

Abstract
Common fragile sites (CFSs) are specific chromosome regions that exhibit an increased frequency of breaks when cells are exposed to a DNA-replication inhibitor such as aphidicolin. PARK2 and DMD, the causative genes for autosomal-recessive juvenile Parkinsonism and Duchenne and Becker muscular dystrophy, respectively, are two very large genes that are located within aphidicolin-induced CFSs. Gross rearrangements within these two genes are frequently observed as the causative mutations for these diseases, and similar alterations within the large fragile sites that surround these genes are frequently observed in cancer cells. To elucidate the molecular mechanisms underlying this fragility, we performed a custom-designed high-density comparative genomic hybridization analysis to determine the junction sequences of approximately 500 breakpoints in germ cell lines and cancer cell lines involving PARK2 or DMD. The sequence signatures where these breakpoints occur share some similar features both in germ cell lines and in cancer cell lines. Detailed analyses of these structures revealed that microhomologies are predominantly involved in rearrangement processes. Furthermore, breakpoint-clustering regions coincide with the latest-replicating region and with large nuclear-lamina-associated domains and are flanked by the highest-flexibility peaks and R/G band boundaries, suggesting that factors affecting replication timing collectively contribute to the vulnerability for rearrangement in both germ cell and somatic cell lines.
Introduction
Common fragile sites (CFSs) are specific chromosome regions that exhibit an increased frequency of gaps or breaks when cells are exposed to a DNA replication inhibitor such as aphidicolin. CFSs are well known to be predisposed to breakages and rearrangements, particularly in cancer cells. Recently, it was reported that aphidicolin-mediated replication stress could induce large submicroscopic deletions at CFSs in a human-mouse cell-hybrid system.1 Many of the aphidicolin-induced CFSs have been found to span extremely large genes, including PARK2 (MIM 602544), DMD (MIM 300377), FHIT (MIM 601153), WWOX (MIM 605131), GRID2 (MIM 602368), LARGE (MIM 603590), CTNNA3 (MIM 607667), NBEA (MIM 604889), and CNTNAP2 (MIM 604569).2 Intriguingly, PARK2 and DMD are both genes responsible for human hereditary diseases, and gross deletions are frequently observed as the causative germline mutations.
PARK2 (chromosome 6: 161,688,580–163,068,824, NCBI build 36.1), encompassing 1.4 Mb, which is embedded in a CFS (FRA6E), is the gene responsible for autosomal-recessive juvenile Parkinsonism (AR-JP [MIM 600116]).3 Among various causative germline mutations in PARK2, gross deletions account for 50 to 60% of causative germline mutations,4 with the deletion hotspots clustering in exons 3 and 4.5 As the consequence of the localization of PARK2 in FRA6E, PARK2 is also frequently targeted by deletions in various cancer cells.6 DMD (chromosome X: 31,047,266–33,139,594), which is also embedded in a CFS (FRAXC),7 encompasses 2.1 Mb and is the gene responsible for Duchenne and Becker muscular dystrophy (DMD and BMD [MIM 310200 and 300376]).8 Similarly to PARK2, DMD is also frequently targeted by gross deletions in patients with DMD or BMD (hereafter DMD/BMD) and in those with various cancers.7,9 Approximately 60% of causative germline mutations are gross deletions, and deletion hotspots are in exons 45 to 52.10 Although it has not drawn much attention, the frequent occurrence of gross rearrangements in the genomic regions corresponding to CFSs in patients with AR-JP or DMD/BMD suggests that a common basis underlies the frequent occurrence of rearrangements in both germ cell and somatic cell lines. CFSs are chromosomal regions that are particularly sensitive to certain forms of replication stress, and there are lines of evidence suggesting that CFSs represent unreplicated DNA resulting from stalled replication forks.11,12 These sites replicate late during the S phase, even under normal culture conditions.13,14 The context of the nucleotide sequences and/or chromosomal structures at these CFSs leading to delay replication, however, has not been well understood. Furthermore, molecular mechanisms responsible for clustering of the breakpoints at these CFSs and those underlying the repair processes of the breakpoints remain to be elucidated.
To explore why these particular genomic regions are prone to rearrangements in germ cells and cancer cells, it is essential to determine the precise positions of the breakpoint-clustering regions and to analyze the junction-sequence signatures in detail. Determination of junction sequences, however, has been extremely laborious by conventional methods, such as the PCR-based genome-walking method, particularly in the case of large-size rearrangements. To date, only a few breakpoints involving PARK2 and DMD have been determined at the nucleotide level in either germ cell or somatic cell mutations.5,15–19 To accomplish an efficient determination of rearrangement breakpoints at the nucleotide level, we have applied a custom-designed high-density array comparative genomic hybridization (array CGH) system, which enabled us to determine approximately 500 breakpoints in patients with AR-JP and DMD/BMD as well as in cancer cell lines. We herein elucidated the clustering of the breakpoints and the sequence signatures at the breakpoint junctions in germ cell and somatic cell mutations in these CFSs. This gives insights into the mechanisms of chromosomal fragility within the CFSs.
Material and Methods
Materials
For the determination of rearrangement breakpoints in the germline mutations of PARK2 or DMD, we enrolled 206 unrelated patients with AR-JP and 208 unrelated male patients with DMD/BMD. The patients with AR-JP were from multiple ethnicities, including 113 Japanese, 15 East Asians, 64 Europeans,20,21 and 14 others, with one or two rearranged PARK2 alleles that have been identified by PCR-based gene-dosage analysis or multiplex ligation-dependent probe amplification (MLPA) analysis. All of the patients with DMD/BMD were males from the Japanese population, with hemizygous deletions or duplications in DMD that have been identified by multiplex PCR analysis or MLPA analysis. For the determination of rearrangement breakpoints in the somatic cell mutations of PARK2 and DMD, we analyzed 125 cancer cell lines obtained from the American Type Culture Collection (ATCC) and the laboratories of D.I.S. or H.A., including 41 gastrointestinal tract cancer cell lines, 26 breast cancer cell lines, 24 urogenital tract cancer cell lines, 14 respiratory tract cancer cell lines, 9 skin cancer cell lines, 7 brain cancer cell lines, and 4 hematological malignancy cell lines (the cancer cell line list is available in Table S1, available online). This study was approved by the institutional review boards of all of the participating institutions.
Array CGH Analysis
High-density microarrays that contain 35,668 probes covering the entire PARK2 gene (chromosome 6: 161,500,000–163,500,000), with an average probe interval of 112 bp, or 40,632 probes that cover the entire DMD gene (chromosome X: 31,000,000–33,500,000), with an average probe interval of 82 bp, were designed on the Agilent platform. The probes were designed by a laboratory-made program (programmed by S.T.), CGH probe version 4.1 (available on request), and were 60-mer oligonucleotides with GC contents ranging from 31% to 39%. We also avoided repetitive sequences.22 For those regions where the probes could not be designed with GC contents between 31% and 39% at appropriate probe intervals, the probes were designed with shorter lengths (45 to 60 oligonucleotides) depending on the GC content, so that their optimal hybridization temperature was close to longer oligonucleotide probes utilized. A single control sample was used for all of the subjects in CGH analysis of PARK2, and a male control sample was used for CGH analysis of DMD. Genomic DNAs were hybridized to the microarrays, followed by scan and analysis using Agilent CGH Analytics software version 4.0.76 (Agilent Technologies, CA, USA). For determining each breakpoint at the nucleotide level, a pair of oligonucleotide primers was designed to amplify each segment across the breakpoint junction. Amplified junction fragments were subjected to direct nucleotide-sequence analysis utilizing an ABI 3100 Genetic Analyzer (Life Technologies, CA, USA). The data on rearrangements of this study are accessible in the NCBI Database of Genomic Structural Variation (dbVAR); the public accession number is nstd36.
Nucleotide-Sequence Analysis
The positions of nucleotide sequences described in this study were based on the human reference sequence of NCBI build 36 version 1. The nucleotide sequences encompassing the breakpoints were subjected to many different computational analyses. The FASTN program of GENETYX version 9.0.6 software (Genetyx, Tokyo, Japan) was used to calculate the amount of sequence homology between the nucleotide sequences encompassing two breakpoints. To investigate the sequence characteristics of the junctions of rearrangements, we searched for extended homologies between the pairs of nucleotide sequences encompassing the breakpoints (100 bp upstream and 100 bp downstream). The RepeatMasker program was used to evaluate interspersed repeat-element content. Origins of inserted sequences at the junctions were determined by the BLAST program and SSEARCH program against the entire human genome. DNA Pattern Find was used for detecting sequence motifs that were abundant at deletion breakpoints.23 High-flexibility regions were identified with the TwistFlex program, which assesses DNA flexibility by measuring the local potential variation in the DNA structure at a twist angle of DNA, and the flexibility parameter is expressed as the fluctuation of this angle.24 All of these programs were used with default settings. The positions of the chromosomal R/G band25 and nuclear-lamina-associated domains (LADs)26 were retrieved from the UCSC Genome Browser (NCBI build 36.1). The replication-timing map of chromosome 6 was retrieved from a previous report, as determined by array CGH analyses of S phase DNA to G1 phase DNA.27 The sex-averaged recombination rate was obtained from the deCODE recombination map.28
Statistical Methods
All statistical analyses were performed by means of StatsDirect statistical software version 2.6.5 (StatsDirect, UK). Means, medians, variances, skewness, and kurtosis were determined for the distributions of breakpoints at PARK2 and DMD loci, in patients and in cancer cell lines. Differences between the mean breakpoint positions in patients and in cancer cell lines were analyzed by means of the Mann-Whitney U test. Differences between the standard deviations of breakpoint positions in patients and in cancer cell lines were analyzed by means of the squared-ranks test. The null hypothesis was rejected at p < 0.05.
