

Abstract
The anthracycline Doxorubicin (DXR) is used widely for the treatment of human malignancies, and drug delivery technologies are under investigation to enhance antitumor selectivity and effectiveness. A liquid chromatography–tandem mass spectroscopy (LC–MS/MS) method was developed to identify and quantify DXR and key metabolites in small-volume biological samples. The assay was linear over the therapeutically relevant concentration range (0.125–10,000 nM); in brain tissue, the lower limit of quantification was 0.247 nM and the sensitivity was 1.4 pg. The ability to quantify DXR and detect metabolite formation may provide insight into the toxicity and bioavailability of drug incorporated into carriers such as liposomes.
Keywords: Doxorubicin, Anthracyclines
1. Introduction
Doxorubicin (DXR) is an anthracycline antibiotic that possesses broad spectrum antineoplastic activity, and is one of the most important anticancer agents in use [1–5]. However, clinical utility is hampered by cumulative, dose-limiting cardiotoxicity, myelosuppression, and the development of drug resistance.
DXR is composed of an aglycone backbone linked to a daunosamine sugar through an O-glycosidic bond at carbon 7 (Fig. 1). The drug undergoes extensive metabolism by the liver and is excreted primarily in the bile. A small fraction (<10%) is eliminated in the urine [6]. In a linked-rat model [7], it was estimated that 22% of the injected dose may undergo entrohepatic recycling.
Fig. 1.

Structure of Doxorubicin (DXR), metabolites, and proposed routes of metabolism.
Enzyme-mediated reduction and deglycosidation of DXR results in the formation of several structurally similar hydroxylated and aglycone metabolites (Fig. 1). The primary metabolite, Doxorubicinol (DXR-ol), is produced by cytosolic carbonyl reductase through NADPH-dependent aldoketo reduction of a carbonyl moiety at the C-13 position [8]. Deglycosidation at the daunosamine sugar at C-7 produces doxorubicinone (DXR-one) and doxorubicinolone (DXR-olone) [8]. The resulting hydroxyl group may be metabolized to 7-deoxydoxorubicinone (7-DXR-one) or 7-deoxydoxorubicinolone (7-DXR-olone). Sulfonated and glucuronidated metabolites have been identified as urinary metabolites, but this biotransformation may be limited to humans [8–10].
The biological activity and toxicity of the DXR metabolites is not fully elucidated. DXR-ol clearly is clinically important; although it is less active than DXR [11], DXR-ol and similar metabolites can promote the formation of reactive free radicals, and may contribute to dose-limiting cardiotoxicity [5].
Emerging drug delivery approaches can alter DXR biodistribution radically; long circulating, sterically stabilized liposomes containing DXR can increase tumor deposition of drug markedly in animal models, and have been approved recently as clinical products in the US (Doxil®, Alza Inc., Mountain View, CA, USA) and abroad (Caelyx®, Schering-Plough Pharmaceutical NV/SA, Brussels, Belgium) [12–16]. For such formulations, neither the tissue bioavailability of the encapsulated drug nor the interplay between altered tissue deposition and the pattern of drug metabolism are well understood. Altered drug distribution, coupled with tissue-dependent differences in biotransformation, could play an important role in therapeutic effects and toxicity. Analytical methodology enabling the rapid quantification of DXR and its metabolites, particularly in small animal tumor models, would assist in the optimization of carrier-based strategies for anthracycline delivery.
A variety of procedures to extract, resolve, and quantify DXR and its metabolites have been published (summarized in Table 1 [17–28]). Many of these techniques are laborious and necessitate long analytical run times to achieve sufficient peak resolution, owing to the spectral and structural similarities of DXR and its metabolites [29]. The instability of DXR and metabolites in plasma or tissue homogenates also limits the utility of otherwise promising extraction strategies [23,29–31].
Table 1.
Select examples of extraction procedures and analytical techniques for quantitative analysis of DXR in plasma or tissue samples
| Species/samples | Extraction method | Recovery (%) | Detection | Linear range | LOQ or LOD (nM) | Ref. |
|---|---|---|---|---|---|---|
| Murine plasma and tissues | Homogenized in PBS, liquid/liquid extraction (chloroform/methanol, 4:1 (v/v)) | 66–98 | HPLC–Fluor | 367–2200 nM | 367 | [17,18] |
| Murine plasma and lung | Homogenized in methanol and Tris buffer (pH 8.5), deproteinized with CAN | 60–98 | HPLC–Fluor | 15–1550 nM | 15 | [19] |
| Murine plasma and tissues | Homogenized and extracted in chloroform-1-propanol | 64 | HPLC–Fluor | 2.2–2155 nM | 1.8–2.4 | [20] |
| Rat, serum and tissues | Homogenized in phosphate buffer, deproteinized and extracted in methanol and ZnSO4 | 94–115 | HPLC–Fluor | 5–5000 ng/mL | 9–18 | [21] |
| Plasma and tissues | Homogenized in sodium phosphate dibasic (pH 7.0) and extracted by SPE | 80–87 | HPLC–Fluor | 25–1000 ng/mL | 9–18 | [22] |
| Rat serum and bile | Deproteinized and extracted with methanol | >89 | HPLC–Fluor | 10–2500 ng/mL | 18 | [23] |
| Human serum | Solid phase extraction (SPE) | 97–105 | HPLC–ES/MS | 5–4000 nM | 4.6 | [24] |
| Dog and rat plasma | Solid phase extraction (SPE) | 70–49 | LC–APCI MS/MS | 0.5–798 ng/mL | 0.9–11 | [25] |
HPLC–Fluor: HPLC with fluorescence detection; HPLC–ES/MS: HPLC with electrospray ionization and single-quadrupole MS analysis; LC–APCI MS/MS: HPLC with atmospheric pressure chemical ionization (heated nebulizer) and a tandem triple quadrupole MS analysis; LOQ: limit of quantification; LOD: limit of detection.
A simplified, rapid extraction procedure and a liquid chromatography–tandem mass spectroscopy (LC–MS/MS) method were developed to identify and quantify DXR and metabolites in plasma and tissue extracts. Tandem mass spectroscopy permits simultaneous determination of ion pairs unique to DXR or to specific metabolites, permitting analysis and quantification in complex biological samples.
