OG, Kelehan P, Reardon W. Double vagina with sex reversal, congenital diaphragmatic hernia, pulmonary and cardiac malformations–another case of Meacham syndrome. Clin Dysmorphol. 2002;11:25–28. doi: 10.1097/00019605-200201000-00005. [DOI] [PubMed] [Google Scholar]
- Lin AE, Herring AH, Scharenberg Amstutz K, Westgate MN, Lacro RV, Al-Jufan M, Ryan L, Holmes LB. Cardiovascular Malformations: Changes in prevalence and birth status, 1972–1990. Am J Med Genet. 1999a;84:102–110. [PubMed] [Google Scholar]
- Lin AE, Neri G, Hughes-Benzie R, Weksberg R. Cardiac anomalies in the Simpson-Golabi-Behmel syndrome. Am J Med Genet. 1999b;83:378–381. [PubMed] [Google Scholar]
- Lin A, Pober B, Mullen M, Slavotinek A. Cardiovascular malformations in Fryns syndrome suggest a role for neural crest. Am J Med Genet Part A. 2005;139A:186–193. doi: 10.1002/ajmg.a.31023. [DOI] [PubMed] [Google Scholar]
- Losty PD, Vanamo K, Rintala RJ, Donahoe PK, Schnitzer JJ, Lloyd DA. Congenital diaphragmatic hernia- Does the side of the defect influence the incidence of the associated malformations? J Pediatr Surg. 1998;33:507–510. doi: 10.1016/s0022-3468(98)90099-9. [DOI] [PubMed] [Google Scholar]
- Martinez-Frias ML, Prieto L, Urioste M, Bermejo E. Clinical/epidemiological analysis of congenital anomalies associated with diaphragmatic hernia. Am J Med Genet. 1996;62:71–76. doi: 10.1002/(SICI)1096-8628(19960301)62:1<71::AID-AJMG15>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Meacham LR, Winn KJ, Culler FL, Parks JS. Double vagina, cardiac, pulmonary, and other genital malformations with 46,XY karyotype. Am J Med Genet. 1991;41:478–481. doi: 10.1002/ajmg.1320410420. [DOI] [PubMed] [Google Scholar]
- Micheletti A, Hislop AA, Lammers A, Bonhoeffer P, Derrick G, Rees P, Haworth SG. Role of atrial septostomy in the treatment of children with pulmonary arterial hype. Heart. 2006;92:969–972. doi: 10.1136/hrt.2005.077669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michielon G, Marino B, Formigari R, Gargiulo G, Picchio F, Digilio MC, Anaclerio S, Oricchio G, Sanders SP, Di Donato RM. Genetic syndromes and outcome after surgical correction of tetralogy of Fallot. Ann Thorac Surg. 2006;81:968–975. doi: 10.1016/j.athoracsur.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Migliazza L, Otten C, Xia H, Rodriguez JU, Diez-Pardo JA, Tovar JA. Cardiovascular malformations in congenital diaphragmatic hernia: human and experimental studies. J Pediatr Surg. 1999;349:1352–1358. doi: 10.1016/s0022-3468(99)90010-6. [DOI] [PubMed] [Google Scholar]
- Momma K, Ando M, Mori Y, Ito T. Hypoplasia of the lung and heart in fetal rats with diaphragmatic hernia. Fetal Diagn Ther. 1992;7:46–52. doi: 10.1159/000263650. [DOI] [PubMed] [Google Scholar]
- Musewe NN, Alexander DJ, Teshima I, Smallhorn JF, Freedom RM. Echocardiographic evaluation of the spectrum of cardiac anomalies associated with trisomy 13 and trisomy 18. J Am Coll Cardiol. 1990;15:673–677. doi: 10.1016/0735-1097(90)90644-5. [DOI] [PubMed] [Google Scholar]
- Nakazawa M, Jarmakani JM, Gyepes MT, Prochazka JV, Yabek SM, Marks RA. Pre and postoperative ventricular function in infants and children with right ventricular volume overload. Circulation. 1977;55:479–484. doi: 10.1161/01.cir.55.3.479. [DOI] [PubMed] [Google Scholar]
- Nelson K, Holmes LB. Malformations due to presumed spontaneous mutations in newborn infants. N Engl J Med. 1989;320:19–23. doi: 10.1056/NEJM198901053200104. [DOI] [PubMed] [Google Scholar]
- Parvari R, Carmi R, Weissenach J, Pilia G, Mumm S, Weinstein Y. Refined genetic mapping of X-linked thoracoabdominal syndrome. Am J Med Genet. 1996;61:401–402. doi: 10.1002/(SICI)1096-8628(19960202)61:4<401::AID-AJMG18>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Pfleghaar KM, Wapner RJ, Kuhlman KA, Spitzer AR. Congenital diaphragmatic hernia: Prognosis and prenatal detection. Fetal Diagn Ther. 1995;10:393–399. doi: 10.1159/000264264. [DOI] [PubMed] [Google Scholar]