Results
Determination of Breakpoints at the Nucleotide Level on the Basis of Custom-Designed Array CGH Analyses
To characterize the breakpoints in PARK2 and DMD located at CFS, we have applied a locus-specific high-density array CGH analysis system to 206 patients with AR-JP, 208 male patients with DMD/BMD, and 125 cancer cell lines. Representative cases of AR-JP with a deletion in PARK2 (Figures 1A–1D) and a case of AR-JP with a duplication in PARK2 (Figures 1E–1G) are shown. Array CGH analyses easily enabled detection of a deletion or a duplication, as shown in Figure 1A or 1E, respectively. For determination of the nucleotide sequences at the deletion breakpoints, a pair of PCR primers flanking the deletion was designed to obtain junction fragments by PCR (Figure 1C). When the PCR products containing the junction segment were obtained (Figure 1B), the nucleotide sequences were easily determined by direct nucleotide-sequence analysis of the PCR products (Figure 1D). For determination of the nucleotide sequences of duplication breakpoints, three pairs of PCR primers were designed, based on the head-to-tail, head-to-head, and tail-to-tail models (Figure 1F). When the PCR products were obtained for either of these configurations (Figure 1G), the nucleotide sequences were determined as described above.
Figure 1.
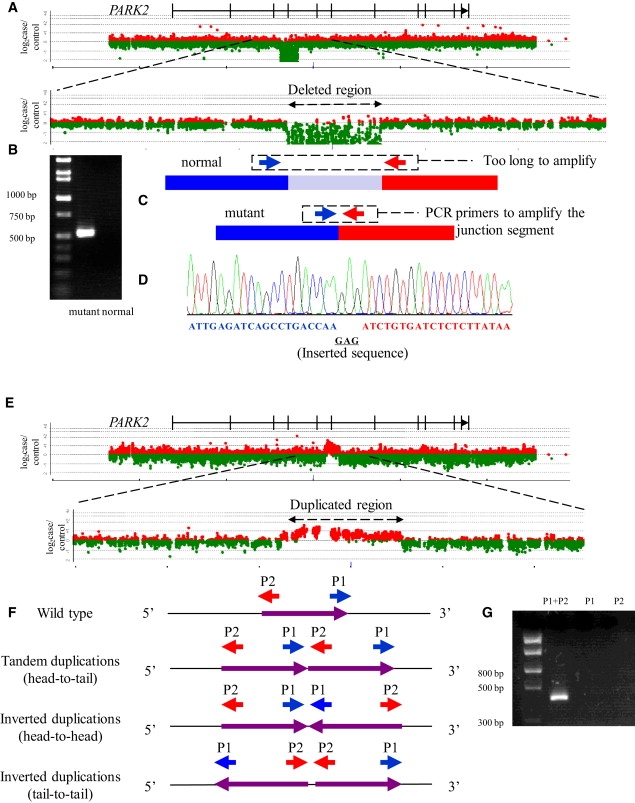
Determination of Breakpoint-Junction Sequences in PARK2 by Custom-Designed High-Density Array CGH Analysis
(A) Scan data of array CGH analysis of a patient with AR-JP with 82 kb homozygous deletions (exon 4 of PARK2). The horizontal axis represents the nucleotide position. The vertical axis represents log2 (ratio of case to reference signal intensities on array CGH). Dots of log2 (ratio of case to reference signal intensities) larger than 0 are shown in red, and those smaller than 0 are shown in green. The physical map of PARK2 is also shown above the scan data.
(B) Agarose gel electrophoresis of PCR products derived from the patient's genomic DNA obtained by employing primer pairs flanking the deletion. Amplifications did not occur in normal alleles because the segment between primers was too large (82 kb), while the band corresponding to the PCR products of 520 bp derived from the deletion allele was clearly visualized.
(C) Design of primer pairs for specific amplification of the deletion allele by PCR. A pair of oligonucleotide primers (denoted by red and blue arrows) was designed to amplify the segment across the breakpoint junction.
(D) Electropherogram of amplified segment encompassing breakpoint junctions. The nucleotide sequence corresponding to the segment upstream of the deletion is shown in blue, and the sequence corresponding to the segment downstream of the deletion is shown in red. The underlined inserted sequence not identical to either the upstream or the downstream segment is shown in black.
(E) Scan data of array CGH analysis of a patient with AR-JP with a homozygous duplication (exons 6 of PARK2) that turned out to be a tandem duplication. The horizontal axis represents the nucleotide position. The vertical axis represents log2 (ratio of case to reference signal intensities on array CGH). Dots of log2 (ratio of case to reference signal intensities) larger than 0 are shown in red, and those smaller than 0 are shown in green.
(F) Design of primer pairs for specific amplification of the duplicated allele by PCR based on head-to-tail, head-to-head, and tail-to-tail models. Oligonucleotide primers are denoted by red and blue arrows.
(G) Agarose gel electrophoresis of the PCR products derived from patient's genomic DNA obtained by employing primer pairs flanking duplicated segment. The PCR products are generated only when appropriate primers are used for amplification of rearranged genomic DNA segments.
We then applied these methods to determine the breakpoints of PARK2 at the nucleotide-sequence level in patients with AR-JP. For this purpose, we selected patients with AR-JP who had previously been determined to have one or two rearranged PARK2 alleles on the basis of PCR-based gene-dosage analysis or MLPA analysis. In array CGH analyses of PARK2 of 206 patients with AR-JP, 268 exonic rearrangements (243 deletions and 25 duplications) and five intronic deletions were detected. Nucleotide sequences of the 252 breakpoint junctions (92.3%) were determined, including 235 deletions (94.8%) and 17 duplications (68.0%). In total, 62 had homozygous exonic rearrangements, 57 had compound-heterozygous exonic rearrangements, and 69 had a heterozygous exonic rearrangement. In contrast to the results obtained by the PCR-based gene-dosage or MLPA analysis, exonic rearrangements were not detected by the array CGH analysis in 18 patients with AR-JP, raising the possibility that the PCR-based conventional analyses may provide false positive results. For comparison of the breakpoints of PARK2 in the germline mutations in patients with AR-JP, we then conducted similar array CGH analyses of PARK2 in 125 cancer-derived cell lines and identified 42 rearrangements (39 deletions and three duplications) in 28 of the cancer cell lines (22.4%). The nucleotide sequences of the 41 breakpoint junctions (97.6%), including 39 deletions (100.0%) and two duplications (66.7%), were determined. Because ten deletions and two duplications were found among multiple cancer cell lines, 32 independent breakpoints (31 deletions and one duplication) were determined. Among 32 independent breakpoints, two (one deletion and one duplication) were also found in patients with AR-JP, raising the possibility that they were derived from germ cell lines or that the identical rearrangements of germ cell lines independently occurred in somatic cell lines. Intriguingly, in one cancer cell line (COLO320), six independent deletions were observed simultaneously (Figure S1).
To compare the breakpoint clustering and the signatures of the breakpoint-junction sequences of PARK2 (FRA6E) with those at other CFSs, we further conducted array CGH analyses of DMD, which is embedded in another CFS, FRAXC,7 in 208 patients with DMD/BMD. All of the patients had hemizygous rearrangements (172 deletions and 36 duplications) involving exons, but three intronic deletions were also identified. We were able to determine nucleotide sequences of 197 breakpoint junctions (93.4%), including 167 deletions (95.4%) and 30 duplications (83.3%). None of the breakpoints determined occurred at the same exact position. We subsequently conducted similar array CGH analyses of DMD in the same 125 cancer cell lines. This analysis identified nine rearrangements (eight deletions and one duplication) in the seven cancer cell lines (5.6%) and determined the nucleotide sequences of six breakpoint junctions (66.7%), including six deletions (75.0%). Although most of the breakpoints demonstrated by the array CGH were identified at the nucleotide level, several breakpoints were not able to be determined. This included 13 of the 248 deletions and eight of the 25 duplications in PARK2 in patients with AR-JP, one of the two duplications in PARK2 in cancer cell lines, eight of the 175 deletions and six of the 36 duplications in DMD in patients with DMD/BMD, and two of the eight deletions and the one duplication in DMD in cancer cell lines. For three deletions in patients with DMD/BMD, breakpoints located outside the region covered by the designed array were not identified. With the exception of these large deletions, the reasons of failed breakpoint identification were not certain. It could be due to the complex structures of rearrangements, such as a deletion coupled with an inversion, or the insertion of the duplicated sequence in a nontandem site, which were difficult to amplify by the strategies shown in Figures 1C and 1F.
