

Abstract
Background
Aromatase inhibitors provide superior disease control when compared with tamoxifen as adjuvant therapy for postmenopausal women with endocrine-responsive early breast cancer.
Purpose
To present the design, history, and analytic challenges of the Breast International Group (BIG) 1-98 trial: an international, multicenter, randomized, double-blind, phase-III study comparing the aromatase inhibitor letrozole with tamoxifen in this clinical setting.
Methods
From 1998–2003, BIG 1-98 enrolled 8028 women to receive monotherapy with either tamoxifen or letrozole for 5 years, or sequential therapy of 2 years of one agent followed by 3 years of the other. Randomization to one of four treatment groups permitted two complementary analyses to be conducted several years apart. The first, reported in 2005, provided a head-to-head comparison of letrozole versus tamoxifen. Statistical power was increased by an enriched design, which included patients who were assigned sequential treatments until the time of the treatment switch. The second, reported in late 2008, used a conditional landmark approach to test the hypothesis that switching endocrine agents at approximately 2 years from randomization for patients who are disease-free is superior to continuing with the original agent.
Results
The 2005 analysis showed the superiority of letrozole compared with tamoxifen. The patients who were assigned tamoxifen alone were unblinded and offered the opportunity to switch to letrozole. Results from other trials increased the clinical relevance about whether or not to start treatment with letrozole or tamoxifen, and analysis plans were expanded to evaluate sequential versus single-agent strategies from randomization.
Limitations
Due to the unblinding of patients assigned tamoxifen alone, analysis of updated data will require ascertainment of the influence of selective crossover from tamoxifen to letrozole.
Conclusions
BIG 1-98 is an example of an enriched design, involving complementary analyses addressing different questions several years apart, and subject to evolving analytic plans influenced by new data that emerge over time.
Introduction
Breast cancer is currently the leading type of cancer among women worldwide and accounts for nearly one in four cases of cancer among women [1]. According to recent data from the World Health Organization, the incidence rates among industrialized countries range from 80–99/100 000 women [1]. Although 90% of patients are initially diagnosed with early and operable breast cancer, more than 50% relapse within 10 years without adjuvant treatment [2].
Since the late 1950s, randomized trials of adjuvant systemic therapy have been conducted in an effort to reduce the number of relapses and to prolong the survival of patients with operable disease. The Early Breast Cancer Trialists' Collaborative Group, summarizing data from 194 randomized trials of women with early breast cancer, found that adjuvant systemic treatments with chemotherapy, endocrine therapy, or combinations of both improved the prognosis of patients with breast cancer. In their meta-analysis, treatment with tamoxifen for 5 years reduced the relative risks of breast cancer death and recurrence by 34% and 41%, respectively, in women with estrogen receptor-positive disease [2]. Treatment with tamoxifen alone or in combination with chemotherapy was shown to be more effective than chemotherapy alone. Through the late 1990s, prolonged endocrine therapy with tamoxifen was considered to be a standard treatment in a post-menopausal patient population with early breast cancer.
During the last several years, the superiority of tamoxifen has been questioned after the development of aromatase inhibitors (AIs). Third-generation aromatase inhibitors include the nonsteroidal inhibitors, letrozole and anastrozole, and the steroidal inhibitor, exemestane. AIs were shown to be efficacious in the treatment of advanced breast cancer [3–5], and multiple clinical trials were developed to evaluate the role of AIs as an adjuvant treatment for women with early breast cancer. One such trial is the Breast International Group (BIG) 1-98 study, which compares 5 years of monotherapy with either tamoxifen or letrozole, and also examines the effect of sequential treatment of 2 years of one agent followed by 3 years of the other. A total of 8028 postmenopausal women with hormone receptor-positive, operable, breast cancer enrolled in the BIG 1-98 trial between March 1998 and May 2003. This article presents the history of the trial, the evolution of the trial design over time, published results, and lessons learned during the study conduct.
History
BIG 1-98 was originally conceived by the pharmaceutical company, Novartis, as the FEMTA Trial, a two-arm, phase-III, randomized, double-blind trial to compare 5 years of treatment with either letrozole or tamoxifen in postmenopausal women with operable, invasive breast cancer that was positive for estrogen receptors, progesterone receptors, or both. Begun in March 1998, this trial was designed as a head-to-head comparison of letrozole versus tamoxifen to satisfy regulatory requirements and to obtain the answer in as short a time frame as possible.
During the FEMTA trial development, the strategy for the development of letrozole in the adjuvant setting was re-evaluated by Novartis and a consensus was reached to place the trial under the auspices of a large network of collaborating cooperative groups specializing in the conduct of breast cancer adjuvant therapy trials, the Breast International Group. Shortly after the trial was underway, the International Breast Cancer Study Group (IBCSG), which is one of 41 cooperative groups that comprise the BIG, became the coordinating group for the BIG 1-98 adjuvant letrozole study and has been responsible for the scientific integrity, operation, and logistics of the trial.
In April 1999, the BIG 1-98 trial was activated with two randomization options, the two-arm option, which incorporated the FEMTA trial, and a new four-arm option, which added a second objective to evaluate the strategy of sequencing one agent after the other, with the switch at 2 years, for a total of 5 years of therapy. The two-arm option remained open to accrual only long enough to permit previously participating institutions to obtain ethics committee approval for activation of the four-arm option (August–September 1999) or to allow previously consented patients to complete their chemotherapy regimens prior to randomization (March 2000).
The BIG 1-98 protocol expanded upon the original hypothesis of treatment superiority to address a second question regarding optimal sequencing of existing therapies. Specifically, for a patient who is disease-free, does switching to an alternative endocrine agent at 2 years reduce the risk of relapse compared with continuing the initial treatment to complete 5 years? The concept behind the sequential therapy was to study whether disease-free survival and overall survival could be improved by suppression or elimination of nonclinically evident micro-metastases, which developed resistance to the first treatment. Previous trials had shown letrozole to be an active agent in patients whose prior treatment with anti-estrogens failed, as well as in patients whose disease was resistant to treatment with tamoxifen.
BIG 1-98 trial design
Patients meeting the eligibility criteria (Table 1) were stratified by institution and by chemotherapy received (i.e., adjuvant and/or neo-adjuvant chemotherapy completed prior to randomization, neither adjuvant nor neo-adjuvant chemotherapy received with none planned, or adjuvant chemotherapy given concurrently with protocol therapy). If an institution participated in the two-arm option, patients were randomly assigned to receive either tamoxifen or letrozole for 5 years. If the institution participated in the four-arm option, patients were randomly assigned to receive one of four treatments: one of the two 5-year monotherapy treatments, letrozole for 2 years followed by tamoxifen for 3 years, or tamoxifen for 2 years followed by letrozole for 3 years (Figure 1). The trial was blinded to patients, physicians, and all study personnel with the exception of personnel at the IBCSG Statistical Center during the preparation of reports submitted to the Data and Safety Monitoring Committee.
Table 1.
Key inclusion and exclusion criteria for the BIG 1-98 trial
| Inclusion |
|
| Exclusion |
|
Figure 1.

BIG 1-98 trial design. The design accommodated its genesis as a two-arm trial that evolved into a four-arm trial enabling both head-to-head comparison of letrozole vs tamoxifen as well as assessment of the role of sequential endocrine therapies.
The primary endpoint was disease-free survival (DFS), which was defined as the time from random assignment to the earliest time of invasive recurrence in local, regional, or distant sites; a new invasive breast cancer in the contralateral breast; any second (non-breast) malignancy; or death from any cause. Secondary outcomes were overall survival, systemic disease-free survival, and safety. Although the duration of treatment was set at 5 years, all patients were to be followed lifelong for disease status, survival, and certain adverse events. An intention-to-treat analysis approach would be implemented.
Sample size considerations
The original target accrual for the BIG 1-98 trial was 5180 patients. We assumed that 1680 patients would be the final accrual figure for the two-arm option, and sought to recruit an additional 3500 patients in the four-arm option. The protocol was amended in 2002 to increase the total sample size to 8000 patients.
Original sample size
The original sample size calculation was based on estimates of 5-year DFS among patients treated with tamoxifen alone from two prior IBCSG trials, one in a node-positive and one in a node-negative population. For the head-to-head comparison of letrozole versus tamoxifen, a sufficient number of patients were sought to provide 80% power using a two-sided, 0.05-level test to detect a 20% reduction in the risk of a DFS event (hazard ratio = 0.80; 75% versus 79.4% 5-year DFS). A total of 642 events were required from a total recruitment of 5180. Included in the analysis, known as the primary core analysis (PCA), were all patients randomized under the two- or four-arm randomization options and events in the sequential therapy arms that occurred prior to the treatment switch.
To test the second primary hypothesis that a sequence of endocrine agents is superior to a single endocrine agent, a combined analysis of two pair-wise comparisons, stratified according to the initial endocrine agent, would be performed for patients enrolled in the four-arm option. A sufficient number of patients was sought to provide 80% power using a two-sided, 0.05-level test to detect a 23% reduction in the risk of a DFS event measured after the switching time point, approximately 2 years after treatment initiation (hazard ratio = 0.77 landmarked from the switching time point, thus including only patients alive and disease-free at 2 years). A total of 470 events were required.
