

Abstract
Streptococcus pyogenes is an important pathogen that causes a variety of diseases. The most common infections involve the throat (pharyngitis) or skin (impetigo); however, the factors that determine tissue tropism and severity are incompletely understood. The S. pyogenes NAD+ glycohydrolase (SPN) is a virulence factor that has been implicated in contributing to the pathogenesis of severe infections. However, the role of SPN in determining the bacterium's tissue tropism has not been evaluated. In this report, we examine the sequences of spn and its endogenous inhibitor ifs from a worldwide collection of S. pyogenes strains. Analysis of average pairwise nucleotide diversity, average number of nucleotide differences, and ratio of nonsynonymous to synonymous substitutions revealed significant diversity in spn and ifs. Application of established models of molecular evolution shows that SPN is evolving under positive selection and diverging into NAD+ glycohydrolase (NADase)-active and -inactive subtypes. Additionally, the NADase-inactive SPN subtypes maintain the characteristics of a functional gene while ifs becomes a pseudogene. Thus, NADase-inactive SPN continues to evolve under functional constraint. Furthermore, NADase activity did not correlate with invasive disease in our collection but was associated with tissue tropism. The ability to cause infection at both the pharynx and the skin (“generalist” strains) is correlated with NADase-active SPN, while the preference for causing infection at either the throat or the skin (“specialist” strains) is associated with NADase-inactive SPN. These findings suggest that SPN has a NADase-independent function and prompt a reevaluation of the role of SPN in streptococcal pathogenesis.
Many bacterial pathogens that are capable of causing infection at multiple tissue sites have considerable underlying genetic diversity that is reflected by the presence or absence of different subsets of virulence genes or by the presence of alternative alleles of specific virulence genes (37, 44, 48). For the latter genes, variation in sequence may arise under pressure to avoid the immune response or reflect proteins whose functions are diverging. Horizontal gene transfer (HGT) events can initially increase diversity through the reassortment of these variant virulence genes and may result in altered pathogenicity or the ability to more efficiently exploit a given ecological niche (37). Continued selection of fitter variants adapted for infection of a specific niche can then lead to a subsequent purging of genetic diversity and a reduction in the types of clinical syndromes a particular lineage can cause (8). As a consequence, genetically discrete subpopulations with strong tropisms for different tissues emerge within the existing species, and this process may represent a key step in the formation of new species (6). Understanding the changes that occur during niche specialization can provide important insights into pathogenic mechanisms required for infection of a specific tissue.
Analysis of tissue-specific adaptation is emerging as an important approach for understanding the pathogenesis of the numerous diseases caused by Streptococcus pyogenes (group A streptococcus [GAS]). This Gram-positive bacterium has a worldwide distribution and is a pathogen of humans exclusively, causing important diseases, which include those that are destructive of tissue and life-threatening (cellulitis, necrotizing fasciitis) and those associated with deregulation of immunity (glomerulonephritis, rheumatic fever) (6, 12). However, most cases of S. pyogenes disease are more superficial and self-limiting and occur at either the throat (pharyngitis) or the skin (impetigo). These two tissue sites also represent the primary reservoirs responsible for dissemination of the organism to new hosts. A large body of epidemiological evidence that suggests that there are distinct subpopulations of strains more adapted for infection of either the throat or the skin has accumulated, suggesting that specific adaptations to these two tissues are driving the evolution of its pan-genome (6). However, the specific adaptations responsible for niche specialization are not well understood.
A frequently used approach for uncovering a common molecular basis behind bacterial phenotype has been to group strains based on sequence variation in housekeeping genes (18). In the case of niche specialization, continued selection for variants more highly adapted to a particular tissue will purge neutral gene diversity in the adapted population relative to the population as a whole. However, a complication in deciphering trends associated with tissue adaptation in S. pyogenes has been that despite some niche separation, there are high rates of recombination relative to mutation within the species as a whole, on par with that of Streptococcus pneumoniae, a species considered to be highly recombinogenic (6, 22, 57). Frequent recombination has resulted in a random segregation of neutral housekeeping haplotypes between S. pyogenes strains from ecologically distinct subpopulations (6). Thus, standard approaches to establishing relationships between strains have been of only limited utility for understanding niche adaptation for S. pyogenes.
A more productive approach for S. pyogenes has been to look for genetic variation outside neutral housekeeping genes that is strongly associated with ecological niche. In this regard, genotypes based on the gene encoding the M protein (emm) provide a significant correlation with tissue tropism (6). The M protein is a fibrillar surface molecule that plays multiple roles in promoting virulence, and serological typing based on M protein diversity has been the traditional method for classifying S. pyogenes strains (35). It is well established that strains with certain M types have a strong preference for infection at either the throat or the skin (9, 40). There are more than 200 known M types (50), which can be divided into 4 major subfamilies based on the sequence of the peptidoglycan-spanning domain at the 3′ end of emm (25). Furthermore, the emm locus can encode one gene or a combination of subfamily genes in a tandem arrangement (7). Analyses of large strain collections have revealed that in ∼99% of strains, the organization of emm genes in the locus can be assigned to one of five patterns (designated A to E) (6). Although strains with each emm pattern may colonize the same tissue types, there is a strong correlation between emm pattern and the ability of the organism to cause disease at specific tissue sites. Strains with emm patterns A to C generally cause pharyngitis; emm pattern D strains are typically the cause of skin diseases, such as impetigo; and emm pattern E strains are “generalists,” which can cause symptomatic infection at either tissue site at approximately equal fractions of the total (6). Since emm pattern is strongly associated with tissue tropism, it is likely that characteristics consistently coinherited with the emm pattern also play a role in determining the tissue tropism of the organism (6, 29).
The S. pyogenes NAD+ glycohydrolase (SPN, also known as Nga) is a virulence factor with characteristics that merit evaluation for a possible role in tissue tropism. This secreted toxin has an enzymatic activity (NADase) that cleaves the glycosidic bond of β-NAD+ to produce nicotinamide and ADP-ribose. All S. pyogenes strains examined to date possess the gene that encodes SPN (spn), but some strains produce a SPN that lacks detectable NADase activity (1, 30, 36, 42). Since there is evidence that SPN′s robust NADase activity contributes to virulence (4, 43, 52, 56), the existence of NADase-deficient SPN has yet to be explained. Epidemiological studies conducted on several limited strain collections have not been informative, as these studies have both found (1, 52) and failed to find (15) an association between NADase activity and whether a lineage has the capacity to cause invasive disease. Whether or not SPN is associated with tissue tropism is not known.
SPN also has multiple complex interactions with other proteins that suggest it has an important, yet incompletely understood role in disease pathogenesis. These interactions also imply that SPN is under considerable coevolutionary pressure with its partners (47). For example, the ability of S. pyogenes to produce NADase-active SPN is absolutely dependent on the presence of an endogenous inhibitor protein, immunity factor for SPN (IFS) (31, 42). IFS is a competitive inhibitor of SPN′s β-NAD+ substrate and apparently acts to inhibit self-toxicity resulting from any presecretory SPN molecules that adventitiously fold prior to their export from the streptococcal cell. In the absence of IFS, SPN is lethal for S. pyogenes. Interestingly, strains that produce NADase-inactive SPN also have a truncated form of IFS (42). Once secreted, both NADase-active SPN and NADase-inactive SPN are injected into the host cell cytoplasm by a process known as cytolysin-mediated translocation (CMT), which requires interaction between multiple domains of SPN and the pore-forming cytolysin streptolysin O (SLO) (11, 20, 39, 41). When in the cytoplasmic compartment, NADase-active SPN can trigger rapid cell death, which is associated with depletion of β-NAD+ pools (10, 11, 39). The genes for SPN (spn), IFS (ifs), and SLO (slo) are encoded in the same operon (31, 42), as is typical of coevolving virulence factor/inhibitor pairs (47). Thus, SPN has multiple complex interactions and is suspected of being important in pathogenesis; however, there is a considerable amount of genetic and functional variation that has yet to be fully defined.
In the present study, we sought to clarify the role of SPN in the infectious process through analysis of the genetic diversity in spn and ifs and the relationship this diversity has with disease severity and ecologic niche. By examining a diverse, worldwide collection of S. pyogenes strains, we identify the SPN domains evolving under positive (diversifying) and negative (purifying) selection, correlate these sites with NADase activity, and demonstrate that NADase activity is associated with tissue tropism but not invasiveness of disease.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and determination of NADase activity.