The results of the array CGH analyses and determination of breakpoints at the nucleotide level are shown in Tables S2A–S2G and are summarized in Table 1. It should be noted that the frequencies of rearrangements in PARK2 and DMD observed in cancer cell lines were quite high (42 rearrangements in 125 cancer cell lines in PARK2 and nine rearrangements in 125 cancer cell lines in DMD), supporting the instability of CFS-associated loci in cancer cell lines. Nucleotide positions of the breakpoints are defined as shown in Figure S2. All the duplications of PARK2 and DMD were tandem duplications, and inverted duplications were not found among the samples in this study. Deletions were more frequently observed than duplications. The ratios of deletions to duplications detected by the array CGH analyses were 9.9 in PARK2 in patients with AR-JP, 13.0 in PARK2 in cancer cell lines, 4.9 in DMD in patients with DMD/BMD, and 8.0 in DMD in cancer cell lines.
Table 1.
Numbers of Rearrangements Determined by Array CGH Analyses and Those of Breakpoints Determined at Nucleotide Levels along with Numbers of Recurrently and Nonrecurrently Observed Breakpoints
| Locus | Sample Sources | No. of Samples |
Breakpoints Determined at Nucleotide Levelb |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Rearrangements Detected by Array CGHa |
Total No. of Breakpoints Determined at Nucleotide Level |
Recurrently Observedc |
Not Recurrently Observedd |
|||||||||||
| Total | Deletion | Duplication | Total | Deletion | Duplication | Total | Deletion | Duplication | Total | Deletion | Duplication | |||
| PARK2 | Patients with AR-JP | 206 | 273 | 248 | 25 | 252 | 235 | 17 | 22 [112] | 20 [107] | 2 [5] | 140 | 128 | 12 |
| Cancer cell lines | 125 | 42 | 39 | 3 | 41 | 39 | 2 | 4 [12] | 3 [10] | 1 [2] | 28 | 28 | 0 | |
| DMD | Patients with DMD/BMD | 208 | 211 | 175 | 36 | 197 | 167 | 30 | 0 [0] | 0 [0] | 0 [0] | 197 | 167 | 30 |
| Cancer cell lines | 125 | 9 | 8 | 1 | 6 | 6 | 0 | 0 [0] | 0 [0] | 0 [0] | 6 | 6 | 0 | |
Number of rearrangements demonstrated by array CGH.
Number of rearrangements determined at the nucleotide level.
Number of independent rearrangements observed in multiple cases. Number in bracket is the number of total recurrently observed rearrangements.
Number of independent rearrangements observed individually.
Multiple Independent Rearrangements Had Frequently Occurred in PARK2 and DMD
The results that 140 of the 252 breakpoints (55.6%) in PARK2 in patients with AR-JP were distinct (Table 1) indicated that recurrent mutations are less frequent than nonrecurrent mutations. This notion is further strengthened by the observation that all of the 192 breakpoints in DMD in patients with DMD/BMD are independent without any identical junctions. Taken altogether, this indicates that multiple independent rearrangements had frequently occurred in PARK2 and DMD. Although the number of cases is limited, there were 22 recurrently observed breakpoints in PARK2 in patients with AR-JP, and the most frequently observed breakpoint (recurrently observed breakpoint no. 1) was present in 22 index patients from different ethnic populations (eight were observed in Asians and 14 in Europeans), and the other 21 recurrently observed breakpoints were found only in a single ethnic population (Table 2). The signatures of the junction sequences are described later in detail.
Table 2.
List of Recurrently Observed Breakpoints in PARK2 in Patients with AR-JP
| No. | No. of Index Patients | Hom. | Het. | Del. or Dup. | Origin | Upstream | Identical Sequence | Inserted Sequence | Downstream | Exon or Intron | Extended Homology |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 22 | 1 | 21 | deletion | 8 Asia and 14 Europe | 162,506,819 | GATTACAGGCATGAGCCACC | - | 162,503,759 | intron 4 | Alu (311 bp)/Alu (307 bp) |
| 2 | 19 | 9 | 10 | deletion | Asia | 162,567,759 | - | GAG | 162,486,065 | exon 4 | |
| 3 | 9 | 2 | 7 | deletion | Japan | 162,857,698 | TTC | - | 162,494,729 | exons 2–4 | |
| 4 | 8 | 1 | 7 | deletion | Asia | 162,660,297 | - | TAAAACTG | 162,658,014 | intron 2 | |
| 5 | 7 | 5 | 2 | deletion | Japan | 162,333,460 | A | - | 162,126,458 | exons 6–7 | |
| 6 | 6 | 4 | 2 | deletion | Asia | 162,612,866 | - | CACAAATATCACAAATATC | 162,489,437 | exons 3–4 | |
| 7 | 5 | 1 | 4 | deletion | Japan | 162,653,100 | TATTT | - | 162,510,558 | exons 3–4 | |
| 8 | 3 | 0 | 3 | deletion | Asia | 162,189,426 | TAAG | - | 162,085,168 | exon 7 | |
| 9 | 3 | 0 | 3 | deletion | France | 162,547,125 | AGCAC | - | 162,536,937 | exon 4 | |
| 10 | 3 | 1 | 2 | deletion | Asia | 162,571,209 | - | TATATAC | 162,225,376 | exons 4–6 | |
| 11 | 3 | 0 | 3 | deletion | Japan | 162,647,230 | T | - | 162,591,063 | exon 3 | |
| 12 | 3 | 2 | 1 | deletion | Japan | 162,697,743 | - | - | 162,502,832 | exons 3–4 | |
| 13 | 3 | 2 | 1 | duplication | Japan | 162,359,815 | - | T | 162,288,602 | exon 6 | |
| 14 | 2 | 0 | 2 | deletion | Vietnam | 162,461,205 | AAAAATA | - | 162,365,391 | exon 5 | Alu (267 bp)/Alu (302 bp) |
| 15 | 2 | 0 | 2 | deletion | Japan | 162,543,628 | - | T | 162,519,459 | exon 4 | |
| 16 | 2 | 1 | 1 | deletion | Japan | 162,561,255 | CTTC | - | 162,508,229 | exon 4 | |
| 17 | 2 | 1 | 1 | deletion | Europe | 162,608,217 | CT | - | 162,548,381 | exon 3 | |
| 18 | 2 | 1 | 1 | deletion | Japan | 162,615,492 | AGG | - | 162,555,347 | exon 3 | |
| 19 | 2 | 0 | 2 | deletion | Korea | 162,623,148 | - | AA | 162,569,292 | exon 3 | |
| 20 | 2 | 0 | 2 | deletion | France | 162,630,240 | GAT | - | 162,288,742 | exons 3–6 | |
| 21 | 2 | 1 | 1 | deletion | Japan | 162,840,504 | C | - | 162,735,373 | exon 2 | |
| 22 | 2 | 0 | 2 | duplication | France | 162,835,997 | - | T | 162,637,347 | exon 2 |
Abbreviations are as follows: Hom., homozygous; Het., heterozygous; Del., deletion; Dup., duplication.
Breakpoints Are Clustered in Specific Genomic Regions in Germ Cell Mutations
The histogram and cumulative-frequency distribution of the positions of breakpoints showed that the breakpoints were obviously clustered at specific genomic regions in PARK2 and DMD in germ cell lines (Figures 2A and 2B). The breakpoint-clustering region in PARK2 in patients with AR-JP closely coincided with the previously reported region in FRA6E prone to DNA double-strand breaks, which has been referred as to the center of FRA6E (Figure 2D).5 Furthermore, the breakpoint-clustering region in DMD in patients with DMD/BMD was embedded in FRAXC (Figure 2D).7 These findings of breakpoint clustering in PARK2 and DMD in germ cell lines were consistent with the previous studies that had identified deletion hotspots in exons 3 and 4 of PARK2 in patients with AR-JP5 and in exons 45–52 of DMD in patients with DMD/BMD (Figure 2).10 The breakpoint distributions in PARK2 and DMD in cancer cell lines seemed to be more dispersed than those observed in germ cell lines. To assess differences and similarities of the breakpoint distributions between germ cell lines and cancer cell lines, statistical data including mean, median, standard deviation, skewness, and kurtosis of breakpoint positions were calculated (Table 3). It was found that the differences in mean and/or median breakpoint positions between germ cell lines and cancer cell lines were relatively small (within 20–90 kb), with no significant differences detected via the Mann-Whitney U test. The differences in standard deviation of breakpoint positions in cancer cell lines were relatively larger than those in germ cell lines. The squared-ranks equality-of-variance test revealed that differences in variance across germ cell and cancer cell lines in PARK2 was significant, whereas that in DMD was not significant, possibly due to the small sample size of somatic rearrangements in DMD. Taken together, the center of breakpoint distribution in PARK2 and DMD may be similar in germ cell and cancer cell lines, but the variance of distribution may be larger in cancer cell lines than that in germ cell lines. Possible explanations of the difference are that the sample selections for patients with AR-JP and with DMD/BMD biased the breakpoint distributions and that the cancer cell lines tended to generate larger rearrangements in these loci as a result of increased genomic instability.