Revised sample size
In August 2002, the target accrual for the trial was increased to 8000 patients. Preliminary calculations of the hazard rate based on data received through March 4, 2002 suggested that the average annual hazard rate (0.0335) was lower than originally assumed (0.0518). In addition, average accrual during the 6 months from September 2001 through February 2002 was 193 patients per month (2316 per year) much higher than originally anticipated (700 patients per year). Therefore, to compensate for the lower-than-anticipated event rate, the decision was made to increase the number of patients accrued to the four-arm randomization option to at least 6100 patients, which was 2600 patients more than the 3500 originally planned for the four-arm option. By the time of this decision, the two-arm option had been closed with a total accrual of 1835 patients. Increasing accrual to the four-arm option to at least 6100 patients thus increased the total sample size to 7935 patients. These modifications were made prior to any assessments of treatment effects.
For the PCA, a sufficient number of patients was sought to provide 80% power using a two-sided, 0.05-level test to detect a 20% reduction in the risk of a DFS event (hazard ratio = 0.80; 84.5% versus 87.4% 5-year DFS). A total of 647 events were required and allowed for two interim analyses based on the O'Brien-Fleming spending function. Events occurring in the sequential treatment arms would be counted if occurring prior to the treatment switch (i.e., up to approximately 2 years after randomization). Increasing the sample size from 5180 to approximately 8000 patients permitted the original timelines to be met despite the re-estimation of the baseline 5-year DFS percent from 75% (original design) to 84.5% (revised design).
To compare single endocrine agent therapy versus sequential endocrine therapy, the increase in sample size permitted an increase in the power to detect a difference. The power was increased to 92% to detect the 23% reduction in risk originally specified. With 80% power, a 20% reduction in the risk of an event measured subsequent to the switching time point could be detected (hazard ratio = 0.80 from the switching time point). A total of 677 events recorded for the landmark analyses following the time of treatment switch was sufficient to achieve the revised statistical considerations.
In 2003, the BIG 1-98 Steering Committee considered evidence emerging from other studies of adjuvant therapies for breast cancer and decided that the originally planned, landmark analysis, comparing a sequence of agents to a single agent, should be separated into its component parts. An amendment in October 2003 specified that two separate, pair-wise, landmark comparisons (tamoxifen vs. tamoxifen followed by letrozole, and letrozole vs. letrozole followed by tamoxifen) would be performed with both measured from the time of the treatment switch. Each pair-wise comparison would be conducted at the 2.5% level of significance and each would be conducted when at least 331 DFS events were observed. This number of events was achievable before the end of 2008 and would provide 80% power to detect a 29.3% reduction in the risk of an event by switching rather than maintaining the same endocrine therapy beyond 2 years from treatment initiation. This magnitude of treatment effect is consistent with that observed from other studies evaluating the reduction in risk of recurrence by switching to an AI 2–3 years after diagnosis [6].
Study conduct
Accrual to BIG 1-98
From March 1998 to March 2000, 1835 patients were randomly assigned in the two-arm option to receive 5 years monotherapy with either letrozole or tamoxifen. Enrollment included patients from a total of 148 hospitals in North and South America, Australia, New Zealand, Europe, and South Africa.
The four-arm option opened to accrual in April 1999 and closed in May 2003. A total of 6193 women were randomly assigned to one of the four treatment groups (four-arm option). The expansion of the trial increased the number and types of participating medical centers with the inclusion of additional academic centers worldwide. The final study enrollment was 8028 patients from a total of 240 participating centers. Eighteen patients withdrew their consent for trial participation prior to receiving any study treatment, resulting in an intention-to-treat sample of 8010 patients.
Oversight, quality assurance, and data monitoring
Although international in scope, all official study communications, meetings, and teleconferences were conducted in English. Informed consent documents were translated into appropriate languages for presentation to the patients and were reviewed and approved by local ethics committees. The IBCSG provided study oversight with respect to the logistics of the trial and day-to-day operations through the Coordinating Center in Bern, Switzerland, the Data Management Center in Amherst, New York, and the Statistical Center in Boston, Massachusetts. An independent Data and Safety Monitoring Committee (DSMC), blinded to treatment assignment, reviewed study data every 6 months and provided recommendations for supplementary analyses and safety summaries. DSMC meetings and documents were closed to everyone except the DSMC members and appropriate IBCSG Statistical Center staff. The DSMC also reviewed two interim efficacy analyses for the PCA (event-driven) and three interim efficacy analyses for the analysis of treatments in sequence (annual assessments). Upon recommendation from the DSMC, an independent medical reviewer, also blinded to treatment assignment, reviewed all study deaths, recurrences, severe adverse events, and bone fractures to insure uniform, and scientifically rigorous, reporting. Any resulting data discrepancies were discussed and confirmed with the investigator at the study center.
Study data monitors visited each of the participating centers at least quarterly to review the data and verify accuracy.
Randomization
Prior to 1999, a pre-packaged randomization was used. Novartis supplied participating centers with pre-packaged, 6-month drug packs with patient IDs and blinded drug codes pre-printed and packaged according to stratum. The center pulled the next package in the correct stratum, tore off a portion of the label containing the patient ID and blinded drug code, and attached this label to the appropriate case report form.
When the IBCSG became responsible for coordination of the BIG 1-98 in 1999, a centralized, computerized, randomization system was used. Each participating center was assigned to one of four BIG 1-98 randomization centers based upon geography and time zone (Bern, Switzerland; Amherst, New York; Copenhagen, Denmark; Sydney, Australia), which were linked to the central computer in Amherst. After verifying patient eligibility and obtaining informed consent, the participating center contacted its assigned randomization center by telephone or fax to obtain the patient's ID and drug code assignment. If the assigned randomization center could not be reached, randomization was completed in Amherst.
For patients randomized under the early, prepackaged system, Novartis provided the drug codes, unblinded, to the central randomization system, so that patients could be correctly re-supplied.
Study medication
The protocol dosage for letrozole was 2.5 mg daily oral administration and for tamoxifen 20 mg daily oral administration. Six-month supplies of study medication were provided to patients according to the drug code assignment. Patients took two tablets once a day. Using a double-dummy scheme, each pack included either active tamoxifen with placebo letrozole or placebo tamoxifen with active letrozole. Re-supply of study drug packs occurred every 6 months using an interactive voice response system (IVRS). Clinical site personnel used the telephone-based IVRS to obtain subsequent drug code assignments. Compliance with distribution of study drug packets to patients was monitored using a study drug administration case report form. Emergency code breaks were permitted through the IVRS. Non-emergency code breaks required prior permission of the medical review staff at the IBCSG Coordinating Center.
For patients assigned to the sequential treatment arms, the drug switch occurred at the time the fifth drug pack was dispensed, approximately 2 years after study randomization.
Adverse-event reporting
For patients undergoing study drug administration, adverse events (AEs) were recorded at each 6-month follow-up visit using check boxes on the case report forms. Severity was classified according to NCI Common Toxicity Criteria Version 2.0. The relationship of the AE to study treatment was assessed by the local investigator. Targeted AEs that were explicitly collected were cardiovascular events (i.e., myocardial infarction, cerebrovascular accident, transient ischemic attack, angina requiring PTCA or CABG, thromboembolic event, hypercholesterolemia, and ‘other’), bone fractures, vaginal bleeding, endometrial pathology, nausea, vomiting, hot flushes, night sweats, and events leading to therapy discontinuation. Other cardiovascular AEs were collected using an open-text comment field for specification by the investigator. Open-text fields were coded according to MedDRA, and MedDRA preferred term [7] codes were further grouped into categories by IBCSG oncologists.
Senior oncologists at the IBCSG Coordinating Center, who were blinded to treatment assignment, reviewed all grade 3, 4, or 5 cardiovascular AEs, other grade 3–5 AEs whose causes were unclear, and all deaths occurring prior to a DFS event. Pre-existing cardiovascular morbidities reported at the time of enrollment were also medically reviewed. After treatment completion or discontinuation, cardiovascular, musculoskeletal, and endometrial AEs were reported yearly. Serious adverse events (SAEs) were defined and reported according to International Conference on Harmonization (ICH) Guidelines, beginning at the time of informed consent until 30 days after stopping study treatment.
Follow-up reporting
Patients were seen for follow-up at clinic visits every 6 months during treatment to gather general safety data, to document predefined toxicity data, and to receive a new supply of study medications. If a patient discontinued trial treatment for any reason, survival, disease status, and cardiac, bone, and endometrial AEs were reported every 6 months for 5 years from randomization, followed by yearly reports. After 5 years, patients were followed yearly.
