

Abstract
Background
Splenic diffuse red pulp small B-cell lymphoma is an uncommon B-cell lymphoma, now recognized as a provisional entity in the 2008 update of the WHO Classification. Additional work is required to review this entity and establish its diagnostic features.
Design and Methods
We have retrospectively analyzed the disease features in a highly selected series of 17 patients diagnosed as splenic diffuse red pulp small B-cell lymphoma.
Results
The median age was 65.5 years (range 40–79 years) and there was a predominance of males (male/female ratio: 2.4). Clinical manifestations were mainly derived from splenomegaly. Splenectomy was the front-line treatment in 11 symptomatic patients; the remaining 6 received chemotherapy initially followed by splenectomy. After a mean follow-up of 72 months, the five-year overall survival was 93%. All cases showed a purely diffuse pattern of splenic infiltration by monomorphous small cells with small round nuclei and pale cytoplasm. All bone marrow biopsies showed tumoral infiltration, with intrasinusoidal infiltration. Peripheral blood cells were small to medium-sized, with clumped chromatin and round nuclear outline and villous cytoplasm. Neoplastic cells had a CD20+, CD23−, bcl6−, Annexin A1- phenotype, with frequent expression of DBA44+ (15/17) and IgG (10/15). FCM data had a B-cell phenotype (CD19+, CD20+, CD22+) with FMC7 (10/11) and CD11c (5/8) expression. Clonal IgH rearrangement studies in 4 cases showed IgVH mutations in all cases, without VH1.2 usage.
Conclusions
Our data suggest that splenic diffuse red pulp small B-cell lymphoma is a distinct entity with morphological and immunophenotypical features that differ from those of other splenic lymphomas.
Keywords: splenic lymphoma, leukemia, villous cells
Introduction
Splenic diffuse red pulp small B-cell lymphoma (SDRPSBCL) is an uncommon B-cell lymphoma with a diffuse pattern of involvement of the splenic red pulp by small monomorphous B lymphocytes now recognized as a provisional entity in the 2008 update of the WHO Classification,1 and initially identified as a potential variant of splenic marginal zone lymphoma (SMZL) with a diffuse pattern of splenic involvement.2 SDRPSBCL cases are characterized by a leukemic course, with relatively low lymphocytosis, massive splenomegaly and infrequent B symptoms. Most cases are diagnosed at clinical stage IV, with spleen, bone marrow (BM) and peripheral blood (PB) involvement.1 Criteria for SDRPSBCL recognition have mainly been based on spleen histology.
The description of the features of this lymphoproliferative disorder requires further investigation to establish whether it is a separate disease with characteristic clinical and diagnostic features. Information about histology and immunophenotype, clinical presentation and response to therapy is very limited and its possible overlap with typical SMZL, hairy cell leukemia-variant (HCL-v) and other B-cell disorders with splenomegaly is uncertain.1,3 Indeed, patients with this diagnosis have frequently been described by alternative terms, such as diffuse variant of splenic marginal zone lymphoma, or splenic red pulp lymphoma with numerous basophilic villous lymphocytes.2–4 The lack of information precludes the establishment of appropriate staging or therapeutic recommendations.
We have collected a series of cases over recent years that could fit this diagnosis. Here we describe the disease features in a highly selected group of 17 cases, with clinical follow-up and a comprehensive histopathology, which may constitute a representative series of cases with this morphology and be useful for a better understanding of the entity. Our data suggest that SDRPSBCL probably constitutes a distinct entity with different morphological and immunophenotypical characteristics.