2. Experimental
2.1. Reagents and chemicals
Doxorubicin (>99% purity) was provided by Vinchem (Chatham, NJ, USA) and Pharmacia Italia S.p.A. (Milan, Italy). Authentic samples of Doxorubicinol, doxorubicinone, doxorubicinolone, and 7-deoxoydoxorubicinone were a gift from Pharmacia Italia S.p.A. Daunorubicin (DNM, Fig. 2) and cholesterol (Chol) were purchased from Sigma (St. Louis, MO, USA); Chol was recrystallized three times from methanol prior to use. The phospholipids distearoylphosphatidylcholine (DSPC) and distearoylphosphatidylethanolamine conjugated with 2 kDa polyethylene glycol (PEG–DSPE) were purchased from Avanti Polar Lipids, Inc., (Alabaster, AL, USA). Water, acetonitrile, and methanol were from Burdick & Jackson (Muskegon, MI, USA). Glacial acetic acid and chloroform were from Fisher Scientific (Fairlawn, NJ, USA), and ammonium acetate was from J.T. Baker (Phillipsburgh, NJ, USA). All chemicals and solvents were of analytical or HPLC grade unless otherwise stated. Male Fisher 344 rats (160–200 g) were obtained from Harlan Sprague–Dawley (Indianapolis, IN, USA). The rat 9L gliosarcoma cell line, designated as 9L-72, was obtained from Dr. D. Deen, Brain Tumor Research Center, University of California at San Francisco, USA, and maintained in Dulbeccos Modified Eagles Medium (Invitrogen, Faraday, CA, USA) supplemented with 10% fetal bovine serum.
Fig. 2.

Structure of Daunorubicin (DNM), the internal standard
2.2. LC–MS/MS instrumentation
Mass analysis was performed on two similarly equipped Applied Biosystems/MDS Sciex API 3000 triple quadrupole tandem mass spectrometers (ABI, Inc., Foster City, CA, USA) following chromatographic separation using an HPLC system consisting of an autosampler and dual pumps. Two HPLC systems were used: a model 200 Perkin-Elmer (Wellesley, MA, USA) or a model 1100 Agilent (Palo Alto, CA, USA). Solvents were vacuum filtered through a 0.45 micron nylon membrane (Gelman Sciences, Ann Arbor, MI, USA), and degassed prior to use. Separation was achieved by automated injection of 10 μL samples onto a reversed phase C18 guard and analytical column (Agilent Zorbax Extend Narrow-Bore Guard Column, 2.1 mm × 12.5 mm 5 μm packing and an Agilent Zorbax Extend Rapid Resolution 4.6 mm × 50 mm 3.5 μm) under isocratic conditions at a flow rate of 250 μL/min. An additional in-line pre-filter (MacMod Analytical, Inc., Chadds Ford, PA, USA) was inserted before the guard column. The column effluent was introduced into the mass spectrometer using a turbo ion-spray ionization (electrospray) source positioned orthogonally to the orifice. High purity nitrogen was used as the curtain and collision gas. Data analysis was performed using Analyst Software (ABI, Inc., version 1.3). The instruments were calibrated biweekly with a mixture of polypropylene glycols (ABI, Inc.) according to the manufacturer’s instructions.
2.3. MS optimization
Doxorubicin was dissolved at a concentration of 0.5 μg/mL in a mobile phase consisting of acetonitrile, water, and ammonium acetate, filtered through a 0.2 μm nylon filter (Gelman Sciences, Ann Arbor, MI, USA). Optimization of the solvent system is described below. Samples were introduced into the mass spectrometer via the ionization source at a flow rate of 6.0–10.0 μL/min using a syringe pump (kdScientific Inc., New Hope, PA, USA). Parent compounds were identified from total ion chromatograms (TIC) acquired in positive ion mode over the range of m/z 100–1,000. Product ion chromatograms (XIC) were acquired for each parent (precursor) ion (i.e., DXR, its metabolites, and DNM) over the range of m/z 50–550. A unique ion pair (precursor and product ion) was identified for each compound. Mass spectrometer parameters for individual ion-pairs were optimized initially with the Analyst software, and optimized further in the manual tune mode (Table 2).
Table 2.
Retention time, ion pairs, and optimized mass spectroscopy parameters
| Compound | Retention time (min) | Selected ion pairs (m/z) | Declustering potential | Focusing potential | Collision energy | Collision exit potential |
|---|---|---|---|---|---|---|
| DXR | 1.94 | 544/361 | 41 | 200 | 35 | 12 |
| DXR-ol | 1.93 | 546/363 | 41 | 210 | 35 | 14 |
| DXR-one | 1.93 | 415/361 | 26 | 150 | 27 | 14 |
| DXR-olone | 1.97 | 417/363 | 16 | 120 | 23 | 14 |
| 7-DXR-one | ND | 399/381.5 | 56 | 240 | 29 | 14 |
| DNR | 2.4 | 528/321 | 41 | 200 | 35 | 12 |
ND, not determined. Pure standard solutions of 0.5 μg/mL DXR or individual metabolites were infused separately into the mass spectrometer to identify unique ion pairs and permit the optimization of MS parameters for each compound. The relative retention time for each of the ion-pairs was obtained by LC–MS/MS after automated injection of 10 μL of pure sample and isocratic separation using a reversed phase C18 analytical and guard column at 250 μL/min.
2.4. Preparation of standards and quality control solutions
Standard and quality control (QC) stock solutions of DXR and DNM (200 μM in methanol) were prepared from individually weighed samples and stored at −20 °C until use. Calibration samples were prepared fresh by serial dilution of drug standard solutions in mobile phase, plasma, or tissue homogenates. The final drug concentration range was 0.06–10,000 nM. Quality control samples were prepared independently in triplicate at concentrations appropriate for the tissue of interest. Stock solutions of DXR and DXR-ol were stable (±10% theoretical) at −20 °C and through three freeze–thaw cycles (Table 3).
Table 3.
Stability of Doxorubicin
| Storage at −20 °C (months) | Nominal concentration (nM) | Measured (nM) (CV%) | Change (%) | |
|---|---|---|---|---|
| DXR | 1 | 500 | 490 (3.6) | −1.9 |
| 15 | 500 | 529 (0.7) | +5.8 | |
| DXR-ol | 1 | 500 | 482 (4.7) | −3.6 |
| 15 | 500 | 312 (1.6) | −37.5 | |
| Peak area (CV%) | ||||
| DXR | Standard | 500 | 43967 (5.2) | N/A |
| Freeze–thaw | 500 | 41353 (4.1) | −5.9 | |
| DXR-ol | Standard | 500 | 42781 (6.7) | N/A |
| Freeze–thaw | 500 | 39272 (5.4) | −8.2 | |
N/A: not applicable. Stock solutions containing DXR were prepared in methanol, diluted to 200 μM and stored at −20 °C. Replicate samples were analyzed after storage and after three freeze–thaw cycles and compared to freshly prepared standards. The concentrations of DXR calculated from peak area were compared and the coefficient of variation was determined; a percent change greater than ±10% was considered an indication of significant change.