Isolates from previously described S. pyogenes strain collections (17, 40) that were selected for this study are listed in Table S1 in the supplemental material. This collection contains 113 genetically diverse, worldwide isolates. The well-studied strains JRS4 (49) and HSC5 (14, 23) were used as a reference since they are known to express SPN that is NADase active and SPN that is NADase inactive, respectively (42). Strains were also collected from the St. Louis Children's Hospital clinical microbiology laboratory (Washington University Human Resources Protection Office approval 08-0236) after isolation from the pharynx, skin, or a body site that is expected to be sterile. These strains were grown in Todd-Hewitt medium (BBL) supplemented with 0.2% yeast extract (BD Biosciences, San Diego, CA) (THY medium). When indicated for NADase activity assays, streptococcal pyogenic exotoxin B (SpeB) cysteine protease activity was inhibited by the addition of E-64 (Sigma, St. Louis, MO) to the culture media at a final concentration of 28 μM. Bacteria were grown to stationary phase in standard culture tubes at 37°C. NADase activity was determined for clinical and reference strains by endpoint titer as previously described (39).
Isolation of chromosomal DNA, PCR, and sequencing.
Chromosomal DNA was isolated from S. pyogenes strains as previously described (24). The spn and ifs genes were amplified by PCR using the following oligonucleotide primer pairs: SPN1 (5′-GAT CTA TTA CTG ATA ACG GTG CTA C-3′) with SPN5 (5′-GAA GCT CCG CTT TCT TTG T-3′), and SPN4 (5′-CAG ATG TCT GCT GTT GCG TCA CG-3′) with IFS2 (5′-TCA TTT GTC GTT GTG GTT TCT GTA-3′). The amplified DNA fragments were purified using a commercial PCR purification kit (Qiagen) according to the manufacturer's recommendations except that sterile water was used to elute the PCR product from the column. Sequencing reactions were performed with each of the oligonucleotide primers that were used for PCR. Additional sequencing reactions were also performed with supplementary oligonucleotide primers so that the complete spn and ifs genes were sequenced in roughly 600-bp overlapping increments along both DNA strands. Primers SPN2 (5′-GCA CAC ATT AGA CGG CTC AAT GAG-3′) and SPN3 (5′-CCC TGA TGG ACC TCT GTT ACC TCA A-3′) were used to determine further sequence from the PCR fragment created by SPN1 and SPN5. Primers SPN6 (5′-CTT CTT CGA TGT TAG CTT TCA ATT G-3′) and IFS1 (5′-GCC AAA GGG TTT AGA ACA TTA CC-3′) were used to determine further sequence from the PCR fragment created by SPN4 and IFS2. Single-pass sequencing reactions were performed on purified PCR products through a genomic services contract research organization (SeqWright, Houston, TX).
Sequence analysis.
Individual overlapping sequence fragments were aligned using DNASTAR Lasergene 8 (DNASTAR Inc., Madison, WI) in order to reassemble the complete spn and ifs gene sequences for each strain. The complete spn and ifs sequences were aligned with the ClustalW algorithm using Molecular Evolutionary Genetics Analysis (MEGA, v4) (34, 55). GenBank accession numbers for the spn and ifs sequences are provided in Table S2 in the supplemental material. Since the spn and ifs sequences obtained from strains MGAS5005, MGAS2096, and MGAS6180 were identical to those found in previously published whole-genome studies, Table S2 contains the GenBank accession numbers for the appropriate regions within these previously published whole-genome sequences (3, 21, 53). Sequences of the internal fragments of the seven housekeeping genes for glucose kinase (gki), glutamine transport ATP-binding protein (gtr), glutamine racemase (murL), DNA mismatch repair (mutS), transketolase (recP), xanthine phosphoribosyltransferase (xpt), and acetoacetyl-coenzyme A (CoA) thiolase (yqiL) were published previously (17).
Measures of gene polymorphism.
The average number of nucleotide differences per nucleotide site (θ), maximum percent divergence, average pairwise nucleotide diversity (π), average pairwise nucleotide diversity for nonsynonymous (πa) and synonymous (πs) polymorphisms, ratio of nonsynonymous to synonymous nucleotide polymorphisms (πa/πs), ratio of the rate of nonsynonymous to synonymous nucleotide polymorphisms (Ka/Ks), and haplotype diversity (Hd) were calculated as measures of genetic diversity. The average number of nucleotide differences per nucleotide site (θ) was calculated using equation 10.3 (45) except 2N was used instead of 4N since S. pyogenes is a haploid organism. The average pairwise nucleotide diversity (π) was calculated using equation 10.5 (45), and the sampling variance was calculated using equation 10.7 (45). The estimation of nucleotide diversity for nonsynonymous (πa) and synonymous (πs) sites for the calculation of πa/πs and the Ka/Ks calculation were performed using methods as previously described (46). The window method for determining πa/πs throughout spn was performed using a window size of 50 bp and a 5-bp step size. All measures except maximum percent divergence were calculated using DnaSP v5 (38). The maximum percent divergence is the number of polymorphic nucleotide sites between the two most dissimilar sequences in the sample population divided by the length of the gene and was calculated using MegAlign in DNASTAR Lasergene 8 (DNASTAR Inc., Madison, WI) after a ClustalW alignment.
Measures of positive selection and Bayesian clustering.
The Hudson, Kreitman, and Aguadé (HKA) test (26) was performed using DnaSP v5 (38). Homologous genes from the published whole-genome sequence of Streptococcus dysgalactiae subsp. equisimilis were used for the outgroup comparison in the HKA test (GenBank accession number, NC_012891; spn nucleotide position, 1964215 to 1965567; gki nucleotide position, 1473212 to 1474183; gtr nucleotide position, 1449866 to 1450600; murL nucleotide position, 389421 to 390218; mutS nucleotide position, 30558 to 2033113; recP nucleotide position, 1666839 to 1669028; and xpt nucleotide position, 882803 to 883384). The calculation of the ratio of the rate of nonsynonymous to synonymous polymorphisms (Ka/Ks) for each codon position was performed using an Internet-based resource (Selecton server at http://selecton.tau.ac.il/) (51) so that specific amino acids undergoing positive or purifying selection could be identified. This resource enables detection of selection through the use of the M8 (58) and M8a (54) evolutionary models. The M8 and M8a models are similar except the M8a model specifies that the distribution of the nonsynonymous/synonymous rate ratio (Ka/Ks) follows a mixture between a beta-distribution and 1, while the M8 model allows Ka/Ks to be greater than 1. The statistical significance for positive selection was evaluated through a goodness-of-fit approach that compares the likelihood ratios of these two models (54). If the likelihood ratio test is found to significantly favor the M8 model, codon sites with Ka/Ks of >1 are identified as evolving under positive selection while codon sites with Ka/Ks of <0.075 are considered to be evolving under purifying selection. Confidence intervals for the Ka/Ks values for each codon are calculated from the posterior distribution. Bayesian clustering of the spn alleles was performed using Bayesian analysis of population structure (BAPS, v5.2) (13). The sequence data were analyzed using the “clustering with linked loci” option in order to perform a genetic mixture analysis for the 113 spn sequences, with the maximum number of populations set to 20. The chi-square test was used to determine if there was a significant association between NADase activity or spn allele cluster and invasiveness, disease category, or emm pattern.
RESULTS
Determination of spn and ifs DNA sequences from a diverse collection of S. pyogenes strains.
Several studies have suggested that heterogeneity in spn and ifs may correlate with specific streptococcal disease processes (10, 52, 56). However, these studies have been based on analysis of strain collections of limited size or diversity and may not have captured the full extent of variation in these genes. Thus, establishing unambiguous correlations requires a more comprehensive analysis of diversity. Furthermore, through the identification of sites in spn and ifs that are evolving under positive and purifying selection and the possible association of these sites with known functional domains, it may be possible to understand how the activities of SPN and IFS are evolving to contribute to different pathogenic processes and/or adapt to fill different ecologic niches. To conduct this analysis, we examined spn and ifs in a well-characterized collection of 113 S. pyogenes strains that was assembled to reflect a high degree of diversity with regard to time (the collection spans 67 years), geography (all continents are represented except Antarctica), and serotype (110 different M types are represented in the sample) (17, 40). In addition, the tissue infected and the disease type caused are documented for 93 of the strains (40), and the sequences of internal fragments from 7 distinct housekeeping genes have been determined for all 113 strains (17, 40). For the present study, the spn and ifs alleles from each of these 113 strains were amplified by PCR and their sequences determined for the regions spanning from the initiation to the termination codon (see Table S2 in the supplemental material).
SPN has a different pattern of polymorphisms than S. pyogenes housekeeping genes.
The spn alleles isolated from our sample population exhibited substantial diversity. In total, the sample population contained 74 unique spn haplotypes. Most of these haplotypes differ due to single nucleotide polymorphisms (SNPs), but indels also occur, none of which induce a frameshift. The spn allele of strain 3850-01 contained an internal duplication of 27 bp within the gene, and 13 other strains contain small terminal duplications or deletions that extend or truncate the coding sequence by 12 bp at the 3′ end. Due to the indels, spn ranges in size from 1,344 bp to 1,383 bp (447 to 460 amino acid residues). The size variation of spn due to indels is intriguing, as this characteristic distinguishes spn from the known housekeeping gene sequences used in this study, which lack indels.