Figure 2.
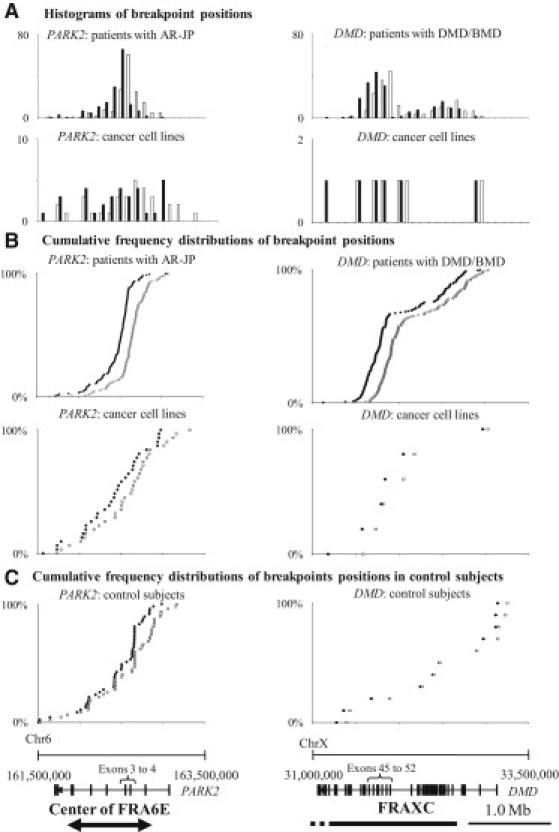
Histograms and Cumulative-Frequency Distributions of Breakpoint Positions
(A) Histograms of breakpoint positions in PARK2 in AR-JP patients or cancer cell lines, and in DMD in patients with DMD/BMD or cancer cell lines. The horizontal axis represents nucleotide positions, and the vertical axis represents the number of breakpoints. The numbers of the positions of the upstream (toward the transcriptional initiation site) breakpoints are shown in white, and those of the downstream breakpoints are shown in black.
(B) Cumulative-frequency distributions of breakpoint positions in PARK2 in patients with AR-JP or cancer cell lines, and those in DMD in patients with DMD/BMD or cancer cell lines: The horizontal axis represents the nucleotide positions of breakpoints. The vertical axis represents cumulative frequencies of breakpoints. The upstream breakpoints are shown in white, and the downstream breakpoints are shown in black.
(C) Cumulative frequency distributions of breakpoint positions (PARK2 and DMD) in control subjects obtained from the Database of Genomic Variants. Physical maps of PARK2 and DMD, along with schematic representations of the center of FRA6E and FRAXC, are shown below.
Table 3.
Distribution of Breakpoint Positions in Germ Cell Lines and in Cancer Cell Lines
| Loci | Breakpoints | Samples | Number | Mean | Median | Mann-Whitney U test | Standard Deviation | Squared-Ranks Test | Skewness | Kurtosis |
|---|---|---|---|---|---|---|---|---|---|---|
| PARK2 | upstream breakpoints | patients with AR-JP | 162 | 162,632,436 | 162,648,608 | p = 0.36 | 203,951 | p < 0.0001 | −1.21 | 4.01 |
| cancer cell lines | 32 | 162,549,884 | 162,626,524 | 401,285 | −0.30 | −0.37 | ||||
| downstream breakpoints | patients with AR-JP | 162 | 162,462,592 | 162,515,588 | p = 0.53 | 210,775 | p < 0.0001 | −1.35 | 3.03 | |
| cancer cell lines | 32 | 162,405,816 | 162,440,014 | 389,800 | −0.38 | −0.56 | ||||
| DMD | upstream breakpoints | patients with DMD/BMD | 197 | 32,151,340 | 31,967,332 | p = 0.49 | 405,283 | p = 0.99 | 0.73 | −0.93 |
| cancer cell lines | 6 | 32,063,805 | 31,932,249 | 518,968 | 1.57 | 2.75 | ||||
| downstream breakpoints | patients with DMD/BMD | 197 | 31,969,561 | 31,796,456 | p = 0.78 | 402,560 | p = 0.81 | 0.80 | −0.71 | |
| cancer cell lines | 6 | 31,900,988 | 31,816,981 | 600,593 | 1.12 | 2.25 |
The Database of Genomic Variants (accessed in March 2010)29 included 48 and 6 copy-number variations (CNVs) (more than 1 kb in length) in the regions in PARK2 (chromosome 6: 161,500,000–163,500,000) and in DMD (chromosome X: 31,000,000–33,500,000), respectively. The distributions of these breakpoints in PARK2 showed similarities with those observed in patients with AR-JP (Figures 2B and 2C).
Junction-Sequence Signatures in Germ Cell and Somatic Cell Mutations
On the basis of the sequences flanking the breakpoints, the junction-sequence signatures were analyzed and then classified into three groups: (1) junctions with extended homologies, (2) junctions with microhomologies, and (3) junctions without extended homologies or microhomologies (Table 4). An extended homology was detected via the FASTN program with an optimum score ≥ 300 by comparing the pairs of 200 bp nucleotide sequences encompassing the breakpoint junctions (100 bp upstream and 100 bp downstream). In this study, we refer to such short stretches of identical sequences (≤ 8 bp) at breakpoint junctions as microhomologies.
Table 4.
Junction-Sequence Signatures
|
PARK2 (Germ Cell Lines) |
PARK2 (Cancer Cell Lines) |
DMD (Germ Cell Lines) |
DMD (Cancer Cell Lines) |
|||||
|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | N | % | |
|
Junctions with Extended Homologies | ||||||||
| Total | 7 | 4.3% | 1 | 3.1% | 2 | 1.0% | 0 | 0.0% |
| Junctions with Microhomologies (Identical Sequences ≤ 8 bp) | ||||||||
| ≥ 9 bp identical sequences | 0 | 0.0% | 0 | 0.0% | 0 | 0.0% | 0 | 0.0% |
| 8 bp identical sequences | 1 | 0.6% | 0 | 0.0% | 0 | 0.0% | 0 | 0.0% |
| 7 bp identical sequences | 1 | 0.6% | 1 | 3.1% | 1 | 0.5% | 0 | 0.0% |
| 6 bp identical sequences | 1 | 0.6% | 0 | 0.0% | 4 | 2.0% | 0 | 0.0% |
| 5 bp identical sequences | 8 | 4.9% | 1 | 3.1% | 6 | 3.0% | 1 | 16.7% |
| 4 bp identical sequences | 9 | 5.6% | 5 | 15.6% | 14 | 7.1% | 1 | 16.7% |
| 3 bp identical sequences | 23 | 14.2% | 4 | 12.5% | 33 | 16.8% | 0 | 0.0% |
| 2 bp identical sequences | 26 | 16.0% | 3 | 9.4% | 30 | 15.2% | 1 | 16.7% |
| 1 bp identical sequences | 28 | 17.3% | 5 | 15.6% | 40 | 20.3% | 0 | 0.0% |
| Total | 97 | 59.9% | 19 | 59.4% | 128 | 65.0% | 3 | 50.0% |
| Junctions without Extended Homologies or Microhomologies | ||||||||
| Insertions of repetitive sequences | 0 | 0.0% | 2 | 6.3% | 1 | 0.5% | 0 | 0.0% |
| Insertions of sequences of undetermined origin | 51 | 31.5% | 8 | 25.0% | 51 | 25.9% | 2 | 33.3% |
| No insertions | 7 | 4.3% | 2 | 6.3% | 15 | 7.6% | 1 | 16.7% |
| Total | 58 | 35.8% | 12 | 37.5% | 67 | 34.0% | 3 | 50.0% |
Search for extended homologies revealed that seven of the 162 junctions (4.3%) in PARK2 in patients with AR-JP, one of the 32 junctions (3.1%) in PARK2 in cancer cell lines (identical to one of the seven junctions observed in patients with AR-JP), two of the 197 junctions (1.0%) in DMD in patients with DMD/BMD, and none of the six junctions (0.0%) in DMD in cancer cell lines had junctions with extended homologies (Table 4), all of which were embedded in the same repetitive sequences: seven Alu/Alu sequences in PARK2 (two were recurrently observed), and one Alu/Alu sequence and one L1/L1 sequence in DMD. Among these nine junctions with extended homologies, seven had identical sequences of 92 bp (L1P1 and L1P1), 28 bp (AluJb and AluSx), 20 bp (AluSq/x and AluSg), 18 bp (AluSq/x and AluY), 15 bp (AluSc and AluSg/x), 8 bp (AluY and AluSg/x), and 7 bp (AluJb and AluSx) flanking the junctions, resulting in formation of completely chimeric L1/L1 or Alu/Alu sequences. The remaining two formed partially chimeric Alu/Alu with inserted sequences of 7 bp (AluSg/x and AluSg/x) and 12 bp (AluY and AluSq) at their junctions (Figure S3). Intriguingly, the majority of the junctions were frequently associated with microhomologies (1–8 bp): 97 of the 162 junctions (59.9%) in PARK2 in patients with AR-JP, 19 of the 32 junctions (59.4%) in PARK2 in cancer cell lines, 128 of the 197 junctions (65.0%) in patients with DMD/BMD in DMD, and three of the six junctions (50.0%) in DMD in cancer cell lines had microhomologies at junctions (Table 4). Note that frequencies of microhomologies were markedly similar between PARK2 and DMD and also between germ cell lines and cancer cell lines. Regarding the junctions without extended homologies or microhomologies, it was revealed that 58 of the 162 junctions (35.8%) in PARK2 in patients with AR-JP, 12 of the 32 junctions (37.5%) in PARK2 in cancer cell lines, 67 of the 197 junctions (34.0%) in DMD in patients with DMD/BMD, and three of the six junctions (50.0%) in DMD in cancer cell lines were without extended homologies or identical sequences (Table 4). Among these, 51 of the 162 junctions (31.5%) in PARK2 in patients with AR-JP, eight of the 32 junctions (25.0%) in PARK2 in cancer cell lines, 51 of the 197 junctions (25.9%) in DMD in patients with DMD/BMD, and two of the six junctions (33.3%) in DMD in cancer cell lines had inserted sequences. We found that four junctions in PARK2 in patients with AR-JP, two junctions in PARK2 in cancer cell lines, and two junctions in DMD in patients with DMD/BMD had inserted sequences of more than 19 bp, whose origins were searched by the BLAST program and SSEARCH programs. It was revealed that two inserted sequences in PARK2 deletions in cancer cell lines and one inserted sequence in DMD deletion in patients with DMD/BMD originated from repetitive sequences (two Alu and one THE1B), which correspond to “67–112 bp of Alu,” “10–30 bp of Alu,” and “83–332 bp of THE1B” (Figure S4). The origins of the other inserted sequences remained undetermined.