Design and Methods
Patient selection and clinical data
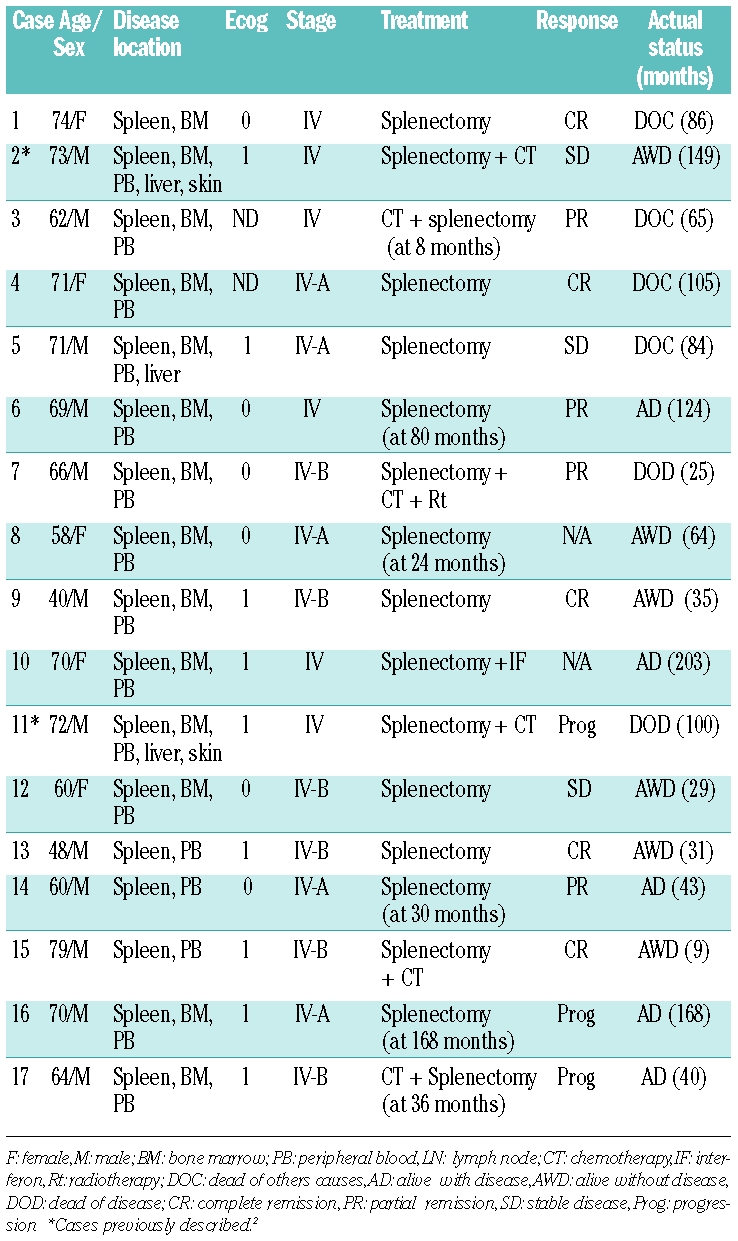
Cases were selected from the routine files (1995–2008) of the Spanish National Cancer Research Centre, Madrid, and Hospital Virgen de la Salud; Toledo, Spain (Table 1). Two of these cases have been previously reported.2
Table 1.
Clinical features of the series.
The selection of cases was based on spleen histology showing features of splenic diffuse red pulp small B-cell lymphoma as described in the 2008 WHO Lymphoma Classification update.1 Cell morphology, immunohistochemical analysis and clinical data were used to exclude entities with similar histological features in the spleen, such as chronic lymphocytic leukemia (CLL), hairy cell leukemia (HCL), lymphoplasmacytic lymphoma (LPL) and prolymphocytic lymphoma (PLL). Research was performed under the supervision of the Institutional Review Board of the Hospital Virgen de la Salud, Toledo, Spain.
The following clinical data were obtained from the referring clinicians: age, clinical manifestations at diagnosis, stage, treatment and outcome. Overall survival (OS) was defined as the time from the date of diagnosis to the date of death or last follow-up. Student’s t-test and χ2 tests of association were used to compare clinical, pathological, and molecular characteristics. Actuarial survival was analyzed by the Kaplan-Meier method, and the curves were compared by the log-rank test. All analyses were carried out with SPSS v15 (SPSS, Chicago, IL, USA).
Morphology and immunohistochemistry
Bone marrow (14 cases) and spleen (17 cases) tissues were processed in the conventional manner for histopathological studies by paraffin embedding and hematoxylin and eosin staining. May-Grumwald-Giemsa-stained peripheral blood smears were used for diagnosis. Subtle changes of the protocols for morphological examination were observed among different institutions.
Immunohistochemical analysis was performed on formalin-fixed, paraffin-embedded tissue. Sections were immunostained using the EnVision method (DAKO, Glostrup, Denmark), with a heat-induced antigen-retrieval step. Sections were immersed in boiling 10 M sodium citrate at pH 6.5 for 2 min in a pressure cooker. The antibody panel used included CD20, CD3, bcl2, bcl6, CD23, IgD, CD38, Ki67, kappa and lambda (Dako), CD10, p53, CD5 (Novocastra, Newcastle, UK), cyclin D1 (Lab Vision, Neo Markers), Annexin A1 (BD Biosciences). Flow cytometry was carried out on the circulating neoplastic lymphocytes from 14 cases on a CD19+ gate with a variety of monoclonal antibodies (McAb) against B cells and with “hairy cell-associated” markers (Table 2). A case was considered positive when more than 30% of the neoplastic cells stained with the McAb.
Table 2.