To evaluate the nucleotide variation in spn, the average number of nucleotide differences per nucleotide site (θ), maximum percent sequence divergence, and average pairwise nucleotide diversity (π) were calculated. Average pairwise nucleotide diversity is a measure of the average number of nucleotide differences per nucleotide site between two randomly chosen DNA sequences. These measures evaluate only SNPs and are not affected by indels (45) or by the length of the gene being studied. Thus, they can be compared reliably between different genes. The θ, maximum percent sequence divergence, and π of spn in this sample were similar to the values for the seven concatenated S. pyogenes housekeeping genes that are used for multilocus sequence typing (MLST) (Table 1). This finding indicates that the degree of polymorphism in spn is roughly equivalent to that for the S. pyogenes housekeeping genes at the nucleotide level.
TABLE 1.
Measures of sequence diversity in the collection of 113 S. pyogenes strains
| Gene(s) and strains | θb | Maximum % divergencec | πd | πae | πsf | πa/πsg | Ka/Ksh | Hdi |
|---|---|---|---|---|---|---|---|---|
| Housekeeping genesa | ||||||||
| All strains | 0.0076 | 2.0 | 0.0074 ± 0.00023 | 0.0025 | 0.023 | 0.105 | 0.047 | 0.999 ± 0.001 |
| NADase-active strains | 0.0062 | 1.9 | 0.0072 ± 0.00032 | 0.0024 | 0.023 | 0.102 | 0.047 | 0.998 ± 0.004 |
| NADase-inactive strains | 0.0057 | 1.7 | 0.0075 ± 0.00033 | 0.0026 | 0.023 | 0.108 | 0.047 | 0.999 ± 0.004 |
| spn | ||||||||
| All strains | 0.0068 | 2.9 | 0.0079 ± 0.004 | 0.0045 | 0.019 | 0.256 | 0.23 | 0.987 ± 0.004 |
| NADase-active strains | 0.0059 | 2.7 | 0.0097 ± 0.00051 | 0.0052 | 0.026 | 0.202 | 0.209 | 0.977 ± 0.01 |
| NADase-inactive strains | 0.0042 | 1.6 | 0.0075 ± 0.00037 | 0.0032 | 0.012 | 0.256 | 0.294 | 0.981 ± 0.008 |
| ifs | ||||||||
| All strains | 0.0099 | 2.5 | 0.0094 ± 0.00038 | 0.0090 | 0.010 | 0.906 | 0.644 | 0.961 ± 0.006 |
| NADase-active strains | 0.0056 | 2.1 | 0.0069 ± 0.00055 | 0.0051 | 0.013 | 0.386 | 0.444 | 0.908 ± 0.02 |
| NADase-inactive strains | 0.0074 | 1.9 | 0.0060 ± 0.00065 | 0.0059 | 0.004 | 1.314 | 0.838 | 0.923 ± 0.019 |
Housekeeping genes include the concatenated sequences from the internal fragments of gki, gtr, murL, mutS, recP, xpt, and yqiL.
θ is the average number of nucleotide differences per nucleotide site (calculated using DnaSP v5.0).
The maximum percent divergence is the number of polymorphic nucleotide sites between the two most dissimilar sequences in the population divided by the length of the gene (calculated using MegAlign in DNASTAR Lasergene 8 after a ClustalW alignment).
π is the average pairwise nucleotide diversity (calculated using DnaSP v5.0).
πa is the average pairwise nucleotide diversity leading to nonsynonymous mutations (calculated using DnaSP v5.0).
πs is the average pairwise nucleotide diversity leading to synonymous mutations (calculated using DnaSP v5.0).
πa/πs is the ratio of nonsynonymous to synonymous mutations (calculated using DnaSP v5.0).
Ka/Ks is the ratio of the rate of nonsynonymous to synonymous mutations (calculated using DnaSP v5.0).
Haplotype diversity (Hd) is the probability that two sequences selected at random from the sample population will be different (calculated using DnaSP v5.0).
In order to evaluate the effect of the nucleotide diversity on the encoded protein, the ratio of nonsynonymous to synonymous nucleotide polymorphisms (πa/πs) was calculated (45). The πa/πs ratio is also a measure of diversity at the nucleotide level, but it indicates the relative number of polymorphisms that cause an alteration in the amino acid sequence. Genes that are evolving with no functional constraint will randomly accumulate nucleotide polymorphisms, and the ratio of nonsynonymous to synonymous polymorphisms (πa/πs) is expected to be roughly 1 in this scenario (16). In contrast, genes that are evolving under purifying selection with few nonsynonymous mutations will have a low πa/πs ratio, and genes evolving under positive selection will have a πa/πs ratio of >1. Previous analysis of 12 published S. pyogenes genome sequences revealed that the overall πa/πs ratio for 875 genes is 0.139 (27). The πa/πs for the 7 concatenated housekeeping genes is 0.105, a ratio that is consistent with genes undergoing purifying selection. Despite the finding that θ and π for spn are similar to those for other S. pyogenes genes, the ratio of nonsynonymous to synonymous polymorphisms (πa/πs) for spn is 0.256, a ratio that is nearly 2× greater than the ratio for the 875 S. pyogenes genes considered together and 2.5× greater than that for the housekeeping genes examined in this study (Table 1). This indicates either that spn is generally evolving under purifying selection but may be under less functional constraint than the housekeeping genes or that portions of the gene may be evolving under positive selection.
This diversity at the amino acid level can be illustrated when the cumulative numbers of nonsynonymous and synonymous nucleotide polymorphisms are compared against the codon position of the encoded protein for each gene. For this comparison, every synonymous or nonsynonymous polymorphism that occurs in the gene of interest from any one of the 113 strains is counted. In two representative housekeeping genes, the glucose kinase (gki) (Fig. 1 A) and glutamine transport ATP-binding protein (gtr) (Fig. 1B) genes, the cumulative number of synonymous mutations is greater than the cumulative number of nonsynonymous mutations across the entire gene. This pattern indicates that the amino acid sequence remains relatively preserved in these two genes in the sample population. In contrast, the cumulative number of nonsynonymous polymorphisms approximates the cumulative number of synonymous polymorphisms throughout the length of the gene when all 113 spn sequences are compared (Fig. 1C). This indicates that the diversity in spn is leading to more variation in the SPN amino acid sequence than in the S. pyogenes housekeeping proteins.
FIG. 1.

spn has a higher ratio of nonsynonymous to synonymous polymorphisms than housekeeping genes. The cumulative numbers of nonsynonymous (Non) and synonymous (Syn) substitutions per codon site are presented for the internal fragments of two representative S. pyogenes housekeeping genes, gki and gtr (A and B), and spn (C). The y axes show the cumulative numbers of substitutions seen in all of the 113 S. pyogenes strains studied, so that if 57 of the alleles contain an adenine at a specific site and the other 56 contain a thymine, 56 is added to the cumulative total. The sliding-window method was used to measure πa/πs throughout spn (D). Areas of the gene undergoing purifying selection have a πa/πs of <1, and areas undergoing positive selection have a πa/πs of >1.
Portions of SPN are undergoing diversifying selection.
Since spn has a higher ratio of nonsynonymous to synonymous polymorphisms than the S. pyogenes housekeeping genes, it was important to determine the cause of this increase. Multiple evolutionary or historical events can result in a relative increase in the proportion of nonsynonymous mutations, such as that observed for spn. If the number of nonsynonymous mutations in spn significantly deviates from a random pattern, it might be evolving under selection. In order to further define the pattern of polymorphism within spn, the sliding-window method was used to calculate the ratio of nonsynonymous to synonymous polymorphisms (πa/πs) in 50-bp overlapping segments throughout the length of the gene. This method allows visualization of specific areas within spn that are diversifying (Fig. 1D) and illustrates that the polymorphisms leading to differences in the amino acid sequence are concentrated in several distinct regions rather than distributed randomly throughout the gene.
Given that the pattern seen in the πa/πs sliding-window test was consistent with portions of spn evolving under diversifying selection, the data were analyzed with the application of neutrality testing in order to determine if there was statistically significant evidence to support that selection is occurring. Evidence for diversifying selection was evaluated by the Hudson, Kreitman, and Aguadé (HKA) test, because it is a powerful measure of selection but still considered conservative in the setting of recombination (26, 59). The HKA test reveals that spn has a significantly greater number of intraspecific observed versus expected segregating (polymorphic) sites (102 observed versus 68 expected) than the concatenated housekeeping gene sequences (222 observed versus 256 expected). Homologous sequences were selected from the whole-genome sequence of Streptococcus dysgalactiae subsp. equisimilis (strain GGS124) and used as the outgroup. This outgroup comparison reveals an inverse pattern in the interspecific divergence, with 29 observed versus 63 expected differences in spn and 271 observed versus 237 expected differences in the housekeeping gene sequences. The greater degree of intraspecific versus interspecific variation in spn than in the housekeeping genes is statistically significant (P = 0.0023 by chi-square test) and indicates that spn is likely under the influence of diversifying selection.