Published efficacy analyses
Primary Core Analysis (2005)
By its innovative design, BIG 1-98 was positioned to address two important questions through two analyses. The first, a protocol-specified primary core analysis (PCA), was presented in 2005 by Thürlimann et al. [8] and provided a head-to-head comparison of letrozole versus tamoxifen. The database was retrieved for this analysis on December 20, 2004. Patients from all four treatment assignments were included in this analysis; however, follow-up information and events for patients enrolled in the two sequential treatment arms were included for up to 30 days after the treatments were switched. The enriched design, including information from the first 2 years of the sequential treatment arms, increased the number of events available for the primary treatment comparison and thus shortened the timeline to conduct the planned analysis. It was noted at that time that the inclusion of patients assigned to the sequential arms reflected early DFS events more strongly due to censoring of their follow-up 30 days after the treatment switch. However, since the median follow-up for the entire PCA was relatively short (25.8 months), the potential bias due to censoring at approximately 2 years in the sequential arms was minimized.
DFS was significantly better in the letrozole group than in the tamoxifen group (HR: 0.81, 95% CI: 0.70–0.93; log-rank p = 0.003). There was no difference in overall survival. Figure 2 displays the overall treatment effect of letrozole relative to tamoxifen in the PCA, and also shows the treatment effect in each of the cohorts defined by the two- and four-arm randomization options. The median follow-up for patients in the two-arm option was 60.5 months compared with 30.0 months for the four-arm monotherapy cohort and 24.9 months for the four-arm sequential cohort. The different durations of follow-up contributed to the different percent of patients having an event in each cohort. Treatment effects within cohorts did not appear to differ from the overall effect in the PCA.
Figure 2.
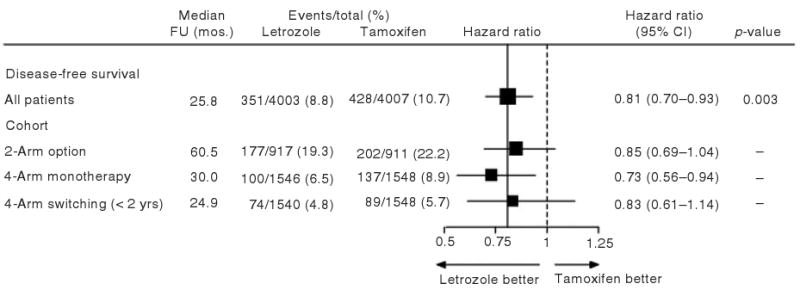
Comparison of treatments for BIG 1-98 PCA cohorts with respect to disease-free survival (DFS). Hazard ratios (HR) and 95% confidence intervals are estimated from a Cox proportional hazards model. The locations of the squares indicate the estimated hazard ratio of a DFS event for letrozole vs tamoxifen, and the size of the squares is proportional to the statistical information available for each comparison. The solid vertical line indicates the overall treatment effect estimate of HR = 0.81, and the dashed vertical line shows the location of the null hypothesis of no treatment difference (HR = 1.0). FU = Follow-up
The BIG 1-98 PCA provided pivotal trial results used for the US Food and Drug Administration's approval of letrozole for the adjuvant treatment of postmenopausal women with hormone receptor-positive early breast cancer in December 2005.
Design revisions following the primary core analysis
The results of the PCA led to the recommendation of the DSMC, and a decision by the BIG 1-98 Steering Committee, to inform the 2459 patients, randomly assigned tamoxifen alone, of their treatment in order to make informed decisions about their future care. The DSMC did not make recommendations about further treatment of these patients; however, the Steering Committee agreed that these patients should be offered the opportunity to stop tamoxifen and receive letrozole. The protocol was amended in April 2005. Patients who were within 4.5 years of randomization could elect to either complete 5 years of treatment with tamoxifen or change to letrozole for the remainder of their 5-year adjuvant therapy. Patients who had been treated for 4.5–5 years with tamoxifen could choose to receive ‘extended’ letrozole for up to an additional 5 years. The remaining three, randomized, treatment groups (i.e., letrozole alone, letrozole followed by tamoxifen, and tamoxifen followed by letrozole) remained blinded.
The unblinding of the tamoxifen-alone group will complicate future analyses, including updates to the PCA. In addition to the intention-to-treat analysis, supplementary analyses will assess the influence of unblinded, selective cross-over to letrozole within the tamoxifen-alone arm.
Analysis of monotherapy cohort (2007)
With more than 1 year of additional follow-up, a protocol-specified analysis was conducted to assess treatment efficacy in the cohort of patients assigned to the monotherapy treatment arms of BIG 1-98 (Figure 1) and was presented by Coates et al. [9] in 2007. This monotherapy analysis was limited to 4922 patients who were assigned to the monotherapy treatments in either the two- or the four-arm randomization options and included protocol-defined updated data, retrieved on February 21, 2006. This analysis compared the efficacy of monotherapy at a median follow-up of 51 months. With the additional follow-up, an adequate number of DFS events made it possible to use only the monotherapy arms, and thus provided adequate power without including information from the first 2 years of the sequential treatment arms. Not including the sequential arms avoided placing extra weight on the early DFS events. Finally, although patients assigned to tamoxifen alone had been unblinded at the time of this analysis, the extent of exposure to letrozole was very short, representing less than 2.5% of the total patient-years of follow-up. Hence, the cross-over to letrozole had little effect on the intention-to-treat analysis of the monotherapy arms at this time. DFS from the monotherapy analysis (HR: 0.82, 95% CI: 0.71–0.95, log-rank p = 0.007) was very similar to that reported for the original PCA, but the results more directly addressed the question of long-term continuous therapy using single endocrine agents (Figure 3).
Figure 3.
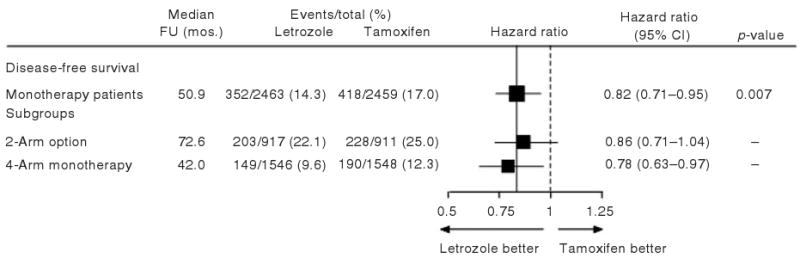
Comparison of treatments for BIG 1-98 monotherapy cohorts with respect to DFS. Hazard ratios and 95% confidence intervals are estimated from a Cox proportional hazards model. The locations of the squares indicate the estimated hazard ratio of a DFS event for letrozole vs tamoxifen, and the size of the squares is proportional to the statistical information available for each comparison. The solid vertical line indicates the overall treatment effect estimate of HR = 0.82, and the dashed vertical line shows the location of the null hypothesis of no treatment difference
Analysis of Sequential Therapy (planned for 2008)
The second, protocol-specified, primary analysis will address the hypothesis that switching endocrine agents is superior to continuing with a single endocrine agent. This sequential therapy analysis (STA) is divided into two parts. The first analysis, known as the STA-S, will use a conditional landmark approach [10] and will be restricted to patients enrolled in the four-arm option who were alive and disease-free 30 days after the fifth drug pack was dispensed (approximately 2 years and 1 month after study randomization).
Two pair-wise comparisons will be performed to assess the effects of: (a) tamoxifen alone versus tamoxifen followed by letrozole, and (b) letrozole alone versus letrozole followed by tamoxifen. The beneficial effect of switching from tamoxifen to an aromatase inhibitor compared with continuing on tamoxifen has already been reported by several trials including the Intergroup Exemestane Study (IES) [11]. Therefore, of particular interest in the STA-S will be the comparison of letrozole for 5 years versus the sequence of letrozole followed by tamoxifen if alive and disease-free at approximately 2 years after treatment initiation, which is yet untested in the adjuvant setting.
Since more than one-third of the patients randomized to tamoxifen alone in the four-arm option have chosen to receive adjuvant letrozole after the release of the initial BIG 1-98 results in 2005, the interpretation of the results of pair-wise comparisons involving tamoxifen alone are complicated in intention-to-treat analyses. As a result, analyses will focus on the remaining three treatment arms, which maintained blinding throughout the conduct of the trial.
Additional pair-wise comparisons among the four treatment groups (four-arm randomization option), starting from the time of randomization, will also be performed to aid in clinical decision-making. Known as the STA-R, these analyses will examine whether or not it is beneficial for a newly diagnosed patient to receive sequential therapy rather than monotherapy, and will also assess which sequence is optimal. Particular attention will be given to two of the comparisons considered to be of greatest clinical relevance: letrozole versus tamoxifen followed by letrozole, and letrozole versus letrozole followed by tamoxifen. Hazard ratios with 99% confidence intervals will be presented to account for multiple comparisons, and estimates of 5-year DFS percent will also be presented as measures for treatment comparison.
To date, the DSMC has reviewed three interim evaluations of the STA-S, with the final review planned for October 2008, approximately 9 years after the start of enrollment into the four-arm option. Updated analyses of the PCA and monotherapy cohort are planned for 10 and 12 years after study initiation.
The timeline for the BIG 1-98 trial, starting with enrollment and ending with the 12-year PCA update, is presented in Table 2. A summary of the study cohorts used for manuscripts and analyses is presented in Table 3.
Table 2.