Among the 22 recurrently observed breakpoints in PARK2 in patients with AR-JP (Table 2), two junctions (9.1%) had extended homologies. One (recurrently observed breakpoint no. 1) was the most frequent and was observed in multiple ethnicities. The other breakpoint (recurrently observed breakpoint no. 14) was found in two patients from Vietnam. These two breakpoints were embedded in the same Alu sequences (approximately 300 bp in length), and chimeric Alu/Alu sequences were formed at the junctions (Figure S5). Among the 20 junctions without extended homologies, 11 junctions (50.0%) had microhomologies (1–5 bp) and nine junctions (40.9%) were without extended homologies or microhomologies, of which eight had inserted sequences (1–19 bp). Importantly, these junction-sequence signatures are similar to those of not recurrently observed breakpoints, as described above.
Breakpoint-Clustering Regions Are Associated with Multiple Factors Affecting Replication Timing
The breakpoint-clustering region in PARK2 in patients with AR-JP coincided with the center of FRA6E, and the breakpoint-clustering region in DMD in patients with DMD/BMD was fully embedded in FRAXC (Figure 2), strongly suggesting that the clustering of the breakpoints is closely related to the mechanisms underlying fragility within the CFSs. Although the mechanisms underlying CFS breakage are still unclear, several factors that may contribute to instability at CFSs have been suggested, including late-replicating regions,13,14,30,31 high-flexibility peaks,32,33 regions rich in nuclear-matrix-attachment regions,32,34,35 and regions located at the interface of G and R bands.36 On the basis of these reports, breakpoint-clustering regions in PARK2 and DMD were analyzed for investigation of the association of these regions with sequence motifs, replication timing, flexibility peaks, nuclear-matrix-attachment regions, and R/G bands. In addition, because there has been a recent report suggesting that a deletion hotspot in PARK2 in patients with AR-JP is associated with a meiotic recombination hotspot,16 breakpoint-clustering regions in PARK2 and DMD were also compared with the deCODE recombination maps.28
We performed a systematic search for 40 different sequence motifs previously associated with DNA breakage using the DNA Pattern Find program to detect sequence motifs reportedly abundant at breakpoints.23 For this search, nucleotide sequences of 200 bp surrounding breakpoints (referred to as a breakpoint region) and 5000 sequences of 200 bp (control sequences) randomly picked from the entire PARK2 and DMD regions were used. Of the 40 sequence motifs, none were overrepresented in the breakpoint regions (Table S2). On the basis of a recent study of a replication-timing map of chromosome 6,27 it was found that one of the latest-replication regions (S phase DNA to G1 phase DNA ratios of less than 1.2) was chromosome 6: 161,884,878–162,579,873, which coincided with the breakpoint-clustering region in PARK2 (Figure 3B). Because there were no reports of replication timing of chromosome X, we were unable to investigate the association of the breakpoint-clustering region in DMD with replication timing. For investigation of flexibility peaks, chromosome 6: 162,370,000–162,870,000 and chromosome X: 31,500,000–32,000,000, corresponding to the breakpoint-clustering regions in PARK2 and DMD, respectively, and the neighboring regions (chromosome 6: 159,870,000–165,370,000 and chromosome X: 29,000,000–34,500,000) were analyzed in terms of AT content, average twist angle, and numbers of flexibility peaks, unified peaks, and cluster of peaks (Table S3). Although there were 25 flexibility peaks in the breakpoint-clustering regions in PARK2 and 26 flexibility peaks in the breakpoint-clustering regions in DMD, both of which were not overrepresented (50 peaks/Mb and 52 peaks/Mb) as compared with their neighboring region, there were regions with high AT content (AT repeats) near the breakpoint-clustering region (Figure 3C). Furthermore, the highest-flexibility peaks with a twist angle of more than 15.5 evidently flanked the breakpoint-clustering region (Figure 3D). On the high-resolution map of the LADs,26 it was revealed that PARK2 and DMD were embedded in large LADs (chromosome 6:161,789,694–163,646,839 and chromosome X: 31,589,326–34,513,733) (Figure 3E). This prompted us to investigate the relationships of LADs with other CFS genes, including FHIT, WWOX, GRID2, LARGE, CTNNA3, NBEA, and CNTNAP2. Intriguingly, all the CFS genes were embedded in large LADs spanning several Mb (approximately 1.7–4.7 Mb). Representative CFS genes are shown in Figure S6. The intron 55 of DMD spans the boundary of the chromosomal R/G band: Xp21.2 (G band) to Xp21.1 (R band).25 The breakpoint-clustering region in DMD was flanked by the boundary of the R/G band and was exclusively in the R band, whose AT content was relatively high. In contrast, there were neither obvious boundaries of the R/G band nor significant changes in AT content within PARK2 (Figure 3F). With the use of the deCODE map, the meiotic recombination rate of the breakpoint-clustering regions in PARK2 (D6S955 to D6S1599) was found to be high (5.0 cM/Mb), as previously reported.16 The recombination rate of the region covering the breakpoint-clustering regions in DMD (DXS1214 to DXS1219) was also higher (2.80 cM/Mb) than the average recombination rate along chromosome X (1.14 cM/Mb), but was similar to those of other regions in DMD (Figure 3G).
Figure 3.
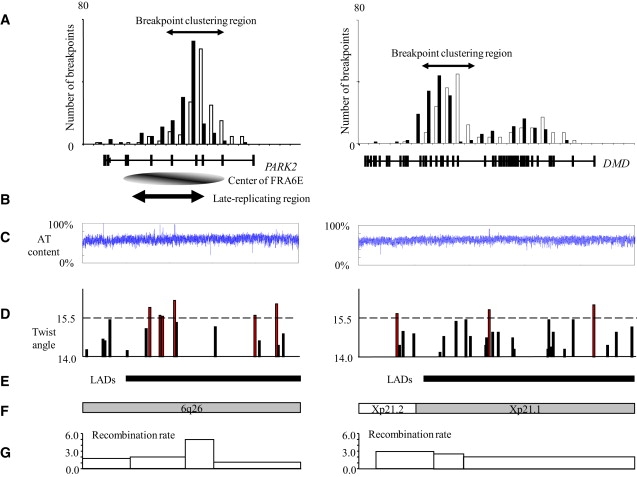
Association of Breakpoint-Clustering Regions in PARK2 and DMD with Replication Timing, Flexibility Peak, R/G Band, and AT Content
(A) Histograms of positional distributions of breakpoints in PARK2 and DMD in germ cell lines. Breakpoint-clustering regions are the regions with high frequencies (70%–78%) shown by arrows. The physical positions of PARK2, DMD, and the center of FRA6E are shown below.
(B) Late-replicating regions,27 defined as S to G1 DNA ratios of 1.1–1.2.
(C) Distributions of AT content (calculated with an average span of 500 bp and an average step of 100 bp).
(D) Distributions of flexibility peaks of more than 13.7° in twist angle and more than 100 bp in length. Red bars are the highest peaks whose twist angles exceed 15.5.
(E) Physical positions of LADs in PARK2 and DMD.26
(F) Chromosomal R and G bands are indicated by open and shaded boxes, respectively.