The degree of polymorphism in IFS is greater than that in SPN.
As the evidence indicated that spn alleles were experiencing diversifying selection, it was also important to evaluate the evolutionary forces operating on its cytoplasmic inhibitor partner ifs. It is expected that spn and ifs experience similar evolutionary forces, given that they are a virulence factor-inhibitor pair that are closely linked in one operon (Fig. 2 A) and should thus coevolve. However, after comparing the sequences of the 113 different ifs alleles, it became evident that ifs has a different evolutionary pattern than spn.
FIG. 2.

ifs is positioned between spn and slo on the bacterial chromosome and has multiple truncated variants. The genetic organization of the spn-slo operon is presented (A). A multiple alignment of ifs genes from several S. pyogenes strains is shown (B). The ifs sequence from the well-studied reference strain JRS4 is provided as an example of the full-length protein. Strain MGAS2109 has a truncation at amino acid site 24 secondary to the substitution of an adenine for a thymine at bp 71. Strain MGAS2111 has a truncation at amino acid site 24 secondary to the deletion of a thymine at bp 71. Strain SS116 has a truncation at amino acid site 19 secondary to the deletion of an adenine at bp 40. Strain 22RS72 has a truncation at amino acid site 24 secondary to the substitution of an adenine for a thymine at bp 71, similarly to MGAS2109, but also contains another truncation in the second open reading frame that begins at codon site 44. Polymorphic amino acid sites are shaded in black. An asterisk (*) indicates a stop codon. The designations of ifs as full length, truncated 1, truncated 2, or truncated 3 correspond to the ifs allele in Table S1 in the supplemental material.
There is significant variation in ifs at the allelic level in the sample population. Fifty-two percent of ifs genes in the sample population are 486 bp in length (161 amino acids) (Fig. 2B, JRS4). The remaining 55 ifs alleles are truncated due to indels of single nucleotides and SNPs that lead to nonsense mutations. The most common polymorphism resulting in ifs truncation is the presence of an adenine instead of a thymine at bp 71, which causes a nonsense mutation at codon 24 in 62% of the shortened alleles (Fig. 2B, MGAS2109). The deletion of a thymine at bp 71 occurs in 35% of the truncated genes and also results in a nonsense mutation at codon 24 (Fig. 2B, MGAS2111). The remaining two truncated ifs alleles have a deletion of an adenine at bp 40, which triggers a frameshift resulting in a stop codon at amino acid position 19 (Fig. 2B, SS116). All truncated ifs alleles contain a potential second start codon after the nonsense mutation (corresponding to codon site 44 of the full-length protein), which results in another open reading frame (ORF). This second ORF is potentially capable of coding for a protein 117 amino acids in length (Fig. 2B, MGAS2109); however, it is uncertain if either of the ORFs is expressed in these truncated alleles. Three of the truncated alleles also contain further polymorphisms within the second ORF that would produce additional stop codons (Fig. 2B, 22RS72). All truncated ifs alleles are incapable of inhibiting the NADase activity of SPN (42).
In order to quantify and compare the levels of diversity in ifs, we again calculated the average nucleotide differences per nucleotide site (θ), average pairwise nucleotide diversity (π), and ratio of nonsynonymous to synonymous nucleotide polymorphisms (πa/πs) (Table 1) (45). For this analysis, a 486-nucleotide (nt) region corresponding to the wild-type length of the ifs gene was used from all haplotypes, and stop codons were ignored. The overall θ, maximum percent divergence, and nucleotide diversity were not significantly different from those of spn (Table 1). However, ifs had a πa/πs of 0.906, which is much higher than that of spn and approaches a value consistent with evolution in the absence of functional constraint. When the πa/πs for the full-length ifs genes is evaluated separately and compared to that of the truncated genes (0.407 and 1.245, respectively), a pattern of greater amino acid sequence degradation is seen in the truncated ifs genes (Fig. 3). This finding suggests that the truncated variants of ifs are undergoing random nucleotide change, as expected for a pseudogene, while the full-length versions of ifs have comparatively little amino acid sequence variation.
FIG. 3.
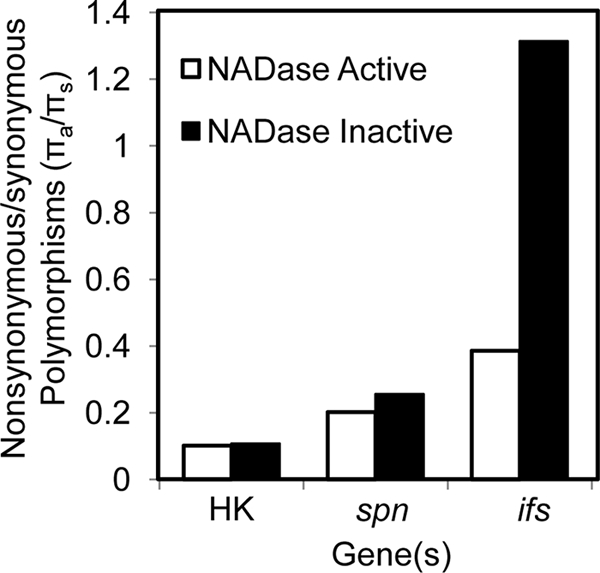
The ratio of nonsynonymous to synonymous nucleotide polymorphisms (πa/πs) reveals different patterns of evolution for NADase-active and NADase-inactive strains. The ratios of nonsynonymous to synonymous polymorphisms (πa/πs) of the concatenated internal fragments of the seven housekeeping genes (HK), spn, and ifs are presented for comparison. Strains with NADase activity and strains without NADase activity are compared. Only ifs from the NADase-inactive strains has a ratio consistent with a nonfunctional gene, while spn remains relatively conserved despite the loss of NADase activity.
IFS polymorphism provides information about the function of SPN.
Given that the evolutionary patterns of spn and ifs revealed an unexpected disassociation and that 48.6% of ifs alleles were severely truncated, we sought to further understand the coevolutionary relationship. Since only full-length IFS would be expected to be an effective inhibitor of NADase function, the pairing of truncated IFS with functional NADase would be fatal to the bacterial cell (42). Therefore, all spn sequences that were paired with truncated ifs were examined for polymorphisms that may result in loss of NADase activity. A single nucleotide polymorphism in spn that results in the presence of an aspartic acid instead of a glycine at amino acid residue 330 (G330D) was found in all of the alleles that are paired with the 55 ifs sequences that encode a truncated protein product. This finding is consistent with previous smaller studies that have associated the G330D polymorphism with loss of SPN NADase activity (42, 56).
In an effort to provide further evidence that the G330D polymorphism and truncated IFS are accurate predictors of NADase inactivity, we evaluated clinical S. pyogenes strains from the local patient population. S. pyogenes strains were obtained from patients presenting to St. Louis Children's Hospital, a large Midwestern metropolitan pediatric tertiary care center. NADase functional activity and spn and ifs nucleotide sequences were obtained from 36 of the S. pyogenes strains. There were 31 strains with the spn and ifs alleles that predicted NADase activity and 5 strains predicted to lack NADase activity. NADase activity was detected in all of the strains containing a glycine at residue 330. In contrast, no NADase activity was detected in strains with the G330D polymorphism. These data add further support for the conclusion that the amino acid residue at position 330 is a reliable predictor of NADase activity.
SPN amino acid sequence remains preserved despite loss of NADase activity.
The above-described findings, in conjunction with previous studies (42, 56), provide evidence that SPN exists in two functionally distinct subtypes, one with NADase activity and one without. Since ifs appears to have evolved without functional constraint and degraded into a pseudogene in strains without NADase activity, the 113 spn sequences were reexamined in order to evaluate whether loss of NADase activity resulted in the same level of sequence degradation. The average nucleotide differences per nucleotide site (θ), maximum percent divergence, and nucleotide diversity (π) were all higher in the NADase-active group than in the NADase-inactive group (Table 1), indicating greater diversity in the spn alleles from the NADase-active strains. The ratios of nonsynonymous to synonymous polymorphisms are similar between the NADase-active and -inactive strains for the housekeeping genes and slightly higher for spn from NADase-inactive strains (Fig. 3). In contrast, the ratio of nonsynonymous to synonymous polymorphisms for ifs is substantially elevated in the NADase-inactive strains (Fig. 3). Thus, while there is a modest increase in the proportion of amino acid-altering polymorphisms occurring in the NADase-inactive spn alleles, it is substantially less than that occurring in the ifs alleles from the same strains. This result indicates that while ifs loses selective constraint and transitions to a pseudogene in the absence of NADase activity, the same degradation does not occur for NADase-inactive spn.