- Phoon CK, Silverman NH. Conditions with right ventricular pressure and volume overload, and a small left ventricle: “Hypoplastic” left ventricle or simply a squashed ventricle? J Am Coll Cardiol. 1997;30:1547–1553. doi: 10.1016/s0735-1097(97)00351-3. [DOI] [PubMed] [Google Scholar]
- Pober BR, Lin AE, Russell M, Ackerman KG, Chakravorty S, Strauss B, Westgate MN, Wilson J, Donahoe PK, Holmes LB. Infants with Bochdalek diaphragmatic hernia: Sibling precurrence and monozygotic twin discordance in a hospital-based malformation surveillance program. Am J Med Genet Part A. 2005;138A:81–88. doi: 10.1002/ajmg.a.30904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SA, Olney RS, Holmes LB, Lin AE, Keppler-Noreuil K, Moore CA The National Birth Defects Prevention Study. Guidelines for case classification for the National Birth Defects Study. Birth Defects Res Part A. 2003;67:193–201. doi: 10.1002/bdra.10012. [DOI] [PubMed] [Google Scholar]
- Robinson WM, Newburger JW. Ethical issues concerning cardiac surgery in patients with syndromic abnormalities. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2003;6:147–151. doi: 10.1053/pcsu.2003.50016. [DOI] [PubMed] [Google Scholar]
- Rosenquist GC, Kelly JL, Chandra R, Ruckman RN, Galioto FM, Jr, Midgley FM, Scott LP. Small left atrium and change in contour of the ventricular septum in total anomalous pulmonary venous connection: a morphometric analysis of 22 infant hearts. Am J Cardiol. 1985;55:777–782. doi: 10.1016/0002-9149(85)90155-9. [DOI] [PubMed] [Google Scholar]
- Rozkovec A, Montanes P, Oakley C. Factors that influence the outcome of primary pulmonary hypertension. Br Heart J. 1986;55:449–458. doi: 10.1136/hrt.55.5.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CA, Perreault T, Johnston-Hodgson A, Finer NN. Extracorporeal membrane oxygenation in infants with congenital diaphragmatic hernia and cardiac malformations. J Pediatr Surg. 1994;29:878–881. doi: 10.1016/0022-3468(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Sandoval J, Gaspar J, Pulido T, Bautista E, Guerra MLM, Zeballos M, Palomar A, Gómez A. Graded balloon dilation atrial septostomy in severe primary pulmonary hypertension. J Am Coll Cardiol. 1998;32:297–304. doi: 10.1016/s0735-1097(98)00238-1. [DOI] [PubMed] [Google Scholar]
- Schaefer GB, Jochar A, Muneer R, Sanger WG. Clinical variability of tetrasomy 12p. Clin Genet. 1997;51:102–108. doi: 10.1111/j.1399-0004.1997.tb02429.x. [DOI] [PubMed] [Google Scholar]
- Skari H, Bjornland K, Haugen G, Egeland T, Emblem R. Congenital diaphragmatic hernia: a meta-analysis of mortality factors. J Pediatr Surg. 2000;35:1187–1197. doi: 10.1053/jpsu.2000.8725. [DOI] [PubMed] [Google Scholar]
- Slavotinek A. Single gene disorders associated with congenital diaphragmatic hernia. Am J Med Genet Part C Semin Med Genet. 2007 doi: 10.1002/ajmg.c.30125. (in press) [DOI] [PubMed] [Google Scholar]
- Sokol J, Bohn D, Lacro RV, Ryan G, Stephens D, Rabinovitch M, Smallhorn J, Hornberger LK. Fetal pulmonary artery diameters and their association with lung hypoplasia and postnatal outcome in congenital diaphragmatic hernia. Am J Obstet Gynecol. 2002;186:1085–1090. doi: 10.1067/mob.2002.122413. [DOI] [PubMed] [Google Scholar]
- Sokol J, Shimizu N, Bohn D, Doherty D, Ryan G, Hornberger LK. Fetal pulmonary artery diameter measurements as a predictor of morbidity in antenatally diagnosed congenital diaphragmatic hernia: a prospective study. Am J Obstet Gynecol. 2006;195:470–477. doi: 10.1016/j.ajog.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Stasik CN, Goldberg CS, Bove EL, Devaney EJ, Ohye RG. Current outcomes and risk factors for the Norwood procedure. J Thorac Cardiovasc Surg. 2006;131:412–417. doi: 10.1016/j.jtcvs.2005.09.030. [DOI] [PubMed] [Google Scholar]
- Stege G, Fenton A, Jaffray B. Nihilism in the 1990s: the true mortality of congenital diaphragmatic hernia. Pediatrics. 2003;112:532–535. doi: 10.1542/peds.112.3.532. [DOI] [PubMed] [Google Scholar]