Peripheral blood and flow cytometry data.

FISH for bcl2, bcl1, del 7q, 17p (TP53)
FISH was performed on paraffin-embedded samples as described by Ferreira et al.5 Briefly, hybridizations were carried out overnight at 37°C after deparaffinization of 4 μm thick sections of tissue, target retrieval by pressure cooking with 1 mM EDTA for 10 min, and pepsin digestion (4 mg/mL at 37°C for 60 min). The probes used were the LSI IGH/BCL2 Dual Fusion Color Probe, LSI BCL6 Dual Color, Break Apart Rearrangement Probe, centromeric probe for chromosome 7 and TP53 (Vysis R, Abbott Park, IL, USA). For 7q deletion we selected seven BAC clones from the BACPAC Resource Center at the Children’s Hospital Oakland Research, CA, USA: clones RP11-10I12, RP11-138A9 covered the 7q32.3 region (genomic bp: 130141293-130377509) and clones RP11-154N21, RP11-597L6, RP11-166D01, RP11-706J21, RP11-140I14 covered the 7q33 region (genomic bp:134704640-135055570).
IgVH somatic mutations assay
The study of IgVH included DNA extracted from paraffin-embedded tissue specimens. Rearranged IgVH genes were amplified and sequenced as previously described.6 Cases were considered mutated when they exhibited less than 98% homology with the closest germ line VH genes. IgVH sequences were submitted to IgBLAST and IMGT analysis to obtain VH family usage and mutation information.
Results
Clinical features
The clinical features are summarized in Tables 1 and 2. The median age was 65.5 years (range 40–79 years); 12/17 (70.5%) were males, giving a male/female ratio of 2.4. Clinical manifestations, such as abdominal pain and/or hypersplenism (cytopenias), were mainly derived from splenomegaly and bone marrow infiltration. B symptoms were recorded in 6/17 (35.3%) cases. In 3 patients the disease was diagnosed as a result of infections or lymphocytosis in routine analysis, or incidentally. All patients were fully active, with a performance status (PS/ECOG) of 0 or 1. One patient had a previous history of rheumatoid arthritis. No other previous autoimmune conditions were recorded in the remaining patients. Serology for HCV or HIV infection was negative in all cases.
All patients had Ann-Arbor stage IV disease, with involvement of the spleen and peripheral blood in all but one case, which had bone marrow involvement. Bone marrow infiltration was present in 14/14 (100%) patients for whom this information was available. Splenic hilar lymph nodes were involved in 3/17 (18%) cases, and liver was involved in 3/17 (18%) cases. CT scans showed abdominal and mediastinal lymphadenopathy in only one case. Two cases showed cutaneous infiltration at diagnosis manifested as erythematous and pruritic papules. The skin biopsy showed dermal lymphoid infiltrates, arranged around the appendages and blood vessels.
Peripheral blood findings were as follows: leukocyte counts ranged from 3.5 to 86×109/L (median 11×109/L), lymphocyte counts from 1 to 28×109/L (median 5.4×109/L), hemoglobin values ranged from 7.8 to 15.5 g/dL (median 12.2 g/dL) and platelet numbers ranged from 13 to 1,572×109/L (median 112×109/L). Eight out of 16 (50%) patients presented with lymphocytosis (> 5×109/L). Anemia (< 10 g/dL) was observed in 2 out of 16 (12.5%) patients and thrombocytopenia (platelets < 100×109/L) in 5 out of 16 (31.2%) patients, 2 of whom (12.5%) with leukocytopenia (leukocytes < 4×109/L). Only one case (N. 13) had a serum monoclonal paraprotein IgM Lambda.
Histological, cytological, immunophenotypical and molecular features
Spleen weight ranged from 518 to 5,500 g (median 2,046 g), with a homogeneously brown-red cut surface, without nodularity. All cases had a purely diffuse pattern of infiltration, replacing both the white and red pulp in the majority; some residual lymphoid follicles were present in 8 cases. Red pulp involvement was seen in both the cords and sinusoids, with some cases showing stuffed cords, filled up by the tumoral cells. Sinusoidal dilatation forming pseudo-blood lakes were seen in 5 cases (Figures 1–3). Neither follicular replacement and biphasic cytology, nor marginal zone infiltration was seen; pseudofollicles (proliferation centers) were not identified.
Figure 1.

(A) Splenic histology, with diffuse infiltration erasing follicles, occupying both cords and sinusoids. (B) Wilder staining highlighting sinusoidal dilatation forming pseudo-blood lakes. (C and D) Neoplastic cells have monomorphous cytology, round nuclei with clumped chromatin and pale cytoplasm.