Analysis at the protein level reveals specific residues evolving under selection.
Since the NADase-inactive SPN sequences appeared relatively preserved despite loss of enzymatic function, this suggests that other areas of the protein may have important functions. In order to address this hypothesis, we sought to identify the areas of the protein that are undergoing positive (diversifying) and negative (purifying) selection in an effort to further define these significant domains. SPN has been studied most extensively in the serotype M6 strain JRS4 and has been shown to be a multidomain virulence factor, with the mature protein consisting of residues 38 to 447 (20, 42). Amino-terminal residues up to position 195 form the “jelly roll” domain, which is involved in translocation into host cells and shares homology with certain carbohydrate-binding proteins, while the remainder of the mature protein forms the NADase domain (20) (Fig. 4 A). To test for selection at the protein level, the spn sequences were evaluated using two evolutionary models in which the ratio of the rate of nonsynonymous (Ka) to synonymous (Ks) polymorphisms is calculated for each codon site. The M8 and M8a evolutionary models were chosen since the M8 model of selection allows for positive, neutral, and purifying selection and permits nesting of the M8a null model, which does not allow for positive selection. Both models are also more reliable than alternatives in the setting of frequent recombination (2). Analysis of spn sequences using the M8 model, allowing positive selection (58), revealed a log likelihood value of −3,462.6, while application of the M8a model, not allowing for positive selection (54), resulted in a lower log likelihood value of −3,484.13, indicating that the M8 model was a more appropriate fit for the data. The likelihood ratio test was significant (P = 0.001), and this finding supports the hypothesis that SPN is evolving under positive selection.
FIG. 4.

Amino acid residues under positive and purifying selection in SPN. The different domains of SPN are shown (A). The first 37 amino acids form the signal sequence (SS, white) and are absent from the mature protein, amino acids 38 to 194 make up the translocation domain (gray), and the remaining 253 amino acids form the NADase domain (black). The amino acid sequence of SPN is shown (B). This sequence is a consensus sequence generated from the alignment of all 113 SPN sequences from this sample population. The residues under selection as determined by the ratio of the rate of nonsynonymous to synonymous substitutions (Ka/Ks) at each site are indicated. Amino acid residue 330 is indicated by a filled star. Catalytic residue 391 is indicated by an open star.
The ratio of the rate of nonsynonymous (Ka) to synonymous (Ks) polymorphisms for each codon site reveals specific residues that are evolving under selection. High Ka/Ks ratios (>1) indicate that positive selection is occurring at that codon site, and low Ka/Ks ratios are indicative of purifying selection (Fig. 4B). Overall, there were 23 amino acid sites identified as evolving under positive selection, while 332 residues demonstrated sufficiently low Ka/Ks ratios to predict that purifying selection is occurring (Fig. 4B). Residues identified as undergoing positive selection occur in both the translocation and the NADase domain and include the functionally important amino acid residue 330 (Fig. 4B).
In addition to the functionally significant positive selection occurring at residue 330 (G330D polymorphism), two other amino acid sites that are important for NADase activity are under positive selection. Alteration of the arginine at position 289 or the isoleucine at position 374 is also associated with a detectable reduction in NADase activity (S. Chandrasekaran, J. Ghosh, and M. G. Caparon, personal communication). All three of these amino acid residues are located in the substrate binding pocket, based on the recently described SPN crystal structure (C. L. Smith, J. Ghosh, J. S. Elam, J. S. Pinkner, S. J. Hultgren, M. G. Caparon, and T. Ellenberger, submitted for publication). Thus, although several residues affecting NADase function are under positive selection, the majority of SPN remains under purifying selection, as indicated by low Ka/Ks ratios. Of note, the glutamic acid at position 391 is one of these amino acids that remains strongly conserved, even when NADase activity is lost through the G330D polymorphism. This finding is intriguing since position 391 has been determined to be the catalytic residue, based on homology with other NADases (19). The large proportion of amino acids with significantly low Ka/Ks ratios indicates that there is continued overall structural preservation in SPN. Similar patterns are seen when NADase-active SPN and NADase-inactive SPN are examined separately (data not shown), which indicates that the protein is generally conserved despite loss of NADase function and supports the earlier conclusions based on analysis at the nucleotide level.
SPN alleles have specific patterns of divergence.
Since the data indicate that SPN exists in two functionally distinct subtypes (NADase active and inactive), it was important to further explore the possibility of there being additional, yet unknown subtypes. Thus, Bayesian analysis of population structure (BAPS) was used to identify haplotype clusters based on the frequencies of SNPs (13). The 74 S. pyogenes spn haplotypes group into 4 Bayesian clusters. The polymorphic amino acid residues that segregate with spn haplotype cluster were identified (Table 2). These segregating amino acid sites are from both the translocation and the NADase domain and represent 10 of the 23 sites identified as evolving under positive selection. The remaining 13 amino acid sites identified as evolving under positive selection did not segregate with the spn haplotype clusters. Clusters 1 and 2 consist of the NADase-positive alleles, while clusters 3 and 4 consist of the NADase-negative alleles. This finding indicates that in addition to loss of NADase function through the G330D polymorphism, SPN is diverging in other ways due to selection at codons positioned throughout the gene.
TABLE 2.
Polymorphic amino acid residues that segregate into spn Bayesian clusters
| Bayesian clustera | NADase activityb | SPN amino acidc |
IFSe | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Translocation domaind |
NADase domaind |
|||||||||||
| 103 | 136 | 143 | 195 | 199 | 232 | 280 | 289 | 330 | 374 | |||
| 1 | + | H | G | H | I | I | I | L | R | G | I | Full length |
| 2 | + | R | G | Q | M | L | V | L | R | G | I | Full length |
| 3 | − | H | R | Q | I | I | V | V | K | D | V | Truncated |
| 4 | − | R | G | Q | M | L | V | L | N | D | I | Truncated |
Bayesian clusters were identified through Bayesian analysis of population structure (BAPS).
NADase activity is predicted based on the presence of a glycine at residue 330 in active strains (+) or an aspartic acid at residue 330 in inactive strains (−).
Residues are numbered based on the SPN consensus sequence shown in Fig. 4.
SPN domains are illustrated in Fig. 4.
Indicates the ifs allele associated with the Bayesian spn cluster. Examples of full-length form and several truncated forms of IFS are illustrated in Fig. 2. Either the truncated 1 or the truncated 2 form of IFS may be paired with either Bayesian spn cluster 3 or Bayesian spn cluster 4. The two ifs alleles that encode a truncated 3 protein are found in strains with Bayesian spn cluster 4.
SPN haplotype clusters display variable degrees of polymorphism.
To further characterize the haplotype clusters identified by BAPS, we investigated the degree of nucleotide polymorphism in each cluster. The πa/πs ratios for each spn allele cluster were calculated and compared to those of the housekeeping genes from the same strains. For all strains in the sample population, the ratio of nonsynonymous to synonymous polymorphisms is roughly 2.5× higher in spn than in the housekeeping genes (Fig. 5, all clusters). When the NADase-active clusters are considered together, the πa/πs for spn is roughly 2× higher than that of the housekeeping genes (Fig. 5, cluster 1 and cluster 2). Dissecting the NADase-active group into Bayesian clusters 1 and 2 reveals that the πa/πs for cluster 1 approaches that of the housekeeping genes (Fig. 5, cluster 1), while the ratio for cluster 2 remains roughly 2.5× greater than that for the housekeeping genes (Fig. 5, cluster 2). This finding indicates that cluster 1 SPN sequences are relatively conserved. When considered together, the two NADase-inactive clusters have a πa/πs that is roughly 2.2× greater than that of the housekeeping genes (Fig. 5, cluster 3 and cluster 4), again illustrating that there is not a disproportionate number of nonsynonymous polymorphisms occurring in this group as a whole compared to that in the NADase-active groups. Considering the NADase-inactive clusters separately, the πa/πs for cluster 3 is roughly 1.8× greater than that for the housekeeping genes (Fig. 5, cluster 3), and the πa/πs for cluster 4 is roughly 3× greater than that for the housekeeping genes (Fig. 5, cluster 4). This indicates that cluster 4 strains display the highest level of diversifying selection of the Bayesian clusters. None of the Bayesian clusters have a πa/πs ratio that approaches 1, supporting the conclusion that SPN remains subject to functional constraint despite the loss of NADase activity. This is in contrast to the previous finding with the truncated ifs gene products from the NADase-inactive strains in which the πa/πs ratio was consistent with random nucleotide change (comparison not shown).
FIG. 5.