2.5. Extraction procedures and efficiencies
2.5.1. Plasma extraction
The solvent for DXR extraction from plasma consisted of 40% 5 mM ammonium acetate, pH 3.5 and 60% acetonitrile (ACN) (v/v). Four hundred microliters of extraction solvent was added to 100 μL of plasma to achieve a final concentration of 20% (plasma/solvent (v/v)). Standards and QC samples were prepared by serial dilution of DXR and DXR-ol in extraction solvent prior to the addition of blank plasma. Twenty microliters of DNM, the internal standard, was transferred into each sample immediately after the addition of the extraction solvent to plasma, thus achieving a final concentration of 200 nM DNM. Samples were mixed, cooled in an ice water bath for 10 min, and centrifuged for 10 min at 15,000 × g at 4 °C. The deproteinized supernatant was recovered and analyzed immediately.
2.5.2. Liver, spleen, lung and heart extraction
Frozen tissue samples were pulverized under liquid nitrogen using a mortar and pestle immersed in liquid nitrogen. The tissue powder was transferred into tared culture tubes and weighed. A sufficient volume of mobile phase was added to each tube to achieve the desired tissue concentration (Table 4). Twenty microliters of the DNM standard was added per 1.0 mL aliquot of tissue homogenate, producing a final concentration of 200 nM DNM. Samples were mixed briefly (<1 min) using a homogenizer (Tekmar, Cincinnati, OH, USA), cooled in an ice water bath, and centrifuged for 10 min at 15,000 × g at 4 °C. The deproteinized supernatant was recovered and analyzed immediately.
Table 4.
Extraction efficiency, linearity and sensitivity of assay in rat tissues
| Sample | Tissue concentration | Linear range (nM) | In vivo sample range (nM) | Extraction efficiency (%) | LOQ (nM) | Sensitivity (pg) | |
|---|---|---|---|---|---|---|---|
| Plasma | DXR | 20% (v/v) | 0.343–10,000 | 0–5000 | 87.2 | 0.343 | 1.87 |
| DXR-ol | 20% (v/v) | 0.945–10,000 | 0–2000 | ND | 1.89 | 10.3 | |
| Brain/tumor | DXR | 5% (w/v)# | 0.125–1000 | 0.6–126 | 91.4 | 0.25 | 1.36 |
| DXR-ol | 5% (w/v)# | 0.500–1000 | 0.4–92 | ND | 0.5 | 2.73 | |
| Liver | DXR | 0.5% (w/v) | 0.686–1000 | 43–240 | 88.9 | 2.06 | 11.2 |
| DXR-ol | 0.5% (w/v) | 2.06–1000 | 4–18 | ND | 2.1 | 11.2 | |
| Spleen | DXR | 1% (w/v) | 2.06–1000 | 91–846 | 91.8 | 2.06 | 11.2 |
| DXR-ol | 1% (w/v) | 2.06–1000 | 18–246 | ND | 2.1 | 11.2 | |
| Heart | DXR | 2.5% (w/v) | 1.37–1000 | 60–680 | 112 | 2.06 | 11.2 |
| DXR-ol | 2.5% (w/v) | 2.06–1000 | 11–55 | ND | 2.1 | 11.2 | |
| Lung | DXR | 2.5% (w/v) | 2.06–1000 | 267–931 | 84.2 | 2.5 | 13.6 |
| DXR-ol | 2.5% (w/v) | 2.06–1000 | 4–42 | ND | 2.1 | 11.2 |
Standard solutions of DXR or DXR-ol were prepared in blank plasma or tissue homogenates at known concentrations ((v/v) or (w/v)), extracted, and analyzed by LC–MS/MS. The extraction efficiency, linearity, and sensitivity of the method were determined. The in vivo sample range was determined after analysis of plasma and tissue samples from rats (n ≥ 12) treated intravenously with 5.67 mg/kg SSL–DXR. LOQ: limit of quantification; ND: not determined;
concentration of brain tumor homogenate ranged from 3–7% (w/v).
2.5.3. Tumor and brain extraction
Tumor and normal brain tissue samples were collected into tared containers, weighed, frozen in liquid nitrogen, and stored at −80 °C. For analysis, frozen brain tissue samples were removed from the freezer, and a 50–100 mg sample was quickly cut and transferred into a fresh, tared tube. A sufficient volume of mobile phase was added to achieve a final concentration of 5% (w/v) tissue in solvent. For the analysis of tumor samples, which weighed approximately 30–70 mg, the entire sample was used; 1 mL of mobile phase was added, producing a final concentration of 3–7% (w/v). All samples were homogenized briefly (<1 min), cooled in an ice water bath for 10 min, and centrifuged for 10 min at approximately 15,000 × g at 4 °C. The deproteinized supernatant was recovered and analyzed immediately.
2.5.4. Extraction efficiency and matrix effects
Extraction efficiencies for plasma and each tissue were determined by comparing the peak area of DXR standards extracted from tissue samples (n ≥ 3) with DXR standards prepared in mobile phase. A small difference in volume (<5%) was observed for tissue-extracted versus solvent-extracted samples but was not corrected.
The effect of different sample matrices on DXR ionization was determined by extracting (in triplicate) three or more different blank tissue samples (plasma and individual tissues). The extracts were spiked with a known concentration of DXR, and the peak areas of these samples were compared to DXR standards using ANOVA. No statistical differences in peak area were observed for plasma or for each of the tissues examined (Table 5).
Table 5.
Effect of matrix on Doxorubicin analysis
| Tissue | Nominal concentration (nM) | Peak area (CV%) |
P-value | ||
|---|---|---|---|---|---|
| Sample 1 | Sample 2 | Sample 3 | |||
| Plasma | 30 | 1985 (3.0) | 1870 (11.0) | 2090 (6) | 0.239 |
| Brain | 65 | 7153 (7.8) | 6853 (2.0) | 6620 (2.0) | 0.276 |
| Liver | 250 | 39767 (5.9) | 33567 (7.8) | 36600 (16) | 0.236 |
| Spleen | 55 | 3740 (6.1) | 3810 (4.1) | 2290 (12.6) | 0.237 |
| Heart | 65 | 5813 (2.3) | 6120 (0.3) | 5797 (4.3) | 0.147 |
| Lung | 65 | 8087 (2.8) | 8023 (2.4) | 7767 (3.7) | 0.302 |
Plasma and tissue samples from three different animals were spiked with DXR and extracted in triplicate. The peak area for DXR was compared and the coefficient of variation was determined; P ≤ 0.05 was considered an indication of significant change.
2.6. Assay performance and validation
Standard and quality control samples were prepared by extracting tissues obtained from untreated animals. Serial dilutions of DXR and DXR-ol were added to blank tissue homogenates and processed as described above.
Standard curves were constructed by calculating the ratio of the analyte peak area to that of the internal standard, and plotting the ratio versus the theoretical concentration; data was fit using weighted least squares; the inverse of the variance (1/χ2) of the data was used as the weighting factor. The standard curve was considered acceptable if: (i) the calculated accuracies of >90% of the standards were within 15% of their theoretical value and (ii) no systematic deviations over the linear range were observed. The lower limit of quantification (LOQ) was determined experimentally to be the lowest concentration that was: (i) within the linear range of the standards; (ii) had an accuracy within 20% of the theoretical concentration; (iii) had a coefficient of variation (CV) of ≤ 20%; and (iv) had a signal to noise ratio ≥ 5. The absolute sensitivity for each tissue was determined experimentally by calculating the amount of analyte that was applied to the column at the LOQ, given the sample injection volume (10 μL).