(G) Recombination rates based on the deCODE map.28
Discussion
We have shown that a locus-specific high-density array CGH analysis system is highly efficient for beginning to localize the exact breakpoints in genomic DNAs in germ cell lines as well as in cancer cell lines. Utilizing this system has enabled us to acquire data on approximately 500 breakpoint junctions involving PARK2 and DMD and to investigate the various breakpoint-sequence features. This study is applied to identifying such a large number of rearrangements at the nucleotide level. The high frequencies of somatic rearrangements observed in cancer cell lines (42 rearrangements in 125 cancer cell lines in PARK2 and nine rearrangements in 125 cancer cell lines in DMD) and the various independent rearrangements for germ cell line rearrangements (140 of the 252 rearrangements in PARK2 and 197 of the 197 rearrangements in DMD) demonstrated how vulnerable these regions are for rearrangements. The difference in the frequency of somatic rearrangements between PARK2 and DMD may be consistent with the relative instability within these two loci: PARK is within one of the most active CFSs,37 whereas DMD is in a very low-expressing CFS.7
Microhomologies Are Predominantly Involved in Rearrangement Processes at CFSs in Germ Cell and Somatic Cell Mutations
The present study demonstrated that microhomologies were notably frequent (59.9% in PARK2 in patients with AR-JP, 59.4% in PARK2 in cancer cell lines, 65.0% in DMD in patients with DMD/BMD, and 50.0% in DMD in cancer cell lines) at the junctions, strongly raising the possibility that the rearrangements are predominantly generated by mechanisms mediated by microhomologies (Table 4, Figures 4Aa and 4Ab). Note that there are similarly high frequencies of microhomologies in PARK2 rearrangements in germ cell and cancer cell lines, which further supports the notion that a common mechanism underlies the generations of rearrangements in germ cell and cancer cell lines. Consistent with our findings, microhomologies at junctions have recently been observed in the rearrangements of human culture cells experimentally induced by aphidicolin, a model of CFS.38 Taken together, the present findings strongly support the concept that the mechanisms mediated by microhomologies play a major role in rearrangement processes within CFSs (Figure 4A). In contrast, rearrangements that can be explained by the homology-dependent nonallelic homologous recombination (NAHR) are relatively rare, because there is only a limited number of rearrangements (4.3% in PARK2 in patients with AR-JP, 3.1% in PARK2 in cancer cell lines, 1.0% in DMD in patients with DMD/BMD, 0.0% in DMD in cancer cell lines) whose junctions show extended homologies (repetitive sequences) (Table 4, Figures 4Aa and 4Ac). Considering the observation that multiple independent rearrangements had frequently occurred in PARK2 and DMD, it is in a striking contrast to other common genomic disorders, such as Charcot-Marie-Tooth disease type 1A39 or Smith-Magenis syndrome,40 whose recurrent mutations are characterized by homologous recombination and unequal crossing over between the flanking repeat elements.
Figure 4.
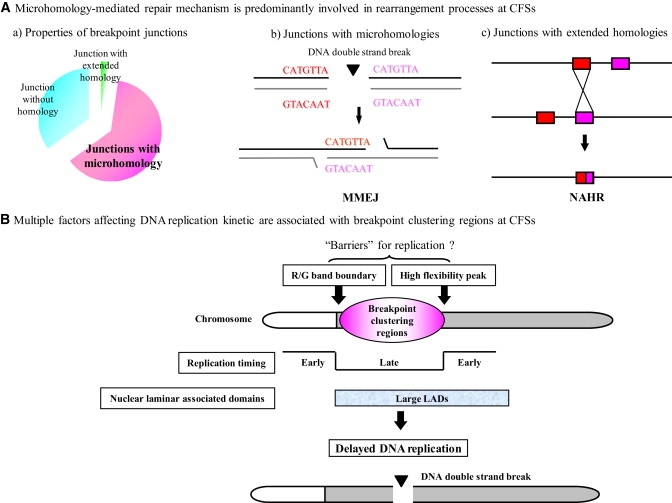
Schematic Representations of Mechanisms Underlying CFSs
(A) The microhomology-mediated mechanism is predominantly involved in rearrangement processes at CFSs. (a) Detailed analysis of the nucleotide-sequence content flanking the breakpoints demonstrated that junctions with microhomologies (pink) are predominantly observed, compared with junctions without any homology (sky blue). Junctions with extended homologies (green) underlying NAHR are infrequent. (b) Schematic representation of MMEJ. (c) Schematic representation of NAHR.
(B) Multiple factors affecting DNA-replication kinetics collectively contribute to fragility as a common molecular basis. The breakpoint-clustering region at CFSs is flanked by the high-flexibility peaks and the R/G band boundaries. The breakpoint-clustering region coincides with the late-replicating region and is embedded in large LADs.
Various mechanisms of rearrangement processes that can result in microhomologies at junctions have been proposed, which include nonhomologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), microhomology-mediated break-induced replication (MMBIR), and/or fork stalling and template switching (FoSTeS). In eukaryotes, NHEJ is the major repair pathway of DNA double-strand breaks, which functions by ligating the two ends together.41 It has the potential to ligate any type of double-strand break end without the requirement for an extended homology. Even when starting with two identical DNA ends, NHEJ is a highly flexible process accounting for the diverse breakpoint junctions, with some ends showing short microhomologies (usually 1–4 bp) and some ends showing inserted sequences without microhomologies.41 In addition, it was shown that replication stress leads to the focus formation of key components of the NHEJ pathway (Rad51 and DNA-PKcs) colocalized with markers of DNA double-strand breaks (MDC1 and gamma H2AX), and the downregulation of the component of the NHEJ pathway (Rad 51, DNA-PKcs, or DNA ligase 4) leads to a significant increase in gaps and breaks at CFSs.42
MMEJ is another distinctive pathway of end-joining repair, which requires microhomologies of terminal ends, in contrast to NHEJ. High frequencies of microhomologies at junctions (60%–65%) observed in this study would favor the involvement of MMEJ at CFSs. Recently, the MMBIR and/or FoSTeS model with emphasis on replication fork collapse and/or stalling has also been proposed to explain the origin of rearrangements on the basis of the findings of complex rearrangements and junction sequences showing microhomologies of 2–5 bp.43 Because delayed replication at CFSs has been implicated to underlie the rearrangements involving CFSs, MMBIR/FoSTeS deserves serious consideration as a possible mechanism underlying the rearrangements at CFSs. Actually, we observed a case of complex rearrangements in DMD comprising short tandem multiplications followed by large deletions (Figure 5), which strongly supports the involvement of multiple MMBIR/FoSTeS events, at least in this case. For other cases, however, it is difficult to deduce, on the basis of breakpoint sequences, whether a replication-based repair mechanism (MMBIR/FoSTeS) is commonly involved in the generation of rearrangements.
Figure 5.
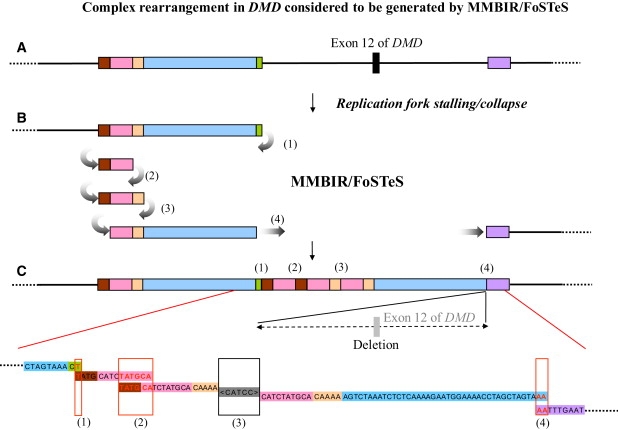
Complex Rearrangements in DMD Considered to be Generated by MMBIR/FoSTeS Observed in One Patient with DMD
An example of complex rearrangements in DMD with microhomology junctions leading to the deletion of approximately 5.7 kb, including exon 12 of DMD, is shown.
(A) A map of a part of DMD. The colored boxes represent blocks of sequences.
(B) A hypothetical series of four template switches leading to rearrangements, indicated by gray curved arrows and numbers; a gray curved arrow indicates resumption of replication on the original template. Numbers corresponding to the sequences are shown in (C).
(C) Rearranged chromosomal region, in which tandem multiplications connect the green sequence to the brown sequence (1), the pink sequence to the brown sequence (2), and the tan sequence to the pink sequence (3), followed by gross deletion between the sky blue sequence and the purple sequence (4). The nucleotide sequences of the colored segments correspond to the colored boxes in (A), (B), and (C). The red boxes indicate the sequences of microhomologies. The gray box represents the inserted sequence of a junction. (1) The junction between the green and the brown sequences shows a 1 bp microhomology. (2) The junction between the pink and the brown sequences shows a 6 bp microhomology. (3) The junction between the tan and the pink sequences shows a 5 bp inserted sequence without microhomology. (4) The junction between the sky blue and the purple sequences shows a 2 bp microhomology. The sizes of the brown, pink, tan, sky blue, and green fragments are 4, 10, 5, 41, and 2 bp, respectively, including the microhomology sequences at both ends.