The ratios of nonsynonymous to synonymous nucleotide substitutions (πa/πs) vary among the S. pyogenes spn haplotype clusters. The ratio of nonsynonymous to synonymous substitutions (πa/πs) is plotted for selected strain categories and spn haplotype clusters. The data for the concatenated internal fragments of the seven housekeeping genes (HK) are provided for each group as a reference for comparison to the data for spn.
SPN allele/NADase activity is not associated with invasive streptococcal disease.
Since the spn and ifs sequences appeared to be a very reliable predictor of NADase activity, this information was used to evaluate the importance of NADase activity in disease type. The disease type caused by each strain was available for 93 of the 113 strains examined in this study (see Table S1 in the supplemental material). Diseases were divided into the following categories: all invasive diseases, impetigo, pharyngitis, carrier disease (pharyngeal carriage), and nonsuppurative sequelae. Two wound infections and one superinfection of an eczematous lesion were excluded from the analysis since the pathophysiology behind these infections is distinct from that for impetigo. NADase-active and -inactive strains were represented almost equally in each disease category (Fig. 6). There were no significant associations between NADase activity and invasiveness or disease category (by chi-square test or Fisher's exact test). There was also no significant association between the four Bayesian spn clusters and invasiveness or disease type (data not shown).
FIG. 6.

The distributions of S. pyogenes strains with and without NADase activity are approximately equal for each disease type. The number of strains present in each category is shown on the y axis, with associated disease categories indicated on the x axis. Invasive diseases are defined by the isolation of S. pyogenes from a body site that is expected to be sterile. Carrier disease is defined as the isolation of S. pyogenes from the pharynx of an asymptomatic patient. Nonsuppurative sequelae are rheumatic fever and glomerulonephritis.
SPN allele/NADase activity is associated with tissue tropism.
Since SPN NADase activity did not correlate with invasive disease in this worldwide sample, analysis was focused on identifying an association between NADase activity and tissue tropism (based on emm pattern genotypes). The NADase-active strains were primarily emm pattern E, “generalists” (85.5%), while only 3.6% were emm patterns A to C, “throat specialists,” and 10.9% were emm pattern D, “skin specialists” (Fig. 7 A). The distribution was inversely skewed in the NADase-inactive strains, with 31% emm patterns A to C, 63.8% emm pattern D, and only 5.2% emm pattern E (Fig. 7B). Thus, the NADase-active strains are strongly associated with the “generalist” emm pattern, while the NADase-inactive strains are primarily “tissue specialists” (P = 0.0001 by chi-square test). This association is unlikely due to linkage, since spn and emm genes are separated by over 300 kb in the MGAS6180 reference strain (roughly 17% of the bacterium's chromosome) and cpa, the gene closest to spn that has been correlated with tissue tropism, is 35 kb apart (6).
FIG. 7.

SPN NADase activity or inactivity is associated with emm pattern. The relative proportions of emm patterns found in NADase-active and NADase-inactive strains are shown. Numbers in parentheses indicate the number of strains in each emm pattern. Eighty-five percent of NADase-active strains are emm pattern E, in comparison to only 5% of NADase-inactive strains found in this pattern. This skewed distribution of NADase-active and -inactive strains is statistically significant (P = 0.0001 by chi-square test).
Since the data indicate that the significant majority of the throat and skin specialist strains possess the NADase-inactive spn allele, we explored the possibility that either of the two NADase-inactive spn allele clusters (3 and 4) may have a stronger association with either emm patterns A to C or emm pattern D. Should such an association exist, it would indicate residues other than 330 that would be involved in adapting to tropism for the pharynx or skin. Although there appears to be a trend for a greater relative proportion of cluster 4 spn alleles to group with emm patterns A to C and a greater relative proportion of cluster 3 spn alleles to group with emm pattern D, this association is not statistically significant when the number of strains containing either of the two alleles in the sample population is taken into account (P = 0.2 and P = 0.6, respectively, by chi-square test). Thus, although NADase-inactive spn has diverged into two allele clusters, these clusters are not significantly associated with throat versus skin tropism. Therefore, our current data set cannot further identify which SPN polymorphisms, other than G330D, are associated with specific tissue preferences. Thus, S. pyogenes strains that act as “generalists” benefit from SPN NADase activity, while the data suggest that the NADase activity of SPN is not beneficial or even detrimental in strains that are largely limited to causing disease at specific tissue sites.
DISCUSSION
By characterizing genetic diversity in the virulence factors spn and ifs, we have discovered that spn is experiencing positive selection and diverging into NADase-active and -inactive subtypes. Both NADase-active and -inactive alleles are equally represented in the sample population, maintained over time, and found over a broad geographic range. These findings indicate that SPN is likely a multifunctional virulence factor, playing a complex role in streptococcal pathogenesis. NADase activity has no correlation with invasive disease in this diverse collection of S. pyogenes strains but is significantly associated with genotypes linked with tissue tropism, hinting at other possible NADase-independent functions for SPN.
Prior studies examining the relationship between invasiveness and NADase activity have reported conflicting results. A correlation between NADase activity and invasive disease was absent in a diverse Australian S. pyogenes strain collection (15) and in our study. In contrast, several studies focusing on the M1 serotype strain or animal models have found NADase activity correlated with increased virulence (10, 52, 53, 56). Thus, NADase activity may contribute to severe diseases in some specific strains, likely due to interactions with other proteins that enhance virulence. However, our data demonstrate that NADase-inactive S. pyogenes strains remain equally capable of causing invasive infections. Taken together, these findings suggest that SPN contributes to disease pathogenesis in multiple ways and likely experiences complex evolutionary pressures.
In addition to revealing that SPN NADase activity does not correlate with disease severity in a diverse strain collection, our data demonstrate that the divergence of SPN into NADase-active and -inactive subtypes correlates with streptococcal tissue site preferences for infection. Multiple previous studies have evaluated the genotypic and phenotypic differences between generalist strains and tissue specialists, but this is the first study to evaluate SPN in this context. Most of the known adaptations associated with tissue preference in S. pyogenes involve the presence/absence of specific genes, divergent gene lineages, and alterations in gene expression. Factors that diverge with tissue tropism include several genes within the FCT region (e.g., cpa, a gene encoding a pilus accessory protein with affinity for collagen in the dermis) (32), characteristics of the M protein (e.g., the plasminogen binding M protein) (28), and transcriptional regulators that control expression of multiple virulence proteins (e.g., rofA or nra transcriptional regulators of the FCT region) (8). The majority of these virulence factors are surface-associated proteins that facilitate binding of the bacteria to host tissues or evasion of the immune response. In contrast, SPN does not have a direct role in the binding of S. pyogenes to the host cell, nor is it known to be a target of the humoral immune system. Because the majority of SPN is translocated into the host cell (39), it is unlikely to be subject to the same selective pressures as the other tissue tropism-associated factors, and positive selection is likely driven by interactions with molecules within the host cell.
Thus, the finding that SPN with and SPN without NADase activity segregate with genetic markers for tissue tropism prompts a reevaluation of the role of SPN in disease pathogenesis. Several reported functions of SPN have previously been proposed to account for its role in virulence. First, the NAD+ glycohydrolase activity of SPN depletes cellular β-NAD+ stores, depriving the host cell of this cofactor essential for redox reactions (4, 42). Second, SPN′s ADP-ribosyltransferase activity (52) may contribute to pathogenesis through inactivation of host cell proteins. Lastly, SPN′s ability to produce cyclic ADP-ribose (cADPR) (30) may mobilize cellular calcium stores and interfere with cell signaling processes. However, each of these functions is dependent on the cleavage of β-NAD+; therefore, they cannot play a role in the pathogenesis of strains with NADase-inactive SPN.
Both the observed high frequency of NADase-inactive spn alleles in the sample population and the finding that these alleles maintain sequence characteristics of a functional gene provide credibility for the hypothesis that SPN has a NADase-independent function. At this point, it is unclear which function is driving the observed pattern of sequence evolution, but a possible secondary function of SPN could be another enzymatic activity or an activity related to host cell carbohydrate binding via the translocation domain. The close interaction of SPN and SLO might provide a potential structural reason for the maintenance of NADase-inactive SPN (20, 41). In this scenario, SPN would be required for facilitating SLO function (43), providing structural support or stabilizing SLO. It could also be postulated that spn sequence fidelity is necessary for transcription of slo; however, strains with various experimental deletions in spn do not have altered slo expression (20). Several additional functions have been attributed to SPN, and they include alteration of neutrophil migration and chemiluminescence response, leukotoxic effects, and inhibition of internalization of S. pyogenes into keratinocytes (5, 11, 52). It is currently unknown which of these functions are dependent on NADase activity and which are independent. Regardless of the exact nature of the NADase-independent secondary function of SPN, any viable model for this function should strive to successfully incorporate the tissue tropism aspect of SPN allele distribution uncovered in our study.