Intra-day variation and accuracy were calculated by comparing the theoretical and experimentally determined concentrations of triplicate QC samples prepared at two or more concentrations that encompassed the linear range of the standard curve. Inter-day variation and accuracy was determined from QC samples analyzed on three or more different days.
2.7. Preparation of Doxorubicin liposomes
Small, unilamellar, sterically stabilized Doxorubicin liposomes (SSL–DXR) were prepared from DSPC:Chol:PEG–DSPE in a 9:5:1 mole ratio utilizing a remote loading procedure [15,16,32] that was driven by combined ammonium sulfate and pH gradients, as previously described [33]. Un-encapsulated drug was removed by dialysis. Doxorubicin concentration was measured at 490 nm using a spectrophotometer (Cary 300, Varian Inc., Palo Alto, CA, USA) assuming εa = 12, 500M × A/cm [13]. The phospholipid concentration was determined by assaying inorganic phosphate following acid hydrolysis [34]. The final concentration of Doxorubicin was typically 2 mg/mL, and the drug:lipid ratio was 0.23:1.0 (mol:mol). Liposomes had a mean particle diameter of 80–110 nm, as determined using a Nicomp model 380 dynamic light scattering particle size analyzer (Santa Barbara, CA, USA). Doxorubicin liposome preparations were purged with nitrogen and stored in the dark at 4 °C until use.
2.8. Brain tumor implantation, treatment, and sampling
Rat 9L gliosarcoma brain tumor cells (4 × 104 cells) were implanted stereotaxically in the caudate/putamen brain region of Fisher 344 male rats as described previously [33]. All animals were housed, cared for, and used following a protocol approved in advance by the Institutional Animal Care and Use Committee of the University at Buffalo, in accordance with the US Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals, updated 1996. Animals were provided a standard rat chow diet (Harlan Teklad Rodent Diet 2016, Indianapolis, IN, USA) and water ad libum.
SSL–DXR was administered at 5.67 mg/kg by tail vein injection 7 days after tumor implantation. Animals were sacrificed at 8, 24, and 48 h post administration. At the time of sacrifice, 500 μl of blood was collected from the inferior vena cava into heparinized syringes. Samples were iced immediately. The remaining blood was flushed from the vascular system by severing the inferior vena cava and infusing 100 ml of heparinized saline (5 U/mL) through the left ventricle. The tumor, contralateral (normal) brain hemisphere, liver, lung, heart, and spleen were excised rapidly, snap-frozen in liquid nitrogen, and stored at −80 °C until analysis. Plasma was obtained by centrifugation of the iced blood samples for 10 min at approximately 1,200 × g at 4 °C and was then stored at −80 °C.
3. Results
3.1. Mass spectroscopy
Two ionization sources were evaluated for the quantification of DXR and its metabolites: turbo ion-spray (electrospray; ESI) and heated atmospheric pressure chemical ionization nebulizer (APCI). The heated nebulizer is considered a “hard-ionization” source and is commonly employed with compounds that are thermally stable and difficult to ionize [35]. DXR is a weak base (pKa: 8.3) and is ionized (H+) under mildly acidic conditions. Preliminary studies established that electrospray ionization provided greater sensitivity for the detection of DXR and resulted in fewer product ions compared to APCI (data not shown). Therefore, the electrospray ionization source was used for all subsequent studies.
Representative TIC and XIC of DXR (m/z: 544) are shown in Fig. 3. Unique precursor/product ion-pairs were identified from TIC and XIC for DXR, DXR-ol, DXR-one, DXR-olone, 7-DXR-one, and DNR (Table 2). Ion-pair selection for 7-DXR-olone was based on fragmentation patterns reported in the literature [24] for DXR and metabolites.
Fig. 3.

(A) Total ion chromatogram (TIC) for 0.5 mg/mL Doxorubicin in 40% (v/v) acetonitrile and 60% 5 mM ammonium acetate, pH 3.5. The LC–MS/MS was operated in positive ion mode using the turbo-spray ionization source. (A): A prominent peak for DXR was observed at m/z 544 [M + H]+ and selected for fragmentation. (B) Product ion chromatogram (XIC); fragmentation of the m/z 544 DXR ion produced prominent fragments of m/z 361 and 321. The m/z 544/361 ion-pair was selected and fragmentation conditions were further optimized for production of this product ion.
Two similarly-equipped API 3000 mass spectrometers were used in these studies, and differences in sensitivity and instrumental parameters were observed. Optimal values for instrument I are reported (Table 2). For the analysis of 7-DXR-olone, the instrumental parameters for 7-DXR-one were used, based on the structural similarities of these two compounds. The instrumentation parameters which were independent of ion-pair and common to both spectrometers are as follows; ion-spray voltage (ISV), 5000 V; temperature, 300 °C; nebulizer gas, 8 (~0.8 L/min); curtain gas, 8 (~0.5 L/min); and collision-assisted dissociation gas, 4.
Multiple reactions monitoring (MRM), in which precursor ions are fragmented and unique product ions are measured, enabled selective detection of all compounds simultaneously, in spite of the fact that some ion-pairs were not well resolved chromatographically. Representative TIC and MRM spectra for DXR, DXR-ol, and DNR (the internal standard) extracted from rat plasma are shown in Fig. 4C and D. Interfering peaks were not observed when DNM alone was spiked into blank plasma (Fig. 4A and B). Similar results were obtained for each of the tissues examined (data not shown).
Fig. 4.

(Panels A and C) The total ion chromatogram (TIC) and (Panels B and D) product ion chromatogram (XIC) for individual ion-pairs observed in MRM mode. (Panel A) The internal standard DNM was spiked into blank plasma. In the first period (0–1.5 min), the ISV was set to 0 and no signal was detected. The ISV was restored in the second period (1.5–5.0 min) and a single peak was observed; (Panel B) MRM mode of period 2 (Panel A) identified the single peak as DNM. Low (<100 cps) background and the absence of interfering peaks was also observed; (Panel C) TIC of DXR, DXR-ol, and DNM spiked into blank plasma. Three peaks were observed in the second period; (Panel D) MRM mode of period 2 (Panel C) identified the peaks as (i) DXR-ol (1.83 min), (ii) DXR (1.96 min), and (iii) DNM (2.37 min).