Timeline of BIG 1-98 trial
| 1998 | March: Enrollment begins for two-arm randomization option |
| 1999 | April: Enrollment begins for four-arm randomization option |
| 2000 | March: Enrollment ends for two-arm randomization option |
| 2003 | May: Enrollment ends for four-arm randomization option |
| 2005 | January: First results of PCA presented at St. Gallen Conference |
| March: Completion of 5 years of treatment for two-arm randomization option | |
| June: Unblind patients randomized to tamoxifen alone | |
| December: PCA published in New England Journal of Medicine | |
| December: FDA approval of letrozole as an adjuvant treatment of HR-positive, early breast cancer in post-menopausal women | |
| 2007 | February: Monotherapy analysis published in Journal of Clinical Oncology |
| 2008 | May: Completion of 5 years of treatment for four-arm randomization option |
| October: Results of STA reviewed by DSMC | |
| October: PCA – 10-year update | |
| December: First results of STA presented at San Antonio Breast Cancer Symposium | |
| 2010 | STA update |
| PCA – 12-year update |
Table 3.
Summary of study cohorts in BIG 1-98 trial
| Population | N | Description |
|---|---|---|
| Randomized patients | 8028 | Total patients enrolled in BIG 1-98. |
| Intention-to-treat patients | 8010 | Eighteen randomized patients withdrew consent to participate before starting treatment. |
| Two-arm randomization option | 1828 | |
| Four-arm randomization option | 6182 | |
| Safety population of PCA | 7963 | Excludes 47 patients from the ITT population who received no study treatment. This population is used for reporting all adverse events. |
| Primary core analysis (PCA) | 8010 | Follow-up for two sequential treatment groups is censored at the date of the switch + 30 days. Maximizes the number of patients and events in head-to-head comparison of L vs T. Gives more weight to early events. |
| Monotherapy cohort | 4922 | Patients randomized to receive 5 years of tamoxifen alone or letrozole alone. |
| Two-arm randomization option | 1828 | Provides additional events with longer follow-up. More appropriate for direct comparison of intended 5-year longer-term use of each agent as monotherapy. |
| Four-arm randomization option | 3094 | |
| Four-arm cohort from randomization | 6182 | Patients in the ITT population enrolled in the four-arm randomization option to compare treatment strategies from the time of randomization. |
| Four-arm cohort from switch (approximately 2 years from randomization) | 5828 | Patients enrolled in four-arm randomization option, who were alive and disease-free 30 days after the fifth drug pack was dispensed (approximately 2 years and 1 month after randomization). Used to assess the value of starting with one agent and switching to the other compared with remaining on the original agent. |
| Central pathology assessment cohort | 6291 | Total number of patients with at least one of four markers (ER, PgR, HER-2, Ki-67) assessable through retrospective, central pathology review. |
| Monotherapy cohort | 3650 |
A lesson learned: The critical role of accurate assessment of steroid hormone receptor status
To determine eligibility for enrollment in BIG 1-98, the primary tumor had to be positive for estrogen receptors (ER) or progesterone receptors (PgR). Steroid hormone receptor status (ER and PgR) was determined by local pathologists before random treatment assignment. Hormone receptor status was assessed using either biochemical (positive defined as greater than or equal to 10 fmol/mg cytosol protein) or immunohistochemical (IHC) assays (positive defined as greater than or equal to 10% of the invasive tumor cells expressing ER and/or PgR). IHC assays were used to determine hormone receptor status in 93% of patients and biochemical assays in the remaining 7%.
During the course of the trial, some centers participated in the pathology substudy by submitting one stained, hematoxylin and eosin (H and E) slide and either 11 unstained slides or one paraffin block of the primary tumor. In April 2005, a retrospective tissue collection of tumor blocks began in accordance with institutional guidelines and national laws. The blocks were used to centrally assess four tumor markers and to prepare H and E slides to increase the number of patients with tumors centrally reviewed for histopathologic features.
The IBCSG Central Pathology Laboratory received tissue material for 6549 patients (82% of the enrolled sample), with 6291 patients having material with at least one of the four markers assessable (79%). Tissue material was reviewed for histopathologic features and four tumor markers: estrogen receptor (ER), progesterone receptor (PgR) [12], and Ki-67 by IHC [13], and HER2/neu (ErbB2) [14] by IHC and florescence in situ hybridization (FISH). Central assessments were performed without knowledge of patients' treatment assignments or outcomes.
The impact and importance of central assessment of ER and PgR was reported for patients receiving monotherapy by Viale et al., [12] in 2007. Local assessment for eligibility classified 99.9% of enrolled patients as having hormone receptor-positive disease. Central assessment of 6291 tumors classified 97.0% as positive. Concordance of local and central assessment of ER and of PgR varied by country, for example, with nine countries (out of 25) having a concordance of 80% or lower. Discordance was more marked for PgR than for ER.
Patients in the monotherapy cohort, whose tumors were classified locally as hormone receptor-positive and reclassified as hormone receptor-negative by central review, had worse outcomes, with an estimated 65% 3-year DFS compared with 91% among patients whose tumors were classified concordantly (Figure 4). Patients who did not have the appropriate hormone receptor status required by this protocol, and, therefore, who did not have the disease being studied, enrolled in a trial with treatments that could not help them. Thus, an important role of central pathology review is to identify the appropriate target population for treatment through correct eligibility screening. While prospective central review involves additional expense and logistical planning, the personal and scientific costs of administering improperly targeted treatment can be higher.
Figure 4.
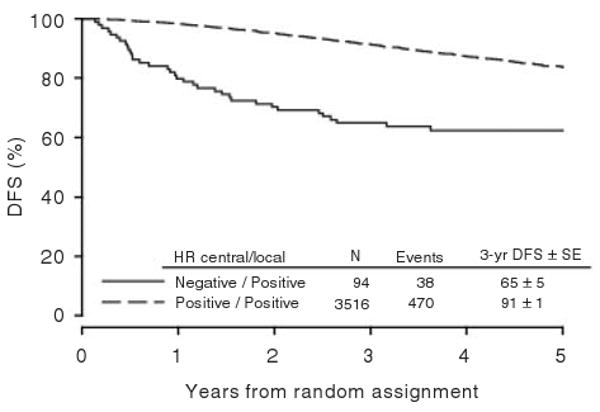
DFS according to central and local classification hormone receptor status. Numbers of patients and events, and estimates of 3-year DFS and standard errors (SE) are summarized. [Reprinted with permission of the Journal of Clinical Oncology 25(25), 2007: 3846–3852].
Conclusion
BIG 1-98 started as a two-arm, randomized, clinical trial, designed to assess the superiority of letrozole over tamoxifen. The trial increased its clinical relevance by expanding to answer two questions concerning how best to use endocrine agents for the treatment of early breast cancer in postmenopausal women with hormone receptor-positive tumors. With the addition of a second primary hypothesis, to assess the benefit of switching as compared with continuing treatment agents, the BIG 1-98 trial will provide information about the effectiveness of letrozole in sequence with tamoxifen.
Random assignment to one of the four possible treatment regimens allowed for two complementary analyses in BIG 1-98. The PCA showed the superiority of letrozole over tamoxifen and was the basis for regulatory approval of letrozole as an adjuvant therapy for early breast cancer. Patients who were assigned the sequential treatments were included in this analysis until the time of the treatment switch at approximately 2 years. The potential for bias was minimized at this relatively early point in the overall follow-up of the trial, when the PCA was reported, but future analyses, comparing 5 years of letrozole versus 5 years of tamoxifen, will focus on the monotherapy cohorts.
The dissemination of these results led to the unblinding of patients assigned tamoxifen alone for 5 years, offering them an option to switch to letrozole for the remainder of their 5 years of adjuvant treatment or choose letrozole as extended adjuvant therapy. Analysis of updated data for the tamoxifen-alone treatment group will be more complex, requiring evaluation of the influence of unblinded, selective cross-over in more than one-third of patients.
Analysis of the second primary hypothesis will complement the PCA. Limited to patients enrolled in the four-arm option, this analysis will use a conditional landmark approach to compare the effects of treatments for patients alive and disease-free approximately 2 years after study enrollment. An additional set of analyses will compare outcomes for the four treatment groups timed from randomization. It is hoped that these results will inform decisions regarding optimal endocrine treatment for postmenopausal women who are newly diagnosed with endocrine responsive, operable, breast cancer.
Acknowledgments
BIG 1-98 collaborative group participants
Steering Committee: B. Thürlimann (Chair), S. Aebi, L. Blacher, M. Castiglione, A. S. Coates, T. Cufer, J. F. Forbes, R. D. Gelber, A. Giobbie-Hurder, A. Goldhirsch, A. Hiltbrunner, S. B. Holmberg, R. Maibach, A. Martoni, L. Mauriac, G. McGrogan, H. T. Mouridsen, R. Paridaens, K. N. Price, M. Rabaglio, B. B. Rasmussen, M. M. Regan, A. Santoro, I. E. Smith, A. Wardley, K. Vantongelen, G. Viale. Novartis: H. A. Chaudri-Ross, D. Emanuel, D. B. Evans, C. Sguotti, U. Trostmann.