The data presented here also have implications related to the evolutionary course of S. pyogenes. It has been hypothesized that S. pyogenes is diverging from a generalist ancestor into two distinct species, one that infects the throat and another that infects the skin (6). The NADase-active SPN allele appears to be the more ancient gene, since other streptococcal species, including the outgroup Streptococcus dysgalactiae subsp. equisimilis used in this study, contain the NADase-active allele. Thus, our data indicate that SPN is evolving from a NADase-active enzyme to a NADase-inactive virulence factor that continues to evolve under functional constraint, while ifs is degrading into a pseudogene in many strains. Since no single NADase-inactive spn allele cluster significantly correlates with emm patterns A to C or emm pattern D, our spn sequence data are consistent with the idea that specialists are derived from generalists, but the data do not provide additional support to the hypothesis that the specialists are further diverging from each other. Therefore, it may be that the NADase activity of SPN was beneficial to S. pyogenes in the past but is becoming irrelevant as the bacterium evolves other roles for SPN or other novel virulence mechanisms.
There are numerous possible models for novel roles of SPN in streptococcal pathogenesis. Since S. pyogenes is exclusively a human pathogen, with no animal reservoir, the finding that generalist strains benefit from NADase-active SPN and tissue specialist strains benefit from NADase-inactive SPN must be related to the interaction between S. pyogenes and its human hosts. The selective advantage may occur at any stage of pathogenesis, including transmission, competition with normal bacterial flora in niche establishment, evasion of the host immune defenses, breaching host anatomic barriers, and/or altering host physiology. However, since SPN is unique among tissue tropism-associated virulence proteins in that it is injected into the host cell, it is most likely that its potential role in tissue tropism involves manipulation of host cell survival or function.
It is also possible that NADase activity is beneficial only in facets of pathogenesis that are unique to generalist strains. There is geographic and temporal partitioning in the incidence of streptococcal diseases, in that pharyngitis is most common in temperate climates during the winter while impetigo is most prevalent in tropical regions and warm, humid climates (6). Thus, in conjunction with other virulence proteins, NADase activity may facilitate transmission and allow the organism to more readily establish infection at whichever tissue site it encounters, thereby helping the generalist strains overcome obstacles to transmission that are not usually faced by the specialist strains. Additionally, the loss of NADase activity may result in limiting the organism to a specific tissue site for infection. In this scenario, the observed positive selection occurring in SPN could be explained only if the NADase-inactive form was consistently coinherited with other traits that enhance the adaptation to specific tissue sites. Another possible model for the role of NADase-inactive SPN in pathogenesis comes from the observation that SPN may have a preventative effect on internalization into host cells (11). Tissue specialists, in contrast to generalists, may have adapted to specific intracellular niches that provide survival advantage (33). In this scenario, inhibition of internalization would be an undesirable property of SPN in specialist strains, whereas generalists that lack these specific adaptations would have a survival advantage if the NADase activity prevented internalization. Unfortunately, the paucity of additional data suggesting NADase-independent functions of SPN limits the further development of comprehensive hypotheses related to the role of SPN in tissue tropism.
Thus, the results of this study shed further light on the role of SPN in streptococcal pathogenesis and lay the foundation for further research in this area. Studies that focus on the role of NADase-inactive SPN in streptococcal pathogenesis and NADase-independent functions of SPN are warranted. Uncovering new data related to the process of how S. pyogenes causes specific diseases and adapts to its ecologic niche will help drive the development of new therapeutic options to combat this important worldwide pathogen.
Supplementary Material
Acknowledgments
D.J.R. was supported by National Institutes of Health grant T32-AI007172. D.E.B. was supported by National Institutes of Health grant R01-AI065572. M.G.C. was supported by National Institutes of Health grant R01-AI064721 from the Public Health Services. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank Gerod Hall and Candace Ford for technical assistance; Nicole C. Riddle, Stuart McDaniel, and the laboratory of Kenneth M. Olsen for comments on the manuscript; and the St. Louis Children's Hospital clinical microbiology personnel for the generous collection of S. pyogenes strains.
Footnotes
Published ahead of print on 21 May 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Ajdic, D., W. M. McShan, D. J. Savic, D. Gerlach, and J. J. Ferretti. 2000. The NAD-glycohydrolase (nga) gene of Streptococcus pyogenes. FEMS Microbiol. Lett. 191:235-241. [DOI] [PubMed] [Google Scholar]
- 2.Anisimova, M., R. Nielsen, and Z. Yang. 2003. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics 164:1229-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beres, S. B., E. W. Richter, M. J. Nagiec, P. Sumby, S. F. Porcella, F. R. DeLeo, and J. M. Musser. 2006. Molecular genetic anatomy of inter- and intraserotype variation in the human bacterial pathogen group A streptococcus. Proc. Natl. Acad. Sci. U. S. A. 103:7059-7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger, F., M. H. Ramirez-Hernandez, and M. Ziegler. 2004. The new life of a centenarian: signalling functions of NAD(P). Trends Biochem. Sci. 29:111-118. [DOI] [PubMed] [Google Scholar]
- 5.Bernheimer, A. W., P. D. Lazarides, and A. T. Wilson. 1957. Diphosphopyridine nucleotidase as an extracellular product of streptococcal growth and its possible relationship to leukotoxicity. J. Exp. Med. 106:27-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bessen, D. E. 2009. Population biology of the human restricted pathogen, Streptococcus pyogenes. Infect. Genet. Evol. 9:581-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bessen, D. E., M. W. Izzo, T. R. Fiorentino, R. M. Caringal, S. K. Hollingshead, and B. Beall. 1999. Genetic linkage of exotoxin alleles and emm gene markers for tissue tropism in group A streptococci. J. Infect. Dis. 179:627-636. [DOI] [PubMed] [Google Scholar]
- 8.Bessen, D. E., A. Manoharan, F. Luo, J. E. Wertz, and D. A. Robinson. 2005. Evolution of transcription regulatory genes is linked to niche specialization in the bacterial pathogen Streptococcus pyogenes. J. Bacteriol. 187:4163-4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bisno, A. L., and D. L. Stevens. 1996. Streptococcal infections of skin and soft tissues. N. Engl. J. Med. 334:240-245. [DOI] [PubMed] [Google Scholar]
- 10.Bricker, A. L., V. J. Carey, and M. R. Wessels. 2005. Role of NADase in virulence in experimental invasive group A streptococcal infection. Infect. Immun. 73:6562-6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bricker, A. L., C. Cywes, C. D. Ashbaugh, and M. R. Wessels. 2002. NAD+-glycohydrolase acts as an intracellular toxin to enhance the extracellular survival of group A streptococci. Mol. Microbiol. 44:257-269. [DOI] [PubMed] [Google Scholar]
- 12.Carapetis, J. R., A. C. Steer, E. K. Mulholland, and M. Weber. 2005. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5:685-694. [DOI] [PubMed] [Google Scholar]
- 13.Corander, J., P. Marttinen, J. Siren, and J. Tang. 2008. Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics 9:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Courtney, H. S., and D. L. Hasty. 1991. Aggregation of group A streptococci by human saliva and effect of saliva on streptococcal adherence to host cells. Infect. Immun. 59:1661-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DelVecchio, A., M. Maley, B. J. Currie, and K. S. Sriprakash. 2002. NAD-glycohydrolase production and speA and speC distribution in group A streptococcus (GAS) isolates do not correlate with severe GAS diseases in the Australian population. J. Clin. Microbiol. 40:2642-2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doyle, J. J., and B. S. Gaut. 2000. Evolution of genes and taxa: a primer. Plant Mol. Biol. 42:1-23. [PubMed] [Google Scholar]