Figure 3.

Bone marrow interstitial/intrasinusoidal infiltration, barely recognizable in some cases by (A) H & E and (B) CD20 staining.
Neoplastic cells were remarkably homogeneous in the tissue sections, being characterized by a small round nucleus with clumped chromatin and a pale or lightly eosinophilic cytoplasm with plasmocytoid features, but lacking cytoplasmic basophilia. Scattered large cells were present.
All bone marrow biopsies evaluated showed tumoral infiltration, which was always partial, with an interstitial or micronodular pattern (4 cases). When present, the nodules were intertrabecular. All cases showed some degree of sinusoidal infiltration, and in 5 cases the tumor was only demonstrated after CD20 staining within bone marrow sinusoids.
Immunohistochemistry in the spleen sections (Figure 4) showed the lymphoid cells to have a CD20+, CD23−, bcl6−, Annexin A1-phenotype (Table 3). Expression of other markers was as follows: DBA44 + in 15 out of 17 (88.2%), p53 + in 5 out of 17 (29.4%), CD5 + in 1 out of 16 (6.25%), CD38 + in 1 out of 12 (8.3%), IgG + in 10 out of 15 (66.7%), IgD + in 4 out of 15 (26.6%) and kappa in 2 out of 6 (33.3%) cases and lambda 4 out of 6 (66.7%) cases analyzable for light-chain expression. CyclinD1 was negative in all but one case, which had moderate expression that was weaker than observed in the macrophages and endothelial cells. In this case the t(11; 14) by FISH was negative. Bcl2 was weakly positive in all cases, with some rare Bcl2-negative germinal centers highlighted with this marker. Proliferation index was low (ki-67 < 10%) in 16 out of 17 cases; only scattered and dispersed tumoral ki-67-positive cells were observed. Case 7 had a slightly higher proliferation index (ki-67 > 10%). An annular or target pattern, a finding typical of SMZL, was not observed in any case in this series. Residual CD3+ T cells were present in peritrabecular locations.
Figure 4.

Illustrations of the characteristic immunophenotype by IHC: (A) CD20. (B) CD3. (C) DBA44. (D) IgD. (E) BCL2. (F) p53. (G) IgG. (H) IgD. (I) MIB1.
Table 3.
Immunohistochemical and molecular features.

The blood-circulating neoplastic cells were small- to medium-sized lymphoid cells, with clumped chromatin and round nuclear outline, with villous cytoplasm, in 11 out of 13 (84.6%) cases. Occasionally, a few large cells with nucleoli were observed but this was not the predominant population.
FCM data were available from blood of 14 cases, showing a B-cell phenotype (CD19+, CD20+, CD22+) with FMC7 expression in 10 out of 11 (91%) cases. CD11c was positive in 6 out of 9 (67%) cases, CD25 in one out of 10 (10%), CD103 in one out of 6 (16.7%) and CD123 in one out of 2 (50%) cases. None had a chronic lymphocytic leukemia immunophenotype (coexpression of CD5+ and CD23+), but 4 out of 11 (36.4%) cases were CD5-positive. Three out of 6 (50%) cases showed kappa light-chain restriction and the remaining 3 had a lambda light chain (Table 2).
FISH analysis of t(11;14) and t(14;18) gave negative results in all cases and deletion 7q was detected in one out of 13 (7%) cases investigated. LOH by FISH of TP53 was present in 2 out of 13 (15.3%) cases, one of whom showed disease progression and finally died of the disease.
We amplified and sequenced 5 clonal IgH rearrangements from 4 cases. All of them were mutated and case 2 had two amplified clonal rearrangements (no clonality study was performed on infiltrated skin). The VH families used were different in each case: VH1.69, VH6.1, VH3.30, VH3.74 and VH5 (Table 3).
Treatment and follow-up
Splenectomy was the front-line treatment in 11 out of 17 (64.7%) symptomatic patients; the remaining 6 out of 17 (35.2%) received initially chemotherapy followed by splenectomy at eight, 24, 30, 36, 80 and 168 months from diagnosis. Eleven patients out of 17 (64.7%) were alive at the last follow-up; 6 out of 11 (54.5%) of them were without disease activity and the other 5 out of 11 (45.5%) had active disease. Two patients (11.8%) died due to disease progression and 4 (23.5%) died from other causes: 2 with acute myeloblastic leukemia and 2 from infection. Two of the latter cases died when in remission from their lymphoma. Four patients showed clinical progression after splenectomy with, skin (# 2 and # 11), liver (# 2, # 5 and # 11), testicular and systemic involvement (# 7). One of these patients was diagnosed with large B-cell lymphoma transformation in a lymph node biopsy. At a mean follow-up of 72 months (range 9–203), the five-year overall survival was 93% (Figure 5). The comparison of the peripheral blood parameters, hemoglobin, leukocyte count, lymphocyte count, platelets and lactate dehydrogenase, with overall survival revealed that hemoglobin values less than 10 g/dL was the only parameter significantly associated with poor survival (P<0.05).