- Strauss B, Lin A, Pober B, Malik S, Curtis M, Kimonis V. Delineation of the type of cardiovascular malformations in DeLange syndrome. Platform presentation at the David W. Smith Workshop on Malformations and Morphogenesis; Iowa City, Iowa. August, 2005. [Google Scholar]
- Suda K, Bigras JL, Bohn D, Hornberger LK, McCrindle BW. Echocardiographic predictors of outcome in newborns with congenital diaphragmatic hernia. Pediatrics. 2000;105:1106–1109. doi: 10.1542/peds.105.5.1106. [DOI] [PubMed] [Google Scholar]
- Sweed Y, Puri P. Congenital diaphragmatic hernia: influence of associated malformations on survival. Arch Dis Child. 1993;69:68–70. doi: 10.1136/adc.69.1_spec_no.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegnander E, Williams W, Johansen OJ, Blaas HG, Eik-Nes SH. Prenatal detection of heart defects in a non-selected population of 30,149 fetuses–Detection rates and outcome. Ultrasound Obstet Gynecol. 2006;27:252–265. doi: 10.1002/uog.2710. [DOI] [PubMed] [Google Scholar]
- Tennstedt C, Chaoui R, Korner H, Dietel M. Spectrum of congenital heart defects and extracardiac malformations associated with chromosomal abnormalities: results of a seven year necropsy study. Heart. 1999;82:34–39. doi: 10.1136/hrt.82.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibboel D, Gaag AV. Etiologic and genetic factors in congenital diaphragmatic hernia. Clin Perinatol. 1996;23:689–699. [PubMed] [Google Scholar]
- Tonks A, Wyldes M, Somerset DA, Dent K, Abhyankar A, Bagchi I, Lander A, Roberts E, Kilby MD. Congenital malformations of the diaphragm: findings of the West Midlands Congenital Anomaly Register 1995–2000. Prenatal Diagn. 2004;24:596–604. doi: 10.1002/pd.908. [DOI] [PubMed] [Google Scholar]
- Trachsel D, Selvadurai H, Adatia I, Bohn D, Schneiderman-Walker J, Wilkes D, Coates AL. Resting and exercise cardiorespiratory function in survivors of congenital diaphragmatic hernia. Pediatr Pulmonol. 2006;41:522–529. doi: 10.1002/ppul.20359. [DOI] [PubMed] [Google Scholar]
- Van Praagh S, Truman T, Firpo A, Bano-Rodrigo A, Fried R, McManus B, Engle MA, Van Praagh R. Cardiac malformations in trisomy-18: a study of 41 postmortem cases. J Am Coll Cardiol. 1989;13:1586–1597. doi: 10.1016/0735-1097(89)90353-7. [DOI] [PubMed] [Google Scholar]
- Van Son JA, Phoon CK, Silverman NH, Haas GS. Predicting feasibility of biventricular repair of right-dominant unbalanced atrioventricular canal. Ann Thorac Surg. 1997;63:1657–1663. [PubMed] [Google Scholar]
- Wilson JG, Warkany J. Cardiac and aortic arch anomalies in the offspring of vitamin A deficient rats correlated with similar human anomalies. Pediatrics. 1950;5:708–725. [PubMed] [Google Scholar]
- Wilson JG, Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency, effects of restoration of vitamin A at various times during gestation. Am J Anat. 1957;92:189–217. doi: 10.1002/aja.1000920202. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Lund DP, Lillehei CW, Vacanti JP. Congenital diaphragmatic hernia a tale of two cities: the Boston experience. J Pediatr Surg. 1997;32:401–405. doi: 10.1016/s0022-3468(97)90590-x. [DOI] [PubMed] [Google Scholar]
- Witters I, Legius E, Moerman Ph, Deprest J, Schoubroeck DV, Timmerman D, Van Assche FA, Fryns JP. Associated malformations and chromosomal anomalies in 42 cases of prenatally diagnosed diaphragmatic hernia. Am J Med Genet. 2001;103:278–282. [PubMed] [Google Scholar]
- Yang W, Carmichael SL, Harris JA, Shaw GM. Epidemiologic characteristics of congenital diaphragmatic hernia among 2.5 million California births, 1989–1997. Birth Defects Res A Clin Mol Teratol. 2006;76:170–174. doi: 10.1002/bdra.20230. [DOI] [PubMed] [Google Scholar]
- Yu J, Gonzalez S, Rodriguez JI, Diez-Pardo JA, Tovar JA. Neural crest-derived defects in experimental congenital diaphragmatic hernia. Pediatr Surg. 2001;17:294–298. doi: 10.1007/s003830100597. [DOI] [PubMed] [Google Scholar]