3.2. Liquid chromatography—mobile phase selection
The composition of the mobile phase was optimized empirically to maximize sensitivity (ionization) and minimize analytical time. Formic acid (5% (v/v)) decreased overall sensitivity and peak area for DXR and DXR-ol (data not shown). Ammonium acetate (5 and 10 mM) adjusted to pH 3.5 with glacial acetic acid consistently yielded greater peak areas (data not shown). The decreased sensitivity of detection with formic acid may be related to the instability of DXR at the more acidic conditions produced (pH < 1), compared to the less acidic conditions produced by ammonium acetate (pH: 3.5).
The optimum mobile phase consisted of 40% acetonitrile (ACN) and 60% 5 mM ammonium acetate, pH 3.5 (v/v). Under isocratic conditions, the retention time for analytes of interest ranged from 1.7 to 2.5 min. Total analysis time was 5 min. Differences in tubing length between the Perkin-Elmer and Agilent LC systems produced small shifts in analyte retention times, but the retention times on a given instrument were reproducible.
Sufficient retardation of analyte peaks from the void front permitted the incorporation of two analytical periods into the method, which improved sensitivity. A large background signal was observed at the void front in extracts of plasma and tissues (data not shown), but was well separated from DXR and metabolites. Therefore, from 0 to 1.5 min following sample injection, the ion-spray voltage was set to 0 V. From 1.5 to 5.0 min, the ISV was increased to 5000 V (Fig. 4). Reduction of the ISV during elution of the void front reduced the amount of ionized species entering the MS and preserved sensitivity of the assay when larger batches of samples were processed.
3.3. Extraction procedure
A simple and rapid extraction procedure was developed which provides a consistent matrix composition between samples and minimizes the potential for concentration-dependent variation in protein binding. Total extraction time was approximately 20 min for batches of 15–20 samples. For tissues, pulverization under liquid nitrogen reduced the opportunity for degradation of analytes. The efficiency of DXR extraction was determined by comparison of DXR standards extracted from plasma and tissue homogenates to DXR standards prepared in mobile phase. For the tissues of interest, the efficiency of DXR extraction was 84–112% over the concentration range of 50–75 nM DXR, which is relevant to in vivo samples (Table 4).
Tissue samples homogenized and extracted in mobile phase and held at a final concentration of 40% ACN were free from precipitates for >24 h at room temperature or 4 °C. However, for plasma samples, the concentration of ACN was an important variable in the overall success of extraction and subsequent analysis. Preparation of plasma samples to a final concentration of 20% (v/v) plasma in mobile phase resulted in an ACN concentration of approximately 32% (v/v); under such conditions, precipitates formed within a few hours of extraction. Variability was also observed in peak height, shape, retention time, and detection sensitivity. Increasing the ACN concentration in the extraction solvent eliminated this delayed precipitation in plasma samples. However, peak splitting and shifts in retention time were observed for final ACN concentrations >50%. The optimum extraction solvent consisted of 60% ACN and 40% 5mM ammonium acetate (v/v). With this solvent composition, the preparation of a 20% (v/v) solution of plasma in extraction solvent produced a final ACN concentration of 48% (v/v). Samples thus prepared were free from precipitates for >24 h at 4–20 °C.
Electrospray ionization is often sensitive to differences that may arise in the sample matrix from different individuals or when different tissues are assayed. No significant differences in DXR peak area was observed after extracting replicates of different tissues obtained from at least three different animals (Table 5).
Reports differ as to the pattern of metabolites produced in liver, and recent studies suggest that tissue damage and non-optimal extraction procedures may promote the formation of aglycone metabolites [30]. Liver samples were stored at −80 °C and fractions were reprocessed three times over a 6 month period. No differences were observed in the tissue content of DXR or in the relative abundance of the detected metabolites, suggesting adequate stability and absence of degradation over this storage period (data not shown).
3.4. Method performance and validation
Standard curves over a broad concentration range (0.06–10,000 nM) were prepared in plasma or various tissues of interest. A linear relationship between peak area and drug concentration was observed; for all tissues, correlation coefficients (r2) typically were >0.990, and accuracies were within 15% of their theoretical values (Tables 6–8). Thus, the assay was linear over the concentration range relevant to in vivo therapeutic use.
Table 6.
Accuracy and precision in rat plasma and brain/tumor tissues
| Tissue | Sample | Quality controls (nM) | Intra-day assay performance |
Inter-day assay performance |
||
|---|---|---|---|---|---|---|
| Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) | |||
| Plasma | DXR | 2500 | 97.2 | 7.1 | 98.7 | 14.1 |
| 250 | 96.4 | 4.6 | 94.6 | 12.6 | ||
| 25 | 99.7 | 10.3 | 98.9 | 7.8 | ||
| DXR-ol | 2500 | 94.5 | 4.4 | 94.9 | 4.9 | |
| 250 | 92.0 | 7.6 | 92.8 | 6.6 | ||
| 25 | 100 | 12.4 | 95.5 | 8.7 | ||
| Brain/tumor | DXR | 250 | 97.3 | 1.5 | 86.6 | 1.1 |
| 25 | 96.3 | 6.2 | 89.1 | 1.6 | ||
| 2.5 | 102 | 17.6 | 100 | 3.8 | ||
| DXR-ol | 250 | 80.1 | 11.7 | 83.1 | 8.2 | |
| 25 | 86.0 | 6.2 | 88.8 | 8.6 | ||
| 2.5 | 105 | 1.9 | 113 | 10.2 | ||
Quality control solutions of DXR and DXR-ol were prepared in blank plasma or brain homogenates at known concentrations ((v/v) or (w/v)), extracted, and analyzed by LC–MS/MS. The intra- (n = 3) and inter-day (n ≥ 3) assay performance was determined.
Table 8.
Accuracy and precision of Doxorubicinol analysis in rat tissues
| Tissue | DXR-ol (nM) | Intra-day assay performance |
|
|---|---|---|---|
| Accuracy (%) | Precision (CV%) | ||
| Liver | 150 | 98.4 | 4.1 |
| 15 | 106 | 3.8 | |
| Spleen | 250 | 111 | 5.0 |
| 25 | 119 | 2.8 | |
| Heart | 150 | 97.6 | 2.8 |
| 15 | 99.8 | 12.4 | |
| Lung | 250 | 82 | 3.4 |
| 25 | 115 | 13.9 | |
Quality control (QC) solutions of DXR-ol were prepared in tissue homogenates at known concentrations (w/v), extracted, and analyzed by LC–MS/MS. The intra-day (n = 3) assay performance was determined.
The limit of quantification, sensitivity, and assay performance were examined for DXR and DXR-ol in plasma (Tables 4 and 6). A lower limit of quantification of 0.343 and 1.89 nM was achieved for DXR and DXR-ol, respectively. This represents an absolute sensitivity of 1.87 pg for DXR and 10.28 pg for DXR-ol. The intra- and inter-day accuracies for DXR and DXR-ol were determined to be within 13 and 20% of their theoretical concentrations, respectively (Table 6). The intra- and inter-day coefficient of variation (CV%) was <20% for both compounds.