Figure 5.
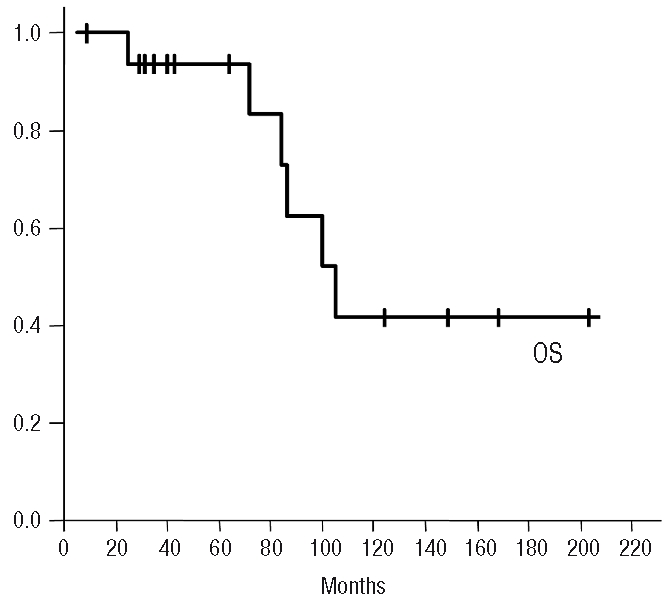
Overall survival probability for the series.
Discussion
Although splenic marginal zone lymphoma (SMZL) is the most frequent splenomegalic small B-cell lymphoma, there is a subset of cases whose clinical presentation mimics SMZL but whose cells have a different morphology and immunophenotype. We have retrospectively analyzed the disease features of a series of patients who, on histological grounds, correspond to the provisional entity recognized in the new WHO classification as splenic diffuse red pulp small B-cell lymphoma (SDRPSBCL), in an attempt to provide a more detailed description of the features of this entity. The frequency of SDRPSBCL is difficult to estimate, but in the series of consultation cases of the Spanish National Cancer Research Centre, it represents 9% of the splenic B-cell lymphomas seen in a 12-year period. Nevertheless, the frequency of the disease in the general population cannot be established here; its determination would require population-based studies. The spectrum of cases described here probably represents those in which splenectomy was considered necessary and in which the pathologists decided to discuss the case due to its unusual infiltration pattern. Most probably, other less aggressive cases which do not require splenectomy also exist; such as the majority of the cases reported by Traverse-Glehen and co-workers.4
This series highlights some potentially useful clinical, morphological and immunophenotypical features of this disease. In most of these cases the clinical features at presentation suggested a diagnosis of SMZL/SLVL, diffuse variant of SMZL or HCL-variant.3,7–11 Nevertheless, it seems that there are some clear differences among these entities. Thus, there is a predominance of males (male/female ratio: 2.4) in SDRPSBCL, in contrast with the even distribution or slight female predominance usually seen in SMZL.12 The degree of lymphocytosis was similar to that of typical SMZL but significantly lower than in HCL-v where median lymphocyte counts are 35×109/L. Additionally, SDRPSBCL cases in this series appear to pursue a relatively favorable clinical course, with 93% of patients alive after 65 months of follow-up, which compares favorably with the 60% survival in SMZL cases.12 Not surprisingly, considering the low aggressivity of this neoplasm and the long follow-up of this series, some of the patients developed second tumors, this being a significant cause of morbidity and mortality. Nevertheless, a caveat must be applied since, as all patients were diagnosed after splenectomy, this series could have been subject to selection bias. A more favorable outcome would be expected if cases at early stages of the disease, when splenectomy is not required, were included.
Figure 2.