IBCSG Scientific Committee: A. Goldhirsch, A. S. Coates (Co-Chairs), L. Blacher, J. F. Forbes, R. D. Gelber, B. A. Gusterson, A. Hiltbrunner, C. Hürny, E. Murray, K. N. Price, M. Rabaglio, R. Studer, G. Viale, A. Wallgren.
IBCSG Foundation Council (members from 1998 to 2008): S. Aebi, A. S. Coates, M. Colleoni, J. P. Collins, H. Cortés Funes, R. D. Gelber, A. Goldhirsch, M. Green, A. Hiltbrunner, S. B. Holmberg, P. Karlsson, I. Kössler, I. Láng, J. Lindtner, F. Paganetti, M. de Stoppani, C.-M. Rudenstam, H.-J. Senn, R. Stahel, B. Thürlimann, A. Veronesi.
Coordinating Center (Berne, Switzerland): A. Hiltbrunner (Director), M. Rabaglio, G. Egli, B. Cliffe, S. Ribeli-Hofmann, F. Munarini, R. Kammler, R. Studer, B. Ruepp, R. Maibach, N. Munarini, M. Castiglione.
Statistical Center (Dana-Farber Cancer Institute, Boston, MA, USA): R. D. Gelber (Group Statistician), K. N. Price (Director of Scientific Administration), A. Giobbie-Hurder (Trial Statistician), A. Keshaviah, H. Litman, M. M. Regan, Z. Sun, H. Huang, L. J. Somos, B. Timmers, L. Nickerson.
Data Management Center (Frontier Science & Technology Research Foundation, Amherst, NY, USA): L. Blacher (Director of Data Management), T. Heckman Scolese (Coordinating Data Manager), M. Belisle, M. Caporale, J. Celano, L. Dalfonso, L. Dooley, S. Fischer, K. Galloway, J. Gould, R. Hinkle, M. Holody, G. Jones, R. Krall, S. Lippert, J. Meshulam, L. Mundy, A. Pavlov-Shapiro, K. Scott, M. Scott, S. Shepard, J. Swick, L. Uhteg, D. Weinbaum, C. Westby, T. Zielinski.
Central Pathology Review Office (University of Glasgow, Glasgow, UK): B. A. Gusterson, E. Mallon; (European Institute of Oncology, Division of Pathology, Milano, Italy): G. Viale, P. Dell'Orto, M. Mastropasqua, B. Del Curto.
Data and Safety Monitoring Committee: J. E. Garber, W. Gradishar, S. W. Lagakos, I. Lindgren.
Study Support (Novartis Corp. Basel, Switzerland): E. Waldie, I. van Hoomissen, M. De Smet, U. Trostmann, W. Schmidt, A. Bolton, W. Hackl.
Breast International Group (BIG)
International Breast Cancer Study Group (IBCSG)
Australian New Zealand Breast Cancer Trials Group (ANZ BCTG): Board Chair: R. D. Snyder, Group Coordinator: J. F. Forbes, Chair Scientific Advisory Committee: A. S. Coates; ANZ BCTG Operations Office (Newcastle, Australia): D. Lindsay (Head Data Management), D. Preece (Senior Study Coordinator), J. Cowell, D. Talbot, A. Whipp.
Australia: The Cancer Council Victoria, Melbourne, VIC: F. Abell, R. Basser, R. Bell, B. Brady, D. Blakey, P. Briggs, I. Burns, P. Campbell, M. Chao, J. Chirgwin, B. Chua, K. Clarke, J. Collins, R. De Boer, J. C. Din, R. Doig, A. Dowling, R. Drummond, N. Efe, S. T. Fan, M. Francis, P. Francis, V. Ganju, P. Gibbs, G. Goss, M. Green, P. Gregory, J. Griffiths, I. Haines, M. Henderson, R. Holmes, P. James, J. Kiffler, M. Lehman, M. Leyden, L. Lim, G. Lindeman, R. Lynch, B. Mann, J. McKendrick, S. McLachlan, R. McLennan, G. Mitchell, S. Mitra, C. Murphy, I. Parker, K. Phillips, I. Porter, G. Richardson, J. Scarlet, S. Sewak, J. Shapiro, R. Snyder, R. Stanley, C. Steer, D. Stoney, A. Strickland, G. Toner, C. Underhill, K. White, M. White, A. Wirth, S. Wong; W P Holman Clinic, Launceston General Hospital, Launceston, Tasmania: D. Byram, I. Byard; Liverpool Hospital, Sydney, NSW: S. Della-Fiorentina, A. Goldrick, E. Hovey, E. Moylan, E. Segelov; Mount Hospital, Perth, WA: A. Chan, M. Buck, D. Hastrich, D. Ingram, G. Van Hazel, P. Willsher; Nepean Cancer Care Centre, Sydney, NSW: N. Wilcken, C. Crombie; Calvary Mater Newcastle, Newcastle, NSW: J. F. Forbes, F. Abell, S. Ackland, A. Bonaventura, S. Cox, J. Denham, R. Gourlay, D. Jackson, R. Sillar, J. Stewart; Prince of Wales Hospital, Sydney, NSW: C. Lewis, B. Brigham, D. Goldstein, M. Friedlander; Princess Alexandra Hospital, Woollongabba, QLD: E. Walpole, D. Thompson; Royal Adelaide Hospital, Adelaide, SA: P. G. Gill, M. Bochner, J. Coventry, J. Kollias, P. Malycha, I. Olver; Royal Brisbane and Women's Hospital, Brisbane, QLD: M. Colosimo, R. Cheuk, L. Kenny, N. McCarthy, D. Wyld; Royal Hobart Hospital, Hobart, Tasmania: R. Young, R. Harrup, R. Kimber, R. Lowenthal; Royal Perth Hospital, Perth, WA: J. Trotter, E. Bayliss, A. Chan, D. Ransom; Sir Charles Gairdner Hospital, Perth, WA: M. Byrne, M. Buck, J. Dewar, A. Nowak, A. Powell, G. Van Hazel; Toowoomba Hospital, Toowoomba, QLD: E. A. Abdi, R. Brodribb, Z. Volobueva; Westmead Hospital, Sydney, NSW: P. Harnett, V. Ahern, H. Gurney, N. Wilcken.
New Zealand: Auckland Hospital, Auckland: V. J. Harvey, B. Evans, W. Jones, M. McCrystal, D. Porter, P. Thompson, M. Vaughan; Christchurch Hospital, Christchurch: D. Gibbs, C. Atkinson, R. Burcombe, B. Fitzharris, B. Hickey, M. Jeffery, B. Robinson; Dunedin Hospital, Dunedin: B. McLaren, S. Costello, J. North, D. Perez; Waikato Hospital, Hamilton: I. D. Campbell, L. Gilbert, R. Gannaway, M. Jameson, I. Kennedy, J. Long, G. Round, L. Spellman, D. Whittle, D. Woolerton.
Brazil: Hospital de Clinicas de Porto Alegre, Porto Alegre: C. Menke, J. Biazús, R. Cericatto, J. Cavalheiro, N. Xavier, A. Bittelbrunn, E. Rabin.
Chile: Chilean Cooperative Group for Oncologic Research, GOCCHI: J. Gutiérrez (Chairman), R. Arriagada (Scientific Adviser), L. Bronfman (Principal Investigator), M. Zuñiga (Data Manager); Clinica Las Condes, Santiago: J. Gutiérrez, J. C. Acevedo, S. Torres, A. León, E. Salazar; Hospital DIPRECA, Las Condes, Santiago: L. Soto Diaz, R. Duval, N. Oddeshede, M. C. Venti; Hospital San Juan de Dios, Santiago: K. Peña, L. Puente, V. Maidana; IRAM/Instituto de Radiomedicina, Vitacura, Santiago: R. Baeza, R. Arriagada, P. Olfos, J. Solé, E. Vinés, C. Mariani.
Hungary: National Institute of Oncology, Budapest: I. Láng, E. Hitre, E. Szabó, Z. Horváth, E. Ganofszky, E. Juhos.
Italy: Centro di Riferimento Oncologico, Aviano: A. Veronesi, D. Crivellari, M. D. Magri, A. Buonadonna, F. Coran, E. Borsatti, E. Candiani, S. Massarut, M. Roncadin, M. Arcicasa, A. Carbone, T. Perin, A. Gloghini; Ospedali Riuniti di Bergamo, Bergamo: C. Tondini, R. Labianca, P. Poletti, A. Bettini; Ospedale degli Infermi, Biella: M. Clerico, M. Vincenti, A. Malossi, E. Seles, E. Perfetti, B. Sartorello; Spedali Civili, Brescia: E. Simoncini, G. Marini, P. Marpicati, R. Farfaglia, A. M. Bianchi, P. Grigolato, L. Lucini, P. Frata, A. Huscher, E. Micheletti, C. Fogazzi; U. O. Medicina Oncologica, Ospedale Carpi, Ospedale Mirandola: F. Artioli, K. Cagossi, L. Scaltriti, E. Bandieri, L. Botticelli, G. Giovanardi; Ospedale di Cattolica ‘Cervesi,’ Cattolica: A. Ravaioli, E. Pasquini, B. Rudnas; Ospedale Civile, Gorizia: L. Foghin; Ospedale “A. Manzoni” Lecco, Lecco: M. Visini, L. Zavallone, G. Ucci; Istituto Europeo di Oncologia, Milano: M. Colleoni, G. Viale, P. Veronesi, G. Peruzzotti, L. Corsetto, R. Ghisini, G. Renne, A. Luini, L. Orlando, R. Torrisi, A. Rocca, T. De Pas, E. Munzone, V. Galimberti, S. Zurrida, M. Intra, F. Nolé, R. Orecchia, G. Martinelli, F. de Braud, A. Goldhirsch; Ospedale Infermi, Rimini: A. Ravaioli, L. Gianni.