The limit of quantification, absolute sensitivity, and intra-day assay performance were also determined for DXR in each tissue of interest. The lower limit of quantification ranged from 43 nM for liver to 0.25 nM for brain/tumor extracts (Table 4), and the absolute sensitivity ranged from 201 to 1.36 pg for those tissues, respectively. Intra- and inter-day accuracies for DXR (Table 7) and intra-day accuracies for DXR-ol (Table 8) were within 82 to 119% of theoretical, with a CV% <20%.
Table 7.
Accuracy and precision of Doxorubicin in rat tissues
| Tissue | DXR (nM) | Intra-day assay performance |
Inter-day assay performance |
||
|---|---|---|---|---|---|
| Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) | ||
| Liver | 150 | 98.4 | 4.9 | 89.1 | 12.1 |
| 15 | 102.2 | 4.0 | 94.0 | 13.3 | |
| Spleen | 250 | 111 | 5.5 | 114 | 4.2 |
| 25 | 110 | 10.0 | 113 | 8.0 | |
| 2.5 | 102 | 4.1 | 103 | 7.3 | |
| Heart | 150 | 98.4 | 5.9 | 104 | 5.9 |
| 15 | 97.1 | 8.7 | 108 | 12.4 | |
| Lung | 250 | 94.8 | 9.6 | 97.4 | 6.7 |
| 25 | 93.5 | 9.3 | 97.2 | 11.9 | |
| 2.5 | 98.4 | 4.6 | 93.7 | 13.9 | |
Quality control (QC) solutions of DXR were prepared in tissue homogenates at known concentrations (w/v), extracted, and analyzed by LC–MS/MS. The intra- and inter-day (n ≥ 3) assay performance was determined.
3.5. DXR quantification and metabolite identification
The utility of the assay to quantify DXR and identify metabolites in small tissue samples was investigated in a rodent therapeutic model of glioblastoma multiforme, a lethal human brain tumor. Small (4 × 104) numbers of 9L rat brain tumor cells were implanted stereotaxically in rat brains and allowed to develop into established tumors. Plasma, brain, tumor, liver, spleen, lung, and heart were harvested at intervals after treatment of animals with SSL–DXR. The time-course of DXR deposition in various tissues is shown in Fig. 5. Plasma concentrations in the range of 60–80 μg/mL were observed over the time period examined, consistent with previous reports on the pharmacokinetics of SSL–DXR [31]. Drug deposition in tissues of the reticuloendothelial system (lung, liver, and spleen), which are the primary determinants of liposome clearance, ranged from 6 to 9 μg DXR/g tissue. The assay was also sufficiently sensitive to quantify DXR in brain tumor samples of 30–50 mg, and in normal brain.
Fig. 5.
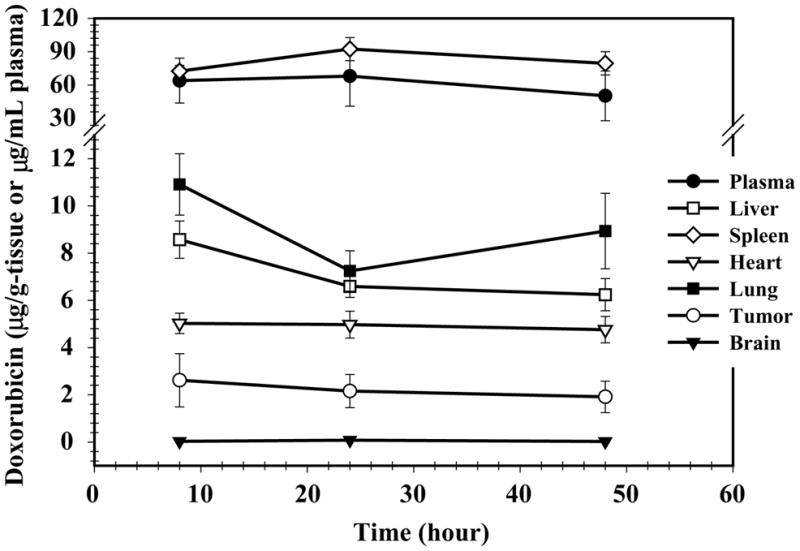
Plasma and tissue concentrations of DXR in rats treated intravenously with 5.67 mg/kg SSL–DXR by tail vein injection. Animals were sacrificed at 8, 24, and 48 h after drug administration and their tissues harvested and analyzed by LC–MS/MS. The symbols represent the mean concentration (n = 4 animals) of DXR in plasma (●), brain tumor (○), brain (▾), liver (□), spleen (⋄), heart (▽), and lung (▪) and the error bars represent the standard error of the mean.
The quantification of DXR metabolites was not performed for all tissues and all metabolites. Differences in ionization efficiency of the various metabolites varied with the turbo ion-spray source, thus preventing simultaneous quantification at high sensitivity in a single run. However, simultaneous analysis was feasible for DXR and DXR-ol, the two pharmacologically-important analytes, because of similarities in molecular structure, ionization efficiency and LC–MS/MS assay performance for the tissues of interest.
In contrast, under the conditions necessary for high-sensitivity analysis of DXR and DXR-ol, the efficiency of ionization for the aglycones (DXR-one, DXR-olone, 7-DXR-one) was poor; aglycone concentrations of 10 μM produced intensity values similar to those produced by 2–10 nM DXR and DXR-ol. Thus, the assay described here permitted the detection of aglycones under conditions in which they are major metabolites, but quantification simultaneous with DXR and DXR-ol was not feasible.
Liver, spleen, heart, lung, brain, and tumor were examined for the presence of the DXR metabolites Dox-ol, Dox-one, Dox-olone, 7-Dox-one, and 7-Dox-olone after SSL–DXR administration. For individual metabolites, peak heights greater than three times the background were used as the threshold criterion to judge presence/absence in each tissue. DXR-ol, DXR-one, and DXR-olone were identified to a varying degree in some or all of the tissues examined, as shown in Table 9.
Table 9.
Metabolite identification after treatment of rats with SSL–DXR
| Tissue | DXR-ol | DXR-one | DXR-olone |
|---|---|---|---|
| Plasma | + | + | − |
| Tumor | + | + | − |
| Brain | − | − | + |
| Liver | + | + | − |
| Spleen | + | + | + |
| Lung | + | + | − |
| Heart | + | + | − |
Rats were treated intravenously with 5.67 mg/kg SSL–DXR, and plasma and tissues samples were harvested. The presence of DXR metabolites was determined by LC–MS/MS. Metabolites were considered present (+) if their peak height was greater than three times their background signal.
No evidence of 7-DXR-one or 7-DXR-olone was observed.