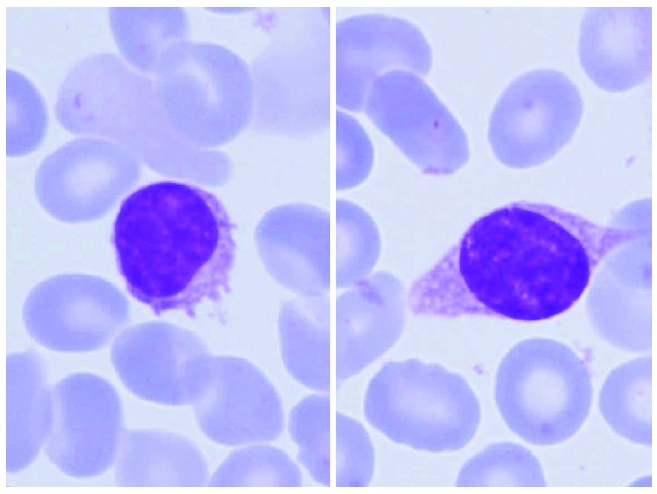
Circulating blood neoplastic lymphoid cells, of small to medium size with condensed chromatin, round or slightly cleaved nuclear outline and villous cytoplasm.
As in other B-cell neoplasms, the diagnosis of SDRPSB-CL was based here on a constellation of features, spleen histology being essential for such a diagnosis in this series. Diagnosis after splenectomy is thus facilitated by the characteristic diffuse infiltration of the splenic red pulp, which contrasts with the usual pattern of infiltration of SMZL, where neoplastic cells in different microenvironments undergo morphological changes (marginal zone and plasmocytoid differentiation being an example), while in SDRPSBCL neoplastic cells are very monomorphous, irrespective of the area of the spleen involved. Thus, diffuse monomorphic splenic histology is the hallmark of this lymphoma and is clearly different from typical SMZL, which is characterized by a distinct micronodular pattern with white pulp replacement and marginal zone differentiation. By contrast, the pattern in SDRPSBCL is very similar to that seen in hairy cell leukemia (HCL). The histology of the spleen in HCL-v has not been described in sufficient detail, but the features of HCL usually overlap with those of SDRPSBCL. However, splenic histology is not always available and a combination of other features, such as bone marrow histology, cell morphology and immunophenotype, can be helpful for recognizing this disease. Intrasinusoidal bone marrow infiltration frequently occurs in this disorder but is also common in other B-cell lymphomas with splenomegaly, particularly SMZL and HCL.13 The immunophenotype of SDRPSBCL is somewhat different from that of typical SMZL as the cells in SDRPSBCL strongly express DBA44 whilst only a fraction of cells are DBA44+ with variable intensity in SMZL. Furthermore, SDRPSBCL cells are frequently IgG-positive and appear to express p53 more frequently than expected in SMZL (29.4% vs. 14% for SMZL). HCL can be distinguished by the frequent severe pancytopenia, bone marrow fibrosis and immunophenotypic profile with expression of Annexin-A1, CD25, CD103 and CD123 markers, which are usually negative in SDRPSBCL. The lack of staining for Annexin A1 is especially important in these cases, since it is a highly specific HCL marker that can be reliably investigated in paraffin-embedded material.14 SDRPSBCL and HCL-v share some features, such as the frequent use of IgG and DBA44 expression, and cutaneous infiltration is present in a minority of cases.11,15 However, unlike SDRPSBCL, HCL-v affects older patients, presents with significantly higher lymphocyte counts and the circulating cells have a distinct morphology with a prominent single nucleolus that mimics prolymphocytes. In addition, the cells frequently express CD103 and the outcome seems less favorable, with a median survival of nine years.1 The frequency of TP53 deletions is lower in SDRPSBCL than in HCL-v.16 It is nevertheless possible that some cases have features shared by both these entities. This is an area that warrants further study.
A diagnosis of lymphoplasmacytic lymphoma (LPL) could be considered because of the pseudoplasmacytic features; indeed, it was frequently proposed by the submitting pathologists. However, a monoclonal component and CD38 expression were only occasionally present, and no single case showed the polymorphous cytological composition in the spleen of small B-lymphoid cells and neoplastic plasma cells seen in LPL. Additionally, immunostaining did not demonstrate the strong cytoplasmic immunoglobulin presence that characterizes lymphoplasmacytic lymphoma.