Associations of Breakpoint-Clustering Regions in CFSs with DNA Replication Kinetics
In this study, we found that regions where breakpoints clustered within CFSs coincided with latest-replicating regions and demonstrated that the highest-flexibility peaks and R/G band boundary flanked a breakpoint-clustering region (Figure 3). The highest-flexibility peaks44 and R/G band boundary45 are considered to affect replication timing. Interestingly, we observed that PARK2 and DMD are embedded in large LADs and furthermore found the colocalizations of other CFS genes, including FHIT, WWOX, GRID2, LARGE, CTNNA3, NBEA, and CNTNAP2, with large LADs (Figure 3E and Figure S6). It was reported that 1344 LADs are aligned on the human genome, comprising approximately 40% of the entire human genome.26 In higher eukaryotic cells, DNA is organized into loops attached to the nuclear matrix. Each loop represents one individual replicon, with the ends of the replicon attached to the nuclear matrix at the bases of the loop. Upon completion of replication of any replicon, the resulting entangled loops of the newly synthesized DNA are resolved by topoisomerase II present in the nuclear matrix, which generate double-strand breaks with the potential risk leading to vulnerability for rearrangements.46 Because LADs comprise approximately 40% of the human genome, as described above, association of CFSs with large LADs does not directly explain the rearrangement clustering of CFSs. Further cytogenetic investigations should be conducted to explore whether LADs are associated with intrinsic replication difficulties in CFSs. It has been shown that recombination rates are relatively high in the regions covering the breakpoint-clustering regions, which may indicate a possibility that genomic instabilities also contribute to meiotic recombination (Figure 3G). In summary, our findings suggest that multiple factors affecting DNA-replication timing collectively contribute to the vulnerability for rearrangements, which include high-flexibility peaks, R/G band boundary, and large LADs (Figure 4B). These factors cause substantial difficulties in replication machineries, and CFSs represent unreplicated regions of the genome that have escaped the replication checkpoints and are visible as gaps and breaks on metaphase chromosomes.
Involvement of CFSs with Rearrangements in Germlines Leading to Human Diseases
To date, several lines of evidence have demonstrated that somatic rearrangements that occur within CFSs are associated with cancer development,47,48 but CFSs have rarely drawn attention as genomic structures associated with germline rearrangements. This study provides evidence that chromosomal instability associated with CFSs plays an important role in gross deletions and duplications in germ cell lines leading to human diseases. Recently, numerous CNVs in the human genome have been identified in control subjects via various platforms, including array CGH, SNP genotyping, and next-generation sequencing.49–52 Because sample-selection bias inevitably affects the distributions of germline rearrangements, unbiased knowledge about CNVs distributions will also be needed to explore whether the common mechanism can underlie CFSs. Such investigations will certainly be essential for better understanding the molecular basis of CFSs and human diseases associated with instabilities in the human genome.
Acknowledgments
We thank the French Parkinson's Disease Genetics Study Group (PDG) and the DNA and cell bank of the CRicm for sample collection and preparation. This work was supported in part by KAKENHI (Grant-in-Aid for Scientific Research) on Priority Areas, Applied Genomics, the 21st Century COE Program, Integrated Database Project, Center for Integrated Brain Medical Science, and Scientific Research (A) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BLAST program, http://blast.ncbi.nlm.nih.gov/Blast.cgi
Database of Genomic Variants, http://projects.tcag.ca/variation
DNA Pattern Find, http://www.bioinformatics.org/SMS/index.html
Leiden Muscular Dystrophy, http://www.dmd.nl
NCBI Database of Genomic Structural Variation (dbVAR), http://www.ncbi.nlm.nih.gov/dbvar
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Parkinson disease mutation database, https://reseq.lifesciencedb.jp/resequence/SearchDisease.do?targetId=2
RepeatMasker program, http://www.repeatmasker.org/
SSEARCH program, http://www-btls.jst.go.jp/cgi-bin/Tools/SSEARCH/index.cgi
TwistFlex program, http://margalit.huji.ac.il/TwistFlex/
University of California Santa Cruz (UCSC) Genome Browser, http://www.genome.ucsc.edu/
Accession Numbers
The NCBI Database of Genomic Structural Variation (dbVAR) accession number for the breakpoint positions reported in this paper is nstd36.
References
- 1.Durkin S.G., Ragland R.L., Arlt M.F., Mulle J.G., Warren S.T., Glover T.W. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc. Natl. Acad. Sci. USA. 2008;105:246–251. doi: 10.1073/pnas.0708097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith D.I., McAvoy S., Zhu Y., Perez D.S. Large common fragile site genes and cancer. Semin. Cancer Biol. 2007;17:31–41. doi: 10.1016/j.semcancer.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 4.Periquet M., Lücking C., Vaughan J., Bonifati V., Dürr A., De Michele G., Horstink M., Farrer M., Illarioshkin S.N., Pollak P., French Parkinson's Disease Genetics Study Group. The European Consortium on Genetic Susceptibility in Parkinson's Disease Origin of the mutations in the parkin gene in Europe: exon rearrangements are independent recurrent events, whereas point mutations may result from Founder effects. Am. J. Hum. Genet. 2001;68:617–626. doi: 10.1086/318791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hedrich K., Eskelson C., Wilmot B., Marder K., Harris J., Garrels J., Meija-Santana H., Vieregge P., Jacobs H., Bressman S.B. Distribution, type, and origin of Parkin mutations: review and case studies. Mov. Disord. 2004;19:1146–1157. doi: 10.1002/mds.20234. [DOI] [PubMed] [Google Scholar]
- 6.Veeriah S., Taylor B.S., Meng S., Fang F., Yilmaz E., Vivanco I., Janakiraman M., Schultz N., Hanrahan A.J., Pao W. Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet. 2009;42:77–82. doi: 10.1038/ng.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McAvoy S., Ganapathiraju S., Perez D.S., James C.D., Smith D.I. DMD and IL1RAPL1: two large adjacent genes localized within a common fragile site (FRAXC) have reduced expression in cultured brain tumors. Cytogenet. Genome Res. 2007;119:196–203. doi: 10.1159/000112061. [DOI] [PubMed] [Google Scholar]
- 8.Koenig M., Monaco A.P., Kunkel L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–228. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- 9.Nancarrow D.J., Handoko H.Y., Smithers B.M., Gotley D.C., Drew P.A., Watson D.I., Clouston A.D., Hayward N.K., Whiteman D.C. Genome-wide copy number analysis in esophageal adenocarcinoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2008;68:4163–4172. doi: 10.1158/0008-5472.CAN-07-6710. [DOI] [PubMed] [Google Scholar]
- 10.White S.J., den Dunnen J.T. Copy number variation in the genome; the human DMD gene as an example. Cytogenet. Genome Res. 2006;115:240–246. doi: 10.1159/000095920. [DOI] [PubMed] [Google Scholar]
- 11.Casper A.M., Nghiem P., Arlt M.F., Glover T.W. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 12.Wang L., Darling J., Zhang J.S., Huang H., Liu W., Smith D.I. Allele-specific late replication and fragility of the most active common fragile site, FRA3B. Hum. Mol. Genet. 1999;8:431–437. doi: 10.1093/hmg/8.3.431. [DOI] [PubMed] [Google Scholar]
- 13.Le Beau M.M., Rassool F.V., Neilly M.E., Espinosa R., 3rd, Glover T.W., Smith D.I., McKeithan T.W. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: implications for the mechanism of fragile site induction. Hum. Mol. Genet. 1998;7:755–761. doi: 10.1093/hmg/7.4.755. [DOI] [PubMed] [Google Scholar]
- 14.Hellman A., Rahat A., Scherer S.W., Darvasi A., Tsui L.C., Kerem B. Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Mol. Cell. Biol. 2000;20:4420–4427. doi: 10.1128/mcb.20.12.4420-4427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clarimon J., Johnson J., Dogu O., Horta W., Khan N., Lees A.J., Hardy J., Singleton A. Defining the ends of Parkin exon 4 deletions in two different families with Parkinson's disease. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2005;133B:120–123. doi: 10.1002/ajmg.b.30119. [DOI] [PubMed] [Google Scholar]