Peru: Instituto de Enfermedades Neoplásicas, Lima: H. Gome.
Slovenia: Institute of Oncology, Ljubljana: T. Cufer, B. Pajk, J. Cervek.
South Africa: Groote Schuur Hospital and University of Cape Town, Cape Town: I. D. Werner, E. Murray, D. Govender, S. Dalvie, T. Erasmus, B. Robertson, B. Read, E. Nel, J. Toop, N. Nedeva, E. Panieri; Sandton Oncology Centre, Johannesburg: D. Vorobiof, M. Chasen, G. McMichael, C. Mohammed. Local funding provided by the Cancer Association of South Africa
Sweden: West Swedish Breast Cancer Study Group: S. B. Holmberg; Sahlgrenska U Hospital, Moelndal: S. B. Holmberg, J. Mattsson; Boras Hospital, Boras; Karlstads Hospital, Karlstads: H. Sellström; Kungalvs Hospital, Kungalvs: B. Lindberg.
Switzerland: Swiss Group for Clinical Cancer Research (SAKK): A. Goldhirsch (up to January 2004), R. Herrmann (from June 2004): Kantonsspital Aarau, Zentrum f. Onkologie, Aarau: A. Schönenberger, W. Mingrone, Ch. Honegger, E. Bärtschi, M. Neter, M. Rederer, G. Schär; University Hospital Basel, Basel: C. Rochlitz, R. Herrmann, D. Oertli, E. Wight, H. Moch; Institute of Oncology of Southern Switzerland: Ospedale San Giovanni, Bellinzona: J. Bernier, L. Bronz, F. Cavalli, E. Gallerani, A. Richetti, A. Franzetti; Ospedale Regionale di Lugano (Civico & Italiano), Lugano: M. Conti-Beltraminelli, M. Ghielmini, T. Gyr, S. Mauri, P. C. Saletti; Ospedale Regionale Beata Vergine, Mendrisio: A. Goldhirsch, O. Pagani, R. Graffeo, M. Locatelli, S. Longhi, P.C. Rey, M. Ruggeri; Ospedale Regionale La Carità, Locarno: E. Zucca, D. Wyss; Istituto Cantonale di Patologia, Locarno: L. Mazzucchelli, E. Pedrinis, T. Rusca; Inselspital, Berne: S. Aebi, M. F. Fey, M. Castiglione, M. Rabaglio; Kantonsspital Olten, Olten: S. Aebi, M. F. Fey, M. Zuber, G. Beck; Bürgerspital, Solothurn: S. Aebi, M. F. Fey, R. Schönenberger; Spital Thun-Simmental AG Thun: J.M. Lüthi, D. Rauch; Hôpital Cantonal Universitaire HCUG, Geneva: H. Bonnefoi; Rätisches Kantons- und Regionalspital, Chur: F. Egli, R. Steiner, P. Fehr; Centre Pluridisciplinaire d'Oncologie, Lausanne: L. Perey, P. de Grandi, W. Jeanneret, S. Leyvraz, J.-F. Delaloye; Kantonsspital St. Gallen, St. Gallen: B. Thürlimann, D. Köberle, F. Weisser, S., Mattmann, A. Müller, T. Cerny, B. Späti, M. Höfliger, G. Fürstenberger, B. Bolliger, C. Öhlschlegel, U. Lorenz, M. Bamert, J. Kehl-Blank, E. Vogel; Kantonales Spital Herisau, Herisau: B. Thürlimann, D. Hess, I. Senn, D. Köberle, A. Ehrsam, C. Nauer, C. Öhlschlegel, J. Kehl-Blank, E. Vogel; Stadtspital Triemli, Zürich: L. Widmer, M. Häfner; Universitätsspital Zürich, Zürich: B. C. Pestalozzi, M. Fehr, R. Caduff, Z. Varga, R. Trüb, D. Fink.
Swiss Private MDs: Private Praxis, Zürich: B. A. Bättig; Sonnenhof-Klinik Engeried, Berne: K. Buser; Frauenklinik Limmattalspital, Schlieren: N. Bürki; Private Praxis, Birsfelden: A. Dieterle; Private Praxis, Biel: L. Hasler; Private Praxis, Baar: M. Mannhart-Harms; Brust-Zentrum, Zürich: C. Rageth; Private Praxis, Berne: J. Richner; Private Praxis, Bellinzona: V. Spataro; Private Praxis, Winterthur: M. Umbricht.
United Kingdom: King's College Hospital/Breast Unit, London: P. Ellis, S. Harris, N. Akbar, H. McVicars, C. Lees, R. Raman, G. Crane.
Danish Group (DBCG)
H. T. Mouridsen; Rigshospitalet, Copenhagen: H. T. Mouridsen; Vejle Hospital, Vejle: E. Jakobsen; Odense University Hospital, Odense: S. Cold; KAS Herlev / Herlev University Hospital, Herlev: C. Kamby; Aalborg Sygehus Syd, Aalborg: M. Ewertz; Hilleroed Hospital, Hilleroed: P.M. Vestlev; Aarhus University Hospital, Aarhus: J. Andersen; Roskilde County Hospital, Roskilde: P. Grundtvig; Esbjerg Central Hospital, Esbjerg: E. Sandberg; Naestved Central Hospital, Naestved: P. Philip; Soenderborg Sygehus, Soenderborg: E. L. Madsen; Herning Central Hospital, Herning: K. A. Moeller; Viborg Sygehus, Viborg: V. Haahr; Landspitali University Hospital, Reykjavik, Iceland: J. Johansson.
French Group (FNCLCC)
Institut Bergonié, Bordeaux: L. Mauriac, M. Debled, P. Campo; Centre Hospitalier de la Côte Basque, Bayonne D. Larregain-Fournier, S. Remy, Centre Jean Perrin, Clermont-Ferrand: H. Auvray; Centre Georges François Leclerc, Dijon: C. De Gislain, F. Delille, M.-C. Porteret; Centre Oscar Lambret, Lille: V. Servent, M. Chapoutier; CHRU, Limoges: N. Tubiana-Mathieu, S. Lavau-Denes, P. Bosc; Centre Léon Bérard, Lyon: J. P. Guastalla, Th. Bachelot, C. Arbault; Centre Hospitalier Meaux, Meaux: G. Netter-Pinon; C.H.G. André Boulloche, Montbéliard: V. Perrin, A. Monnier, Y. Hammoud; Centre Paul Lamarque, Montpellier: G. Romieu, L. Culine, V. Pinosa; Clinique Francheville, Périgueux: L. Cany, C. Maguire; Hôpital de la Milétrie, Poitiers: A. Daban, M. Le Saux, C. Grandon; Centre Eugène Marquis, Rennes: P. Kerbrat, C. Catheline; Centre Henri Becquerel, Rouen: C. Veyret, E. Jugieau, V. Talon; Centre René Gauducheau, Saint-Herblain: A. Le Mevel, S. Maury; Centre Claudius Régaud, Toulouse: L. Gladieff, N. Lignon.
North Yorkshire Group
D. Dodwell; Harrogate District Hospital, Harrogate, North Yorkshire: D. Dodwell; Huddersfield Royal Infirmary, Huddersfield: J. Joffe; Castlehill Hospital, Hull: P. Drew; Airedale General Hospital, Keighley, W. Yorkshire: A. Nejim; Leeds General Infirmary, Leeds: D. Dodwell, K. Horgan; St. James's University Hospital, Leeds: M. Lansdown, T. Perren; Weston Park Hospital, Sheffield: R. E. Coleman.