No evidence of 7-DXR-one or 7-DXR-olone was found in any of the tissues investigated (Table 9). Thus, although the sensitivity of the assay for these aglycone metabolites was relatively poor, it is possible to conclude they are not major products of DXR biotransformation; we estimate from the data that they were present at concentrations <0.03–12.5 ng/g tissue. These results appear to agree with recent findings that suggest tissue damage and extraction procedures may elevate the levels of the aglycone metabolites ex vivo [30], but further investigation will be required.
4. Discussion
New drug delivery methods may alter radically the pharmacokinetics and tissue deposition of the encapsulated drug. For DXR and other anthracyclines incorporated in long-circulating liposomes, effects that differ from the unencapsulated drug have been observed [12–15,33,36]. Carrier-mediated alterations in drug biodistribution, bioavailability, and tissue-specific metabolism may underlie these overall changes in pharmacology. Thus, the optimization of therapy and the anticipation of new toxicities for carrier-based dosage forms necessitates quantitative evaluation of the parent compound and metabolites in plasma and small tissue samples over a concentration range relevant to therapeutic effects. Furthermore, the detection of metabolites may provide insight into the bioavailability of carrier-associated drugs.
Mass spectroscopy has been used widely for structural analysis and has become a dominant technique to identify and quantify pharmacologically active compounds in complex biological samples [35]. LC–MS was applied recently to quantify four anthracyclines, including DXR and DXR-ol, in human serum [24]. In that study, mass spectroscopy improved sensitivity, but selectivity was limited; although single quadrupole instruments monitor single ions (m/z ratios) at high sensitivity, complete peak resolution frequently is necessary to avoid interference from metabolites or contaminants of similar m/z. Thus, extended run times may be required to achieve chromatographic separation.
Here, we describe a LC–MS/MS method for the quantification of DXR, DXR-ol and the identification of its metabolites in rat plasma and tissue samples. By the use of multiple reactions monitoring (MRM), a key capability of tandem mass spectrometers, the selection and quantification of compound-specific ion-pairs enabled a reduction in the interference by co-eluting substances and a considerable improvement in assay selectivity. As complete chromatographic resolution of analytes was unnecessary, analysis time was reduced. The method was validated and employed to quantify the deposition of drug in plasma, liver, spleen, heart, lung, and brain, and brain tumors of rats administered DXR encapsulated in liposomes. Interference peaks resulting from sample matrix effects were not observed. Such interferences limit the utility of otherwise sensitive but poorly selective methods such as fluorescence, for which long analytical run times may be necessitated in order to analyze biological samples.
Marked differences in the plasma pharmacokinetics and tissue biodistribution were observed after intravenous administration of SSL–DXR compared to free drug, as observed previously [13,14]. Free DXR was eliminated rapidly in a polyexponential manner, while drug encapsulated in SSLs exhibited prolonged systemic circulation times and a monoexponential decline in plasma concentrations. A large difference in DXR plasma concentrations were observed after administration of free- versus SSL–DXR; at 4 h post administration the DXR plasma concentration was 3,300-fold higher for SSL–DXR than for free drug (165 μg/mL versus 0.05 μg/mL, respectively) and at 48 h the plasma concentration was approximately 5,900-fold higher for SSL–DXR (58.8 μg/mL versus 0.01 μg/mL, respectively). Administration of SSL–DXR increased the peak DXR deposition in brain tumors 6.5-fold compared to free drug (i.e., 1.04 μg/g at 24 h for SSL–DXR versus 0.16 μg/g-tumor at 0.5 h for free drug). The overall effect is increased efficacy and reduced toxicity of SSL–DXR [16]. The ability to quantify changes in drug biodistribution and detect the formation of metabolites in the nanomolar concentration range is necessary for a better understanding of the carrier system and development of optimal dosing regimes.
The LC–MS/MS assay described here permitted simultaneous quantitative analysis of DXR-ol, a primary metabolite, in plasma and tissues. DXR-ol was not resolved chromatographically from DXR, but was nonetheless quantified without interference. Detection of changes in the pattern of active metabolite formation mediated by novel delivery vehicles may be important for predicting drug toxicity or determining bioavailability.
A simple and rapid extraction method was developed to expedite sample processing and to limit the exposure of the analyte to conditions known to be associated with degradation. Pulverization of frozen tissues in liquid nitrogen was a simple and reproducible method to avoid extended homogenization steps that may promote degradation. No evidence of the 7-deoxy aglycone metabolites were found, suggesting that the extraction and analysis procedures may limit ex vivo degradation.
To our knowledge, this assay represents the first application of electrospray ionization tandem mass spectroscopy for the quantification of Doxorubicin and Doxorubicinol in rat tissues. It provides a significant increase in assay sensitivity, and a considerable reduction in analysis time. Given the structural similarities among the clinically-used anthracyclines, this assay and extraction procedure may be easily adapted for use with other such agents.
Acknowledgments
We thank Antonio Suarato, Pharmacia Italia S.p.A., Nerviano (Milano), Italy, for the donation of the DXR metabolites. We thank R. DiFrancesco, D. Ruszaj and Drs. V. Frerichs and W. Conway for helpful discussions. We thank the Pharmaceutical Sciences Instrumentation Facility at the University at Buffalo for the use of the liquid chromatograph/tandem mass spectrometers. The LC–MS/MS was obtained by a Shared Instrumentation Grant (S10RR14573) from the National Center for Research Resources, National Institutes of Health (USA). RDA was supported in part by an American Foundation for Pharmaceutical Education (AFPE) Pre-Doctoral Fellowship and a University at Buffalo—Merck Graduate Fellowship. JES was supported by a Pfizer Summer Undergraduate Research Fellowship and an AFPE Gateway Research Scholarship.