Other cytogenetic and IgVH mutational data can also be useful for recognizing this disease. For instance, deletion of 7q, a relatively recurrent chromosome abnormality in SMZL,17 was found in only one out of 13 cases (7.6%) of SDRPSBCL, a notably lower frequency than in SMZL, in which it occurs in around 45% of cases. Although initially proposed to be a specific marker for SMZL,17 7q losses have been found in a small proportion of other B-cell lymphoma cases using CGH arrays.5 In 5 cases of this series, we were able to assess the mutational status of the clonal IgH rearrangements. All cases were mutated; none of the cases showed usage of the VH1.2 gene that is characteristic of SMZL.18
The cases described here seem to overlap partially with the series of 37 cases reported by Traverse-Glehen and coworkers, denominated splenic red pulp lymphoma with numerous basophilic villous lymphocytes.4 These patients were older men, with moderate lymphocytosis and splenomegaly without pancytopenia. Neoplastic cells showed an immunophenotype IgM + D, IgM + G, IgM or IgG, as well as CD76 (DBA44) and CD11c, frequently CD103+, and rarely CD123+. Spleen histology was only available in some of these cases and described as “atrophic white pulp with a monomorphic diffuse lymphoma infiltration in a congested red pulp”. Bone marrow infiltration was described as interstitial and intrasinusoidal without extensive fibrosis. Most cases (79%) were IgVH-mutated, with an overrepresentation of VH3 and VH4 gene families. In this series, splenic lymphoma with basophilic villous cells also exhibited a significant difference in time to progression compared with SMZL cases.4 The two studies cannot be compared directly since in our study the cases were included on the basis of spleen morphology and IHC, while those in the Traverse-Glehen study were mainly diagnosed on the basis of peripheral blood morphology and FCM.4 In this series of cases initially studied in different clinical centers, we had some problems applying the cytological criteria used by Traverse-Glehen and co-workers. Review of consecutive case series in the future will probably allow the features of SDRPSBCL to be refined.
In summary, SDRPSBCL is emerging as a distinct disease with some specific features that allow it to be distinguished from SMZL and other splenic lymphomas, especially the diffuse splenic involvement and the IgG and DBA44 frequent expression. More work is needed to establish more precise diagnostic markers that would allow this lymphoma to be identified on the basis of peripheral blood and bone marrow features, without requiring information about spleen histology. Identification of specific molecular markers should also facilitate disease-specific therapeutic strategies.
Acknowledgments
we would like to thank Sonsoles Opazo from Hospital Virgen de la Salud for her technical assistance, and Ma Encarnación Castillo from the CNIO for her excellent work as data manager for this project. We also thank the following pathologists for referring cases and providing clinical and follow-up information: Dr C Alberola, Dr T Medina, Dr MA Limeres, Dr JL Ribera, Dr M Hamblin, Dr I Blanco, Dr C Cinta, Dr C Febles, Dr JF García, Dr I Campos, Dr T Flores, Dr A Dotor, Dr Y Reyes, Dr J Busteros and Dr A Jerez.
Footnotes
Funding: this study is supported by grants from the Instituto de Salud Carlos III (PI052742, PI052800, INT07/028; RETICS, Acción Transversal), MICIN (SAF2008/03871) and the Servicio de Salud de Castilla la Mancha (SESCAM 06047-00, FISCAM GCS-2006-C/06), Spain.
Authorship and Disclosures
GK and MM: designed research, analyzed and interpreted data, performed research, wrote the manuscript. SM-M, S-MR-P, and JCC: performed research, analyzed and interpreted data. PA, and CM: analyzed and interpreted data. EM, and AW: contributed cases, interpreted data, wrote the manuscript; MAP: designed research, analyzed and interpreted data, wrote the manuscript.
All authors reported no conflicts of interest.
References
- 1.Piris MA, Foucar KM, Mollejo M, Campo E, Falini B. Splenic lymphoma/leukemia, unclassifiable. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. WHO classification of Tumours of Haematopoietic and Lymphoid Tissues. 4 ed. Lyon: IARC Press; 2008. pp. 191–3. [Google Scholar]