- 17.Enright, M. C., B. G. Spratt, A. Kalia, J. H. Cross, and D. E. Bessen. 2001. Multilocus sequence typing of Streptococcus pyogenes and the relationships between emm type and clone. Infect. Immun. 69:2416-2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feil, E. J., E. C. Holmes, D. E. Bessen, M. S. Chan, N. P. Day, M. C. Enright, R. Goldstein, D. W. Hood, A. Kalia, C. E. Moore, J. Zhou, and B. G. Spratt. 2001. Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc. Natl. Acad. Sci. U. S. A. 98:182-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh, J., P. J. Anderson, S. Chandrasekaran, and M. G. Caparon. 2009. Characterization of Streptococcus pyogenes beta-NAD+ glycohydrolase: re-evaluation of enzymatic properties associated with pathogenesis. J. Biol. Chem. 285:5683-5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh, J., and M. G. Caparon. 2006. Specificity of Streptococcus pyogenes NAD(+) glycohydrolase in cytolysin-mediated translocation. Mol. Microbiol. 62:1203-1214. [DOI] [PubMed] [Google Scholar]
- 21.Green, N. M., S. Zhang, S. F. Porcella, M. J. Nagiec, K. D. Barbian, S. B. Beres, R. B. LeFebvre, and J. M. Musser. 2005. Genome sequence of a serotype M28 strain of group a streptococcus: potential new insights into puerperal sepsis and bacterial disease specificity. J. Infect. Dis. 192:760-770. [DOI] [PubMed] [Google Scholar]
- 22.Hanage, W. P., C. Fraser, and B. G. Spratt. 2006. The impact of homologous recombination on the generation of diversity in bacteria. J. Theor. Biol. 239:210-219. [DOI] [PubMed] [Google Scholar]
- 23.Hanski, E., P. A. Horwitz, and M. G. Caparon. 1992. Expression of protein F, the fibronectin-binding protein of Streptococcus pyogenes JRS4, in heterologous streptococcal and enterococcal strains promotes their adherence to respiratory epithelial cells. Infect. Immun. 60:5119-5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hauser, A. R., D. L. Stevens, E. L. Kaplan, and P. M. Schlievert. 1991. Molecular analysis of pyrogenic exotoxins from Streptococcus pyogenes isolates associated with toxic shock-like syndrome. J. Clin. Microbiol. 29:1562-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollingshead, S. K., T. L. Readdy, D. L. Yung, and D. E. Bessen. 1993. Structural heterogeneity of the emm gene cluster in group A streptococci. Mol. Microbiol. 8:707-717. [DOI] [PubMed] [Google Scholar]
- 26.Hudson, R. R., M. Kreitman, and M. Aguade. 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116:153-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hughes, A. L., R. Friedman, P. Rivailler, and J. O. French. 2008. Synonymous and nonsynonymous polymorphisms versus divergences in bacterial genomes. Mol. Biol. Evol. 25:2199-2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalia, A., and D. E. Bessen. 2004. Natural selection and evolution of streptococcal virulence genes involved in tissue-specific adaptations. J. Bacteriol. 186:110-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalia, A., B. G. Spratt, M. C. Enright, and D. E. Bessen. 2002. Influence of recombination and niche separation on the population genetic structure of the pathogen Streptococcus pyogenes. Infect. Immun. 70:1971-1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karasawa, T., K. Yamakawa, D. Tanaka, Y. Gyobu, and S. Nakamura. 1995. NAD(+)-glycohydrolase productivity of haemolytic streptococci assayed by a simple fluorescent method and its relation to T serotype. FEMS Microbiol. Lett. 128:289-292. [DOI] [PubMed] [Google Scholar]
- 31.Kimoto, H., Y. Fujii, S. Hirano, Y. Yokota, and A. Taketo. 2006. Genetic and biochemical properties of streptococcal NAD-glycohydrolase inhibitor. J. Biol. Chem. 281:9181-9189. [DOI] [PubMed] [Google Scholar]
- 32.Kratovac, Z., A. Manoharan, F. Luo, S. Lizano, and D. E. Bessen. 2007. Population genetics and linkage analysis of loci within the FCT region of Streptococcus pyogenes. J. Bacteriol. 189:1299-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kreikemeyer, B., M. Klenk, and A. Podbielski. 2004. The intracellular status of Streptococcus pyogenes: role of extracellular matrix-binding proteins and their regulation. Int. J. Med. Microbiol. 294:177-188. [DOI] [PubMed] [Google Scholar]
- 34.Kumar, S., M. Nei, J. Dudley, and K. Tamura. 2008. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 9:299-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lancefield, R. C. 1962. Current knowledge of type-specific M antigens of group A streptococci. J. Immunol. 89:307-313. [PubMed] [Google Scholar]
- 36.Lazarides, P. D., and A. W. Bernheimer. 1957. Association of production of diphosphopyridine nucleotidase with serological type of group A streptococcus. J. Bacteriol. 74:412-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levin, B. R., and O. E. Cornejo. 2009. The population and evolutionary dynamics of homologous gene recombination in bacterial populations. PLoS Genet. 5:e1000601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Librado, P., and J. Rozas. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451-1452. [DOI] [PubMed] [Google Scholar]
- 39.Madden, J. C., N. Ruiz, and M. Caparon. 2001. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in gram-positive bacteria. Cell 104:143-152. [DOI] [PubMed] [Google Scholar]
- 40.McGregor, K. F., B. G. Spratt, A. Kalia, A. Bennett, N. Bilek, B. Beall, and D. E. Bessen. 2004. Multilocus sequence typing of Streptococcus pyogenes representing most known emm types and distinctions among subpopulation genetic structures. J. Bacteriol. 186:4285-4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meehl, M. A., and M. G. Caparon. 2004. Specificity of streptolysin O in cytolysin-mediated translocation. Mol. Microbiol. 52:1665-1676. [DOI] [PubMed] [Google Scholar]
- 42.Meehl, M. A., J. S. Pinkner, P. J. Anderson, S. J. Hultgren, and M. G. Caparon. 2005. A novel endogenous inhibitor of the secreted streptococcal NAD-glycohydrolase. PLoS Pathog. 1:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michos, A., I. Gryllos, A. Hakansson, A. Srivastava, E. Kokkotou, and M. R. Wessels. 2006. Enhancement of streptolysin O activity and intrinsic cytotoxic effects of the group A streptococcal toxin, NAD-glycohydrolase. J. Biol. Chem. 281:8216-8223. [DOI] [PubMed] [Google Scholar]
- 44.Musser, J. M., and S. A. Shelburne III. 2009. A decade of molecular pathogenomic analysis of group A streptococcus. J. Clin. Invest. 119:2455-2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nei, M. 1987. Molecular evolutionary genetics. Columbia University Press, New York, NY.
- 46.Nei, M., and T. Gojobori. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3:418-426. [DOI] [PubMed] [Google Scholar]
- 47.Potempa, J., J. Travis, E. Golonka, and L. Shaw. 2006. Poison-antidote systems in bacteria: the co-evolution of functional counterparts. Cell. Mol. Biol. (Noisy-le-grand) 52:18-22. [PubMed] [Google Scholar]
- 48.Qiu, X., B. R. Kulasekara, and S. Lory. 2009. Role of horizontal gene transfer in the evolution of Pseudomonas aeruginosa virulence. Genome Dyn. 6:126-139. [DOI] [PubMed] [Google Scholar]
- 49.Scott, J. R., P. C. Guenthner, L. M. Malone, and V. A. Fischetti. 1986. Conversion of an M− group A streptococcus to M+ by transfer of a plasmid containing an M6 gene. J. Exp. Med. 164:1641-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steer, A. C., I. Law, L. Matatolu, B. W. Beall, and J. R. Carapetis. 2009. Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect. Dis. 9:611-616. [DOI] [PubMed] [Google Scholar]
- 51.Stern, A., A. Doron-Faigenboim, E. Erez, E. Martz, E. Bacharach, and T. Pupko. 2007. Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res. 35:W506-W511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stevens, D. L., D. B. Salmi, E. R. McIndoo, and A. E. Bryant. 2000. Molecular epidemiology of nga and NAD glycohydrolase/ADP-ribosyltransferase activity among Streptococcus pyogenes causing streptococcal toxic shock syndrome. J. Infect. Dis. 182:1117-1128. [DOI] [PubMed] [Google Scholar]
- 53.Sumby, P., S. F. Porcella, A. G. Madrigal, K. D. Barbian, K. Virtaneva, S. M. Ricklefs, D. E. Sturdevant, M. R. Graham, J. Vuopio-Varkila, N. P. Hoe, and J. M. Musser. 2005. Evolutionary origin and emergence of a highly successful clone of serotype M1 group A streptococcus involved multiple horizontal gene transfer events. J. Infect. Dis. 192:771-782. [DOI] [PubMed] [Google Scholar]
- 54.Swanson, W. J., R. Nielsen, and Q. Yang. 2003. Pervasive adaptive evolution in mammalian fertilization proteins. Mol. Biol. Evol. 20:18-20. [DOI] [PubMed] [Google Scholar]
- 55.Tamura, K., J. Dudley, M. Nei, and S. Kumar. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596-1599. [DOI] [PubMed] [Google Scholar]
- 56.Tatsuno, I., J. Sawai, A. Okamoto, M. Matsumoto, M. Minami, M. Isaka, M. Ohta, and T. Hasegawa. 2007. Characterization of the NAD-glycohydrolase in streptococcal strains. Microbiology 153:4253-4260. [DOI] [PubMed] [Google Scholar]
- 57.Vos, M., and X. Didelot. 2009. A comparison of homologous recombination rates in bacteria and archaea. ISME J. 3:199-208. [DOI] [PubMed] [Google Scholar]
- 58.Yang, Z., R. Nielsen, N. Goldman, and A. M. Pedersen. 2000. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhai, W., R. Nielsen, and M. Slatkin. 2009. An investigation of the statistical power of neutrality tests based on comparative and population genetic data. Mol. Biol. Evol. 26:273-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.