Abbreviations
- ACN
acetonitrile
- CHOL
cholesterol
- DNM
Daunorubicin
- DSPC
distearoylphosphatidylcholine
- DXR
Doxorubicin
- DXR-ol
Doxorubicinol
- DXR-one
doxorubicinone
- DXR-olone
doxorubicinolone
- 7-DXR-one
7-deoxydoxorubicinone
- 7-DXR-olone
7-deoxydoxorubicinolone
- LC
liquid chromatography
- MS/MS
tandem mass spectroscopy
- PEG–DSPE
methoxy-polyethylene glycol (mPEG-2000) conjugated to distearoylphosphatidylethanolamine
- SSL–DXR
Doxorubicin encapsulated in long circulating sterically stabilized liposomes
References
- 1.DeVita VT, Rosenberg SA, Hellman S. Cancer, Principles and Practice of Oncology. Lippincott, Williams & Wilkins; Philadelphia: 2001. [Google Scholar]
- 2.Di Marco A, Gaetani M, Scarpinato B. Cancer Chemother Rep. 1969;53:33. [PubMed] [Google Scholar]
- 3.Hortobagyi GN. Drugs. 1997;54(Suppl 4):1. doi: 10.2165/00003495-199700544-00003. [DOI] [PubMed] [Google Scholar]
- 4.Lown JW. Pharmacol Ther. 1993;60:185. doi: 10.1016/0163-7258(93)90006-y. [DOI] [PubMed] [Google Scholar]
- 5.Licata S, Saponiero A, Mordente A, Minotti G. Chem Res Toxicol. 2000;13:414. doi: 10.1021/tx000013q. [DOI] [PubMed] [Google Scholar]
- 6.Robert J. Bull Cancer. 1988;75:167. [PubMed] [Google Scholar]
- 7.Behnia K, Boroujerdi M. Cancer Chemother Pharmacol. 1998;41:370. doi: 10.1007/s002800050753. [DOI] [PubMed] [Google Scholar]
- 8.Takanashi S, Bachur NR. Drug Metab Dispos. 1976;4:79. [PubMed] [Google Scholar]
- 9.Innocenti F, Iyer L, Ramirez J, Green MD, Ratain MJ. Drug Metab Dispos. 2001;29:686. [PubMed] [Google Scholar]
- 10.Le Bot MA, Begue JM, Kernaleguen D, Robert J, Ratanasavanh D, Airiau J, Riche C, Guillouzo A. Biochem Pharmacol. 1988;37:3877. doi: 10.1016/0006-2952(88)90069-x. [DOI] [PubMed] [Google Scholar]
- 11.Schott B, Robert J. Biochem Pharmacol. 1989;38:4069. doi: 10.1016/0006-2952(89)90688-6. [DOI] [PubMed] [Google Scholar]
- 12.Siegal T, Horowitz A, Gabizon A. J Neurosurg. 1995;83:1029. doi: 10.3171/jns.1995.83.6.1029. [DOI] [PubMed] [Google Scholar]
- 13.Gabizon A, Shiota R, Papahadjopoulos D. J Natl Cancer Inst. 1989;81:1484. doi: 10.1093/jnci/81.19.1484. [DOI] [PubMed] [Google Scholar]
- 14.Papahadjopoulos D, Allen TM, Gabizon A, Mayhew E, Matthay K, Huang SK, Lee KD, Woodle MC, Lasic DD, Redemann C, et al. Proc Natl Acad Sci USA. 1991;88:11460. doi: 10.1073/pnas.88.24.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lasic DD, Martin FJ, Gabizon A, Huang SK, Papahadjopoulos D. Biochim Biophys Acta. 1991;1070:187. doi: 10.1016/0005-2736(91)90162-2. [DOI] [PubMed] [Google Scholar]
- 16.Gabizon AA. Cancer Invest. 2001;19:424. doi: 10.1081/cnv-100103136. [DOI] [PubMed] [Google Scholar]
- 17.Shinozawa S, Mimaki Y, Araki Y, Oda T. J Chromatogr. 1980;196:463. doi: 10.1016/s0021-9673(00)84748-2. [DOI] [PubMed] [Google Scholar]
- 18.Shinozawa S, Mimaki Y, Tomano H, Araki Y, Oda T. J Chromatogr. 1980;190:489. doi: 10.1016/s0021-9673(00)84748-2. [DOI] [PubMed] [Google Scholar]
- 19.Rose LM, Tillery KF, el Dareer SM, Hill DL. J Chromatogr. 1988;425:419. doi: 10.1016/0378-4347(88)80049-5. [DOI] [PubMed] [Google Scholar]
- 20.van Asperen J, van Tellingen O, Beijnen JH. J Chromatogr B. 1998;712:129. doi: 10.1016/s0378-4347(98)00165-0. [DOI] [PubMed] [Google Scholar]
- 21.Alvarez-Cedron L, Sayalero ML, Lanao JM. J Chromatogr B. 1999;721:271. doi: 10.1016/s0378-4347(98)00475-7. [DOI] [PubMed] [Google Scholar]
- 22.Cox SK, Wilke AV, Frazier D. J Chromatogr. 1991;564:322. doi: 10.1016/0378-4347(91)80098-w. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Q, Chowbay B. J Pharm Biomed Anal. 2002;30:1063. doi: 10.1016/s0731-7085(02)00442-9. [DOI] [PubMed] [Google Scholar]
- 24.Lachatre F, Marquet P, Ragot S, Gaulier JM, Cardot P, Dupuy JL. J Chromatogr B. 2000;738:281. doi: 10.1016/s0378-4347(99)00529-0. [DOI] [PubMed] [Google Scholar]
- 25.Mazuel C, Grove J, Gerin G, Keenan KP. J Pharm Biomed Anal. 2003;33:1093. doi: 10.1016/s0731-7085(03)00434-5. [DOI] [PubMed] [Google Scholar]
- 26.Andersen A, Warren DJ, Slordal L. Ther Drug Monit. 1993;15:455. doi: 10.1097/00007691-199310000-00016. [DOI] [PubMed] [Google Scholar]
- 27.Zagotto G, Gatto B, Moro S, Sissi C, Palumbo M. J Chromatogr B. 2001;764:161. doi: 10.1016/s0378-4347(01)00346-2. [DOI] [PubMed] [Google Scholar]
- 28.de Bruijn P, Verweij J, Loos WJ, Kolker HJ, Planting AS, Nooter K, Stoter G, Sparreboom A. Anal Biochem. 1999;266:216. doi: 10.1006/abio.1998.2943. [DOI] [PubMed] [Google Scholar]
- 29.Chan KK, Harris PA. Res Commun Chem Pathol Pharmacol. 1973;6:447. [PubMed] [Google Scholar]
- 30.Vrignaud P, Londos-Gagliardi D, Robert J. Eur J Drug Metab Pharmacokinet. 1986;11:101. doi: 10.1007/BF03189834. [DOI] [PubMed] [Google Scholar]
- 31.Barenholz Y, Amselem S, Goren D, Cohen R, Gelvan D, Samuni A, Golden EB, Gabizon A. Med Res Rev. 1993;13:449. doi: 10.1002/med.2610130404. [DOI] [PubMed] [Google Scholar]
- 32.Madden TD, Harrigan PR, Tai LC, Bally MB, Mayer LD, Redelmeier TE, Loughrey HC, Tilcock CP, Reinish LW, Cullis PR. Chem Phys Lipids. 1990;53:37. doi: 10.1016/0009-3084(90)90131-a. [DOI] [PubMed] [Google Scholar]
- 33.Sharma US, Sharma A, Chau RI, Straubinger RM. Pharm Res. 1997;14:992. doi: 10.1023/a:1012136925030. [DOI] [PubMed] [Google Scholar]
- 34.Bartlett GR. J Biol Chem. 1959;234:466. [PubMed] [Google Scholar]
- 35.Rossi DT, Sinz MW. Mass Spectrometry in Drug Discovery. Marcel Dekker, Inc; New York: 2001. [Google Scholar]
- 36.Muggia FM. Curr Oncol Rep. 2001;3:156. doi: 10.1007/s11912-001-0016-5. [DOI] [PubMed] [Google Scholar]