Independent Centers/Groups
Argentina: Centro Oncológico Confidence, Buenos Aires: D. Campos; Hospital Allemán, Buenos Aires: F. Cóppola; Hospital Británico, Buenos Aires: J. Martinez; Hospital Evita, Buenos Aires: M. Freue; Hospital Posadas, Buenos Aires: C. Wainstein; Hospital Zubizarreta, Buenos Aires: A. Zori Comba; Instituto Dr. Estevez, Buenos Aires: E. Cazap; Instituto Oncológico Dr. Angel H. Roffo, Buenos Aires: E. Mickiewicz; Sanatorio Municipal Julio A. Mendez, Buenos Aires: L. Balbiani; Centro Privado de Ginecología, Córdoba: A. Osuna; Hospital Privado de Córdoba, Córdoba: E. Palazzo; Instituto Modelo de Ginecología y Obstetricia, Córdoba: M. de Romedis; Fundación Mainetti-Centro Oncológico de Excelencia, La Pllata: S. Cagnolati; Hospital Privado de la Comunidad, Mar del Plata: C. A. Delfino, G. Caccia; Escuela de Medicina Nuclear (COIR), Mendoza: R. L. de Angelis; Centro Oncológico de Rosario, Rosario: L. Fein, R. Sala; Hospital Provincial de Rosario, Rosario: C. Nassurdi, A. Colombo Berra; Clínica Especializada ISIS, Santa Fe: R. Viroglio, C. Blajman; Hospital Regional de Concepción, Tucumán: H. Requejo; Instituto de Maternidad y Ginecología Nuestra Señoras de las Mercedes, Tucumán: L. Silberman.
Australia: Flinders Medical Centre, Adelaide, SA: S. Birrell, M. Eaton, C. Hoffman; Queen Elizabeth Hospital, Adelaide, SA: V. Humeniuk; The Canberra Hospital, Canberra, ACT; P. Craft, R. Stuart-Harris, D. Yip; The Geelong Hospital, Geelong, VIC: R. Bell, F. Abell, M. Francis, J. Kiffer, R. Lynch, R. McLennan, K. White; Royal Melbourne Hospital, Melbourne, VIC: M. Green, R. Basser, J. Collins, R. De Boer, J. C. Din, N. Efe, S. T. Fan, G. Lindeman, S. Wong; Western General Hospital, Melbourne, VIC: M. Green, R. Basser, J. Collins, R. De Boer, J. C. Din, N. Efe, S. T. Fan, G. Lindeman, S. Wong; Newcastle Mater Hospital, Newcastle, NSW: J. Stewart, F. Abell, S. Ackland, A. Bonaventura; Royal Perth Hospital, Perth, WA: J. Trotter, E. Bayliss, A. Chan, D. Ransom, A. Redfern; St. George Hospital, Sydney, NSW: P. de Souza, M. Links; St. Vincent's Hospital, Sydney, NSW: D. Dalley, J. Grygiel, R. Ward; Murray Valley Private Hospital, Wodonga, VIC: C. Underhill, K. Clarke, C. Steer; Princess Alexandra Hospital, Woolloongabba, QLD: E. Walpole, D. Thompson.
Belgium: Institut Jules Bordet, Bruxelles: J. M. Nogaret; University Hospitals Leuven, Leuven: M.R. Christiaens, P. Neven, R. Paridaens, A. Smeets, I. Vergote, C. Weltens, H. Wildiers; Les Cliniques Saint-Joseph ASBL, Liège: C. Focan; Clinique du Parc Léopold, Bruxelles: L. Marcelis; C. H. Etterbeek - Ixelles, Bruxelles: J. P. Kains; Service d'Oncologie Clinique Notre-Dame, Charleroi: J.-L. Canon; C. H. U. André Vèsale, Montigny-Le Tilleul: D. Brohèe.
Canada: Cambridge Memorial Hospital, Cambridge: J. Gowing; CHUM- Campus Notre-Dame, Montreal: L. Yelle; Hôpital Maisonneuve-Rosemont, Montreal: P. Dubé.
Chile: Fundacion Lopez Perez, Santiago: C. Vogel; Hospital Carlos Van Buren, Valparaiso: M. León Prieto.
Czech Republic: Institute of Oncology, Brno: K. Petrakova, M. Palacova, R. Demlova; Dept. of Clinical and Radiation Oncology, Ceske Budejovice: H. Siffnerova, J. Fischer, I. Bustova; Centre of Breast Diseases, Prague: H. Kankova, M. Pintova; Institute of Radiation Oncology, Prague: P. Vitek; University Hospital, Prague: J. Abrahamova, D. Kordikova; University Hospital Prague: L. Petruzelka, E. Sedlackova, H. Honova.
Germany: Onkologische Gemeinschaftspraxis, Augsburg: B. Heinrich; Zentralklinikum/Frauenklinik, Augsburg: A. Wischnik; Universi-tätsklinikum Essen, Essen: C. Oberhoff, A. E. Schindler; Universitäts-Frauenklinik d. JLU Giessen, Giessen: K. Münstedt; Onkologische Gemeinschaftspraxis, Göttingen: D. Meyer; Martin-Luther-Universität Halle-Wittenberg, Halle: R. Grosse, H. Kölbl; Universitätskliniken des Saarlandes, Homburg: W. Schmidt, D. Mink; Universitäts-Frauenklinik und Poliklinik Universitätskrankenhaus Eppendorf, Hamburg: F. Jänicke; Kliniken d. Med. Hochschule, Frauenklinik, Hannover: H. J. Lück; Krankenanstalt Mutterhaus der Borromäerinnen, Trier: W. Dornoff; Gynäkologische Abteilung des St. Josefshospital, Wiesbaden: G. Hoffmann; Gynäkologische Abteilung d. Marienhospitals, Universität Witten-Herdecke, Witten: J. Hackmann, W. Bader.
Hungary: SZOTE Onkoterápiás Klinika, Szeged: Z. Kahan; BM Központi Kórház, Budapest: G. Pajkos, K. Kristo; SOTE Radiológiai és Onkoterápiás Klinika, Budapest: M. Dank; Uzsoki Utcai Kórház, Budapest: T. Nagykalnai, L. Landherr; Almási Balogh Pál Kórház, Ózd: E. Kner; Területi Kórház Onkologia, Szentes: M. Kispál; Szent Borbála Kórház, Megyei Onkológiai Gondozó, Tatabánya: Á. Dani.
Italy: Policlinico S. Orsola-Malpighi, Bologna: A. Martoni, C. Zamagni, S. Giaquinta, E. Piana; Ospedale S. Croce, Fano: R. Mattioli, L. Imperatori; Istituto Clinica Humanitas, Milan/Rozzano: A. Santoro, C. Carnaghi, L. Rimassa; Azienda Ospedaliera San Filippo Neri, Rome: G. Gasparini, G. Sciarretta, A. Morabito; Az. Ospedaliera Treviglio-Caravaggio, Treviglio: S. Barni, M. Cazzaniga, M. Cabiddu; Policlinico Universitario (PUDG), Udine: F. Puglisi; Ospedale di Torrette, Ancona: R. Cellerino, S. Antognoli, F. Freddari; Universitiy of Cagliari, Policlinico Universitario, Cagliari: G. Mantovani, E. Massa, G. Astara; Ospedale Civile Feltre, Feltre: R. Segati; Istituto Nazionali Ricerca Cancro, Genova: R. Rosso, L. Del Mastro, M. Venturini, C. Bighin; Istituto Nazionale dei Tumori, Milano: E. Bajetta, N. Zilembo, D. Paleari, G. Procopio; Azienda Ospedaliera di Parma, Parma: S. Salvagni, M. A. Perrone, V. Franciosi; Azienda Ospedaliera “S. Salvatore”, Pesaro: G. Catalano, S. Luzi Fedeli; Azienda Ospedaliera “Ospedale di Circolo e Fondazione Macchi” Varese: G. Pinotti, G. Giardina, I. Vallini; Universitiy of Cagliari, Policlinico Universitario, Cagliari: B. Massidda, M. T. Ionta, M. C. Deidda; Ospedale Maggiore, Lodi: G. Nalli, G. Sita; Policlinico Universitario, Palermo: I. Carreca, S. Cucciarré, D. Burgio; Ospedale Civile dello Spirito Santo, Pescara: M. Lombardo, G. Pandoli, P. Di Stefano; Azienda Ospedaliera Santa Maria Nuova, Reggio Emilia: C. Boni, G. Bisagni, M. C. Banzi, P. Linarello; Azienda Ospedaliera Desenzano del Garda, Manerbio: G. Colosini, A. Spasiano, A. Caldonazzo; Ospedale Civile ASL 20, Tortona: M. G. Pacquola.
Netherlands: Ziekenhuis Leyenburg, Den Haag: H. P. Sleeboom; Catharina Ziekenhuis, Eindhoven: H. J. T. Rutten; St. Anna Ziekenhuis, Geldrop: E. J. T. Luiten; Tweesteden Ziekenhuis, Tilburg: H. Th. J. Roerdink; Maxima Medisch Centrum, Veldhoven: R. H. M. Roumen.
New Zealand: Dunedin Hospital, Dunedin: B. McLaren, S. Costello, J. North, D. Perez, K., Bayston, M. Pfieffer; Waikato Hospital, Hamilton: I. Kennedy, I. D. Campbell, L. Gilbert, R. Gannaway, M. Jameson, J. Long, G. Round, L. Spellman, D. Whittle, D. Woolerton.