- 2.Mollejo M, Algara P, Mateo MS, Sanchez-Beato M, Lloret E, Medina MT, et al. Splenic small B-cell lymphoma with predominant red pulp involvement: a diffuse variant of splenic marginal zone lymphoma? Histopathology. 2002;40(1):22–30. doi: 10.1046/j.1365-2559.2002.01314.x. [DOI] [PubMed] [Google Scholar]
- 3.Papadaki T, Stamatopoulos K, Belessi C, Pouliou E, Parasi A, Douka V, et al. Splenic marginal-zone lymphoma: one or more entities? A histologic, immunohistochemical, and molecular study of 42 cases. Am J Surg Pathol. 2007;31(3):438–46. doi: 10.1097/01.pas.0000213419.08009.b0. [DOI] [PubMed] [Google Scholar]
- 4.Traverse-Glehen A, Baseggio L, Bauchu EC, Morel D, Gazzo S, Ffrench M, et al. Splenic red pulp lymphoma with numerous basophilic villous lymphocytes: a distinct clinicopathologic and molecular entity? Blood. 2008;111(4):2253–60. doi: 10.1182/blood-2007-07-098848. [DOI] [PubMed] [Google Scholar]
- 5.Ferreira BI, Garcia JF, Suela J, Mollejo M, Camacho FI, Carro A, et al. Comparative genome profiling across subtypes of low-grade B-cell lymphoma identifies type-specific and common aberrations that target genes with a role in B-cell neoplasia. Haematologica. 2008;93(5):670–9. doi: 10.3324/haematol.12221. [DOI] [PubMed] [Google Scholar]
- 6.Algara P, Mateo MS, Sanchez-Beato M, Mollejo M, Navas IC, Romero L, et al. Analysis of the IgV(H) somatic mutations in splenic marginal zone lymphoma defines a group of unmutated cases with frequent 7q deletion and adverse clinical course. Blood. 2002;99(4):1299–304. doi: 10.1182/blood.v99.4.1299. [DOI] [PubMed] [Google Scholar]
- 7.Franco V, Florena AM, Iannitto E. Splenic marginal zone lymphoma. Blood. 2003;101(7):2464–72. doi: 10.1182/blood-2002-07-2216. [DOI] [PubMed] [Google Scholar]
- 8.Catovsky D, Matutes E. Splenic lymphoma with circulating villous lymphocytes/splenic marginal-zone lymphoma. Semin Hematol. 1999;36(2):148–54. [PubMed] [Google Scholar]
- 9.Pittaluga S, Verhoef G, Criel A, Wlodarska I, Dierlamm J, Mecucci C, et al. “Small” B-cell non-Hodgkin’s lymphomas with splenomegaly at presentation are either mantle cell lymphoma or marginal zone cell lymphoma. A study based on histology, cytology, immunohistochemistry, and cytogenetic analysis. Am J Surg Pathol. 1996;20(2):211–23. doi: 10.1097/00000478-199602000-00010. [DOI] [PubMed] [Google Scholar]
- 10.Melo JV, Robinson DS, Gregory C, Catovsky D. Splenic B cell lymphoma with “villous” lymphocytes in the peripheral blood: a disorder distinct from hairy cell leukemia. Leukemia. 1987;1(4):294–8. [PubMed] [Google Scholar]
- 11.Matutes E, Wotherspoon A, Catovsky D. The variant form of hairy-cell leukaemia. Best Pract Res Clin Haematol. 2003;16(1):41–56. doi: 10.1016/s1521-6926(02)00086-5. [DOI] [PubMed] [Google Scholar]
- 12.Matutes E, Oscier D, Montalban C, Berger F, Callet-Bauchu E, Dogan A, et al. Splenic marginal zone lymphoma proposals for a revision of diagnostic, staging and therapeutic criteria. Leukemia. 2008;22(3):487–95. doi: 10.1038/sj.leu.2405068. [DOI] [PubMed] [Google Scholar]
- 13.Franco V, Florena AM, Campesi G. Intrasinusoidal bone marrow infiltration: a possible hallmark of splenic lymphoma. Histopathology. 1996;29(6):571–5. doi: 10.1046/j.1365-2559.1996.d01-536.x. [DOI] [PubMed] [Google Scholar]
- 14.Falini B, Tiacci E, Liso A, Basso K, Sabattini E, Pacini R, et al. Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1) Lancet. 2004;363(9424):1869–70. doi: 10.1016/S0140-6736(04)16356-3. [DOI] [PubMed] [Google Scholar]
- 15.Gruszka-Westwood AM, Hamoudi RA, Matutes E, Tuset E, Catovsky D. p53 abnormalities in splenic lymphoma with villous lymphocytes. Blood. 2001;97(11):3552–8. doi: 10.1182/blood.v97.11.3552. [DOI] [PubMed] [Google Scholar]
- 16.Vallianatou K, Brito-Babapulle V, Matutes E, Atkinson S, Catovsky D. p53 gene deletion and trisomy 12 in hairy cell leukemia and its variant. Leuk Res. 1999;23(11):1041–5. doi: 10.1016/s0145-2126(99)00127-7. [DOI] [PubMed] [Google Scholar]
- 17.Mateo M, Mollejo M, Villuendas R, Algara P, Sanchez-Beato M, Martinez P, et al. 7q31-32 allelic loss is a frequent finding in splenic marginal zone lymphoma. Am J Pathol. 1999;154(5):1583–9. doi: 10.1016/S0002-9440(10)65411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Algara P, Mateo MS, Sanchez-Beato M, Mollejo M, Navas IC, Romero L, et al. Analysis of the IgV(H) somatic mutations in splenic marginal zone lymphoma defines a group of unmutated cases with frequent 7q deletion and adverse clinical course. Blood. 2002;99(4):1299–304. doi: 10.1182/blood.v99.4.1299. [DOI] [PubMed] [Google Scholar]