- 16.Asakawa S., Hattori N., Shimizu A., Shimizu Y., Minoshima S., Mizuno Y., Shimizu N. Analysis of eighteen deletion breakpoints in the parkin gene. Biochem. Biophys. Res. Commun. 2009;389:181–186. doi: 10.1016/j.bbrc.2009.08.115. [DOI] [PubMed] [Google Scholar]
- 17.Sironi M., Pozzoli U., Cagliani R., Giorda R., Comi G.P., Bardoni A., Menozzi G., Bresolin N. Relevance of sequence and structure elements for deletion events in the dystrophin gene major hot-spot. Hum. Genet. 2003;112:272–288. doi: 10.1007/s00439-002-0881-5. [DOI] [PubMed] [Google Scholar]
- 18.Nobile C., Toffolatti L., Rizzi F., Simionati B., Nigro V., Cardazzo B., Patarnello T., Valle G., Danieli G.A. Analysis of 22 deletion breakpoints in dystrophin intron 49. Hum. Genet. 2002;110:418–421. doi: 10.1007/s00439-002-0721-7. [DOI] [PubMed] [Google Scholar]
- 19.Toffolatti L., Cardazzo B., Nobile C., Danieli G.A., Gualandi F., Muntoni F., Abbs S., Zanetti P., Angelini C., Ferlini A. Investigating the mechanism of chromosomal deletion: characterization of 39 deletion breakpoints in introns 47 and 48 of the human dystrophin gene. Genomics. 2002;80:523–530. [PubMed] [Google Scholar]
- 20.Rawal N., Periquet M., Lohmann E., Lücking C.B., Teive H.A., Ambrosio G., Raskin S., Lincoln S., Hattori N., Guimaraes J., French Parkinson's Disease Genetics Study Group. European Consortium on Genetic Susceptibility in Parkinson's Disease New parkin mutations and atypical phenotypes in families with autosomal recessive parkinsonism. Neurology. 2003;60:1378–1381. doi: 10.1212/01.wnl.0000056167.89221.be. [DOI] [PubMed] [Google Scholar]
- 21.Lohmann E., Thobois S., Lesage S., Broussolle E., du Montcel S.T., Ribeiro M.J., Remy P., Pelissolo A., Dubois B., Mallet L., French Parkinson's Disease Genetics Study Group A multidisciplinary study of patients with early-onset PD with and without parkin mutations. Neurology. 2009;72:110–116. doi: 10.1212/01.wnl.0000327098.86861.d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrett M.T., Scheffer A., Ben-Dor A., Sampas N., Lipson D., Kincaid R., Tsang P., Curry B., Baird K., Meltzer P.S. Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc. Natl. Acad. Sci. USA. 2004;101:17765–17770. doi: 10.1073/pnas.0407979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abeysinghe S.S., Chuzhanova N., Krawczak M., Ball E.V., Cooper D.N. Translocation and gross deletion breakpoints in human inherited disease and cancer I: Nucleotide composition and recombination-associated motifs. Hum. Mutat. 2003;22:229–244. doi: 10.1002/humu.10254. [DOI] [PubMed] [Google Scholar]
- 24.Sarai A., Mazur J., Nussinov R., Jernigan R.L. Sequence dependence of DNA conformational flexibility. Biochemistry. 1989;28:7842–7849. doi: 10.1021/bi00445a046. [DOI] [PubMed] [Google Scholar]
- 25.Furey T.S., Haussler D. Integration of the cytogenetic map with the draft human genome sequence. Hum. Mol. Genet. 2003;12:1037–1044. doi: 10.1093/hmg/ddg113. [DOI] [PubMed] [Google Scholar]
- 26.Guelen L., Pagie L., Brasset E., Meuleman W., Faza M.B., Talhout W., Eussen B.H., de Klein A., Wessels L., de Laat W., van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- 27.Woodfine K., Beare D.M., Ichimura K., Debernardi S., Mungall A.J., Fiegler H., Collins V.P., Carter N.P., Dunham I. Replication timing of human chromosome 6. Cell Cycle. 2005;4:172–176. doi: 10.4161/cc.4.1.1350. [DOI] [PubMed] [Google Scholar]
- 28.Kong A., Gudbjartsson D.F., Sainz J., Jonsdottir G.M., Gudjonsson S.A., Richardsson B., Sigurdardottir S., Barnard J., Hallbeck B., Masson G. A high-resolution recombination map of the human genome. Nat. Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 29.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 30.Hansen R.S., Canfield T.K., Fjeld A.D., Mumm S., Laird C.D., Gartler S.M. A variable domain of delayed replication in FRAXA fragile X chromosomes: X inactivation-like spread of late replication. Proc. Natl. Acad. Sci. USA. 1997;94:4587–4592. doi: 10.1073/pnas.94.9.4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Handt O., Baker E., Dayan S., Gartler S.M., Woollatt E., Richards R.I., Hansen R.S. Analysis of replication timing at the FRA10B and FRA16B fragile site loci. Chromosome Res. 2000;8:677–688. doi: 10.1023/a:1026737203447. [DOI] [PubMed] [Google Scholar]
- 32.Mishmar D., Rahat A., Scherer S.W., Nyakatura G., Hinzmann B., Kohwi Y., Mandel-Gutfroind Y., Lee J.R., Drescher B., Sas D.E. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc. Natl. Acad. Sci. USA. 1998;95:8141–8146. doi: 10.1073/pnas.95.14.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zlotorynski E., Rahat A., Skaug J., Ben-Porat N., Ozeri E., Hershberg R., Levi A., Scherer S.W., Margalit H., Kerem B. Molecular basis for expression of common and rare fragile sites. Mol. Cell. Biol. 2003;23:7143–7151. doi: 10.1128/MCB.23.20.7143-7151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L., Paradee W., Mullins C., Shridhar R., Rosati R., Wilke C.M., Glover T.W., Smith D.I. Aphidicolin-induced FRA3B breakpoints cluster in two distinct regions. Genomics. 1997;41:485–488. doi: 10.1006/geno.1997.4690. [DOI] [PubMed] [Google Scholar]
- 35.Morelli C., Karayianni E., Magnanini C., Mungall A.J., Thorland E., Negrini M., Smith D.I., Barbanti-Brodano G. Cloning and characterization of the common fragile site FRA6F harboring a replicative senescence gene and frequently deleted in human tumors. Oncogene. 2002;21:7266–7276. doi: 10.1038/sj.onc.1205573. [DOI] [PubMed] [Google Scholar]
- 36.El Achkar E., Gerbault-Seureau M., Muleris M., Dutrillaux B., Debatisse M. Premature condensation induces breaks at the interface of early and late replicating chromosome bands bearing common fragile sites. Proc. Natl. Acad. Sci. USA. 2005;102:18069–18074. doi: 10.1073/pnas.0506497102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Denison S.R., Callahan G., Becker N.A., Phillips L.A., Smith D.I. Characterization of FRA6E and its potential role in autosomal recessive juvenile parkinsonism and ovarian cancer. Genes Chromosomes Cancer. 2003;38:40–52. doi: 10.1002/gcc.10236. [DOI] [PubMed] [Google Scholar]
- 38.Arlt M.F., Mulle J.G., Schaibley V.M., Ragland R.L., Durkin S.G., Warren S.T., Glover T.W. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet. 2009;84:339–350. doi: 10.1016/j.ajhg.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lupski J.R. Charcot-Marie-Tooth disease: lessons in genetic mechanisms. Mol. Med. 1998;4:3–11. [PMC free article] [PubMed] [Google Scholar]
- 40.Chen K.S., Manian P., Koeuth T., Potocki L., Zhao Q., Chinault A.C., Lee C.C., Lupski J.R. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat. Genet. 1997;17:154–163. doi: 10.1038/ng1097-154. [DOI] [PubMed] [Google Scholar]
- 41.Lieber M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 42.Schwartz M., Zlotorynski E., Goldberg M., Ozeri E., Rahat A., le Sage C., Chen B.P., Chen D.J., Agami R., Kerem B. Homologous recombination and nonhomologous end-joining repair pathways regulate fragile site stability. Genes Dev. 2005;19:2715–2726. doi: 10.1101/gad.340905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang F., Khajavi M., Connolly A.M., Towne C.F., Batish S.D., Lupski J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lukusa T., Fryns J.P. Human chromosome fragility. Biochim Biophys Acta. 2008;1779:3–16. doi: 10.1016/j.bbagrm.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 45.Takebayashi S., Sugimura K., Saito T., Sato C., Fukushima Y., Taguchi H., Okumura K. Regulation of replication at the R/G chromosomal band boundary and pericentromeric heterochromatin of mammalian cells. Exp. Cell Res. 2005;304:162–174. doi: 10.1016/j.yexcr.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 46.Anachkova B., Djeliova V., Russev G. Nuclear matrix support of DNA replication. J. Cell. Biochem. 2005;96:951–961. doi: 10.1002/jcb.20610. [DOI] [PubMed] [Google Scholar]
- 47.Sutherland G.R., Baker E. The clinical significance of fragile sites on human chromosomes. Clin. Genet. 2000;58:157–161. doi: 10.1034/j.1399-0004.2000.580301.x. [DOI] [PubMed] [Google Scholar]
- 48.Smith D.I., Huang H., Wang L. Common fragile sites and cancer (review) Int. J. Oncol. 1998;12:187–196. [PubMed] [Google Scholar]
- 49.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korbel J.O., Urban A.E., Affourtit J.P., Godwin B., Grubert F., Simons J.F., Kim P.M., Palejev D., Carriero N.J., Du L. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perry G.H., Ben-Dor A., Tsalenko A., Sampas N., Rodriguez-Revenga L., Tran C.W., Scheffer A., Steinfeld I., Tsang P., Yamada N.A. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 2008;82:685–695. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.