Poland: Department of Oncology and Radiotherapy, Medical University of Gdansk, Gdansk: J. Jassem, M. Welnicka-Jaskiewicz, E. Senkus-Konefka, K. Matuszewska; Rydygier's Memorial Hospital, Krakow-Nova Huta: P. Koralewski, J. Pernal; Klinika Nowotworów Piersi i, Chirurgii Rekonstrukcyjnej-Warszawa, Warszawa: T. Pienkowski, E. Brewczynska, B. Bauer-Kosinska, R. Sienkiewicz-Kozlowska, A. Jagiello-Gruszfeld, K. Sudol.; Centrum Onkologii w Bydgoszczy, Oddzial Onkologii Klinicznej, Bydgoszcz: J. Tujakowski, B. Zurawski; Collegium Medicum Jagiellonian University, Krakow: J. Pawlega, E. Jablonska, A. Zygulska; Oddzial Kliniczny Onkologiczny, Centralnego Szpitala Klinicznego Wojskowej, Akademii Medycznej-Warszawa, Warszawa: M. Górnasiowa; Dolnoslaskie Centrum Onkologii, Wroclaw: E. Filypczyk-Cisarz, K. Pajak.
Portugal: Hospital de S. João, Porto: M. Damasceno; Instituto Português de Oncologia de Coimbra, Coimbra: J. Q. Albano; Hospital de Santa Maria, Lisboa: B. da Costa, L. Costa; Instituto Português de Oncologia de Lisboa, Lisboa: A. Henriques, H. Amaral; Hospital Geral de Santo António, Porto: F. Marques.
Russia: Cancer Research Centre, Moscow: D. V. Komov, S. B. Polikarpova; Moscow Municipal Hospital No. 62, Moscow: A. N. Makhson, N. V. Zabaznyi; Moscow Research Institute of Diagnostics and Surgery, Moscow: E. K. Vozny, N. Y. Dobrovolskaya, S. Bolshakova, O. V. Yurgina; N. M. Emmanuel Institute of Biochemical Physics, Moscow: D. B. Korman, I. A. Maslova; N.N. Petrov Research Institute of Oncology, St. Petersburg: V. Semiglazov, V. Ivanov; Saint-Petersburg City Oncological Dispensary, St. Petersburg: G. Manikhas, G. Dolmatov.
South Africa: Mamma Clinic, Tygerberg Hospital, Cape Town: J. Apffelstaedt; Southern Cross Hospital, Cape Town: D. Eedes; Pretoria Academic Hospital, Pretoria: C. Slabber; Pretoria East Hospital, Pretoria: M. A. Coccia-Portugal; Eastern Cape Oncology Centre, Port Elizabeth: K. Maart.
Spain: Hospital Ruber Internacional, Madrid: J. E. Alés Martinez, P. Aramburo, R. Sánchez; Hospital Son Dureta, Palma del Mallorca: J. Rifa, J. Martin; Centro Oncológico Integral de Madrid (CONIM), Madrid: R. Pérez-Carrión, J. L. González Larriba, A. Cubillo; Hospital Universitario San Carlos, Madrid: M. M. Jiménez, A. Casado; Hospital Central de Asturias, Oviedo: J. Fra, J. M. Vieitez, E. Esteban, A. J. Lacave.
Switzerland: Universitätsfrauenklinik, Basel: E. Wight, S. Bartens, R. Decio, U. Güth; Klinik am Park, Zürich: U. Breitenstein.
Turkey: Ankara University Ibni Sina Hospital, Ankara: F. Icli, D. Dincol; Hacettepe University Oncology Institute, Ankara: E. Baltali, Y. Ozisik; Istanbul University Oncology Institute, Istanbul: E. Topuz, M. Basaran, A. Aydiner; Ege University Medical School, Izmir: E. Ozdedeli; 9 Eylul University Medical School, Izmir: O. Harmancioglu, A. U. Yilmaz.
United Kingdom: The Royal Marsden Hospital, London, Royal Marsden NHS Trust, Surrey: I. E. Smith; University of Dundee, Dundee: A. M. Thompson; Christie Hospital NHS Trust, South Manchester University Hospital Trust, Manchester: A. Wardley; Royal Bournemouth Hospital, Bournemouth: T. Hickish; North Middlesex Hospital, London: F. Neave.
Uruguay: Hospital de Clinicas Dr. Manuel Quintela, Montevideo, Uruguay: G. Sabini.
Funding support: The IBCSG Statistical Center is partially supported by US National Cancer Institute grant CA-75362. The BIG 1-98 trial is financed by Novaris and coordinated by IBCSG. Support for the IBCSG: Swedish Cancer Society, The Cancer Council Australia, Australian New Zealand Breast Cancer Trials Group, Frontier Science and Technology Research Foundation, Swiss Group for Clinical Cancer Research (SAKK), Cancer Research Switzerland/Oncosuisse, and the Foundation for Clinical Cancer Research of Eastern Switzerland (OSKK).
References
- 1.World Health Organization, GLOBOCAN. Cancer incidence, mortality, and prevalence worldwide. [6 March 2008];2002 Available at: http://www.dep.iarc.fr.
- 2.Early Breast Cancer Trialists' Cooperative Group. Effects of chemotherapy and hormone therapy on recurrence and 15-year survival: an overview of the randomized trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 3.Smith IE, Dowsett M. Aromatase inhibitors in breast cancer. N Engl J Med. 2003;348:2431–42. doi: 10.1056/NEJMra023246. [DOI] [PubMed] [Google Scholar]
- 4.Mouridsen H, Gershanovich M, Sun Y, et al. Superior efficacy of letrozole versus tamoxifen as first-line therapy for post menopausal women with advanced breast cancer: results of a phase III study of the International Letrozole Breast Cancer Group. J Clin Oncol. 2001;19:2596–2606. doi: 10.1200/JCO.2001.19.10.2596. Erratum, J Clin Oncol 2001; 19: 3302. [DOI] [PubMed] [Google Scholar]
- 5.Mouridsen H, Gershanovich M, Sun Y, et al. Phase III study of letrozole versus tamoxifen as first-line therapy of advanced breast cancer in post-menopausal women: analysis of survival and update of efficacy from the International Letrozole Breast Cancer Group. J Clin Oncol. 2003;21:2101–09. doi: 10.1200/JCO.2003.04.194. [DOI] [PubMed] [Google Scholar]
- 6.Ingle IN, Dowsett M, Cuzick J, et al. Aromatase inhibitors versus tamoxifen as adjuvant therapy for postmenopausal women with estrogen receptor-positive breast cancer: meta-analyses of randomized trials of monotherapy and switching strategies (abstract) Cancer Res. 2009;69(Suppl. 2):12. [Google Scholar]
- 7.Northrop Grumman Corporation: MedDRA. Available at: http://meddramsso.com.
- 8.The Breast International Group (BIG) 1-98 Collaborative Group. A comparison of letrozole and tamoxifen in post-menopausal women with early breast cancer. N Engl J Med. 2005;353:2747–57. doi: 10.1056/NEJMoa052258. [DOI] [PubMed] [Google Scholar]
- 9.Coates AS, Keshaviah A, Thurlimann B, et al. Five years of letrozole compared with tamoxifen as initial adjuvant therapy for post-menopausal women with endocrine-responsive early breast cancer: update of study BIG 1-98. J Clin Oncol. 2005;25:486–92. doi: 10.1200/JCO.2006.08.8617. [DOI] [PubMed] [Google Scholar]
- 10.Anderson JR, Cain KC, Gelber RD. Analysis of survival by tumor response. J Clin Oncol. 1983;1:710–19. doi: 10.1200/JCO.1983.1.11.710. [DOI] [PubMed] [Google Scholar]
- 11.Coombes RC, Hall E, Gibson LJ, et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in post-menopausal women with primary breast cancer. N Engl J Med. 2004;350:1081–1892. doi: 10.1056/NEJMoa040331. Erratum in: N Engl J Med 2004; 351: 2461. N Engl J Med 2006; 355:1746. van de Velde, Cornelius [added] [DOI] [PubMed] [Google Scholar]
- 12.Viale G, Regan MM, Maiorano E, et al. Prognostic and predictive value of centrally reviewed expression of estrogen and progesterone receptors in a randomized trial comparing letrozole and tamoxifen adjuvant therapy for post-menopausal early breast cancer: BIG 1-98. J Clin Oncol. 2007;25:3846–52. doi: 10.1200/JCO.2007.11.9453. [DOI] [PubMed] [Google Scholar]
- 13.Viale G, Giobbie-Hurder A, Regan MM, et al. Prognostic and predictive value of centrally reviewed Ki-67 labeling index in post-menopausal women with endocrine-responsive breast cancer: results from trial BIG 1-98 comparing adjuvant endocrine therapy with tamoxifen versus letrozole. J Clin Oncol. 2008;26(34):5569–75. doi: 10.1200/JCO.2008.17.0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rasmussen BB, Regan MM, Lykkesfeldt AE, et al. Adjuvant letrozole versus tamoxifen according to centrally-assessed ERBB2 status for post-menopausal women with endocrine-responsive early breast cancer: supplementary results from the BIG 1-98 randomised trial. Lancet Oncol. 2008;9:23–28. doi: 10.1016/S1470-2045(07)70386-8. [DOI] [PubMed] [Google Scholar]