

Abstract
Chronic chagasic cardiomyopathy is a leading cause of heart failure in Latin American countries. About 30% of Trypanosoma cruzi-infected individuals develop this severe symptomatic form of the disease, characterized by intense inflammatory response accompanied by fibrosis in the heart. We performed an extensive microarray analysis of hearts from a mouse model of this disease and determined significant alterations in expression of ∼12% of the sampled genes. Extensive upregulations were associated with immune-inflammatory responses (chemokines, adhesion molecules, cathepsins and MHC molecules) and fibrosis (extracellular matrix components, lysyl oxidase and Timp-1). Our results indicate potentially relevant factors involved in the pathogenesis of the disease that may provide new therapeutic targets in chronic Chagas' disease.
Keywords: Chagas' disease, Gene expression, Cardiomyopathy, Inflammation, Fibrosis
Introduction
Chagas' disease, caused by infection with the protozoan Trypanosoma cruzi, is still a major health problem in Latin America, where it affects 16-18 million people [1]. The most common chronic form, the chagasic cardiomyopathy (CCM), is a fatal disease for which there is no effective treatment available other than heart transplantation. CCM is characterized by focal or disseminated inflammation causing myocytolysis, necrosis, and progressive fibrosis [2-4].
The pathological basis of CCM is multifactorial [5,6]. It may in part result from inflammatory responses directed to T. cruzi antigen, to cardiac autoantigens, or to both types of antigens [7]. A prominent role of parasite antigens in this pathology has been supported by the demonstration that a decrease in parasite load caused a reduction in myocarditis and cardiac disturbances in mice chronically infected with T. cruzi [8].
The identification of factors involved in the establishment of chronic heart lesions is of great interest for the development of new therapeutic strategies for patients with this fatal disease. In this study we have performed a DNA microarray analysis to determine alterations in gene expression in the myocardium of mice chronically infected with the Colombian strain of T. cruzi compared to uninfected counterparts. Our results indicate a profound effect on expression of a number of genes related to inflammation and fibrosis in hearts of mice with CCM.
Methods
Trypomastigotes of Colombian Trypanosoma cruzi strain [9] were obtained from culture supernatants of infected LCC-MK2 cells. C57Bl/6 male/female mice were infected by intraperitoneal injection of T. cruzi trypomastigotes. Parasitemia was evaluated at various times after infection by counting the number of trypomastigotes in peripheral blood aliquots. Animals were raised and maintained at the Gonçalo Moniz Research Center/FIOCRUZ and provided with rodent diet and water ad libitum. Animals were handled according to the NIH guidelines for animal experimentation. All procedures described here had prior approval from the local animal ethics committee.
Mice were sacrificed after 8 months of infection and hearts removed and fixed in 10% buffered formalin. Morphometrical analyses were performed in hematoxylin/eosin or Sirius red-stained heart sections captured using a digital camera adapted to a BX41 microscope (Olympus, Japan). Images were analyzed using Image-Pro Program version 5.0 (Media Cybernetics, San Diego, CA).
Frozen heart sections were used for detection of CD4, CD8, CD11b, ICAM-1 and MHC class II expression by immunofluorescence, using specific antibodies (BD Biosciences, San Jose, CA) followed by streptavidin Alexa 568 (Molecular Probes, Carlsbad, CA). The myocardium was stained with phalloidin (Molecular Probes) or using an anti-cardiac myosin antibody (Sigma). Nuclei were stained with 4,6-diamidino-2-phenylindole (VectaShield Hard Set mounting medium with DAPI H-1500; Vector Laboratories, Burlingame, CA). Sections were analyzed using a BX61 microscope equipped with epifluorescence and appropriate filters (Olympus) and a system to enhance the fluorescence resolution (Optigrid, Thales Optem Inc., Fairport, NY).
SDF-1, TNFα, and IFNγ concentrations were measured in total heart extracts. Heart proteins were extracted from 100 mg tissue/ml PBS to which 0.4 M NaCl, 0.05% Tween 20 and protease inhibitors (0.1 mM PMSF, 0.1 mM benzethonium chloride, 10 mM EDTA and 20 KI aprotinin A/100 ml) were added. The samples were centrifuged for 10 min at 3000 g and the supernatant was kept frozen at -70°C. Cytokine levels were estimated using commercially available Immunoassay ELISA kits for mouse SDF-1, TNFα, and IFNγ (R&D system, Minneapolis, MN), according to the manufacturer's instructions. Reaction was revealed after incubation with streptavidin–horseradish peroxidase conjugate followed by detection using 3,3′,5,5′-tetramethylbenzidine (TMB) peroxidase substrate and reading at 450 nm.
Hearts of normal and T. cruzi-infected mice were extracted and quickly frozen in liquid nitrogen for five minutes. The material was ground and RNA extraction was performed using RNAEasy Minikit (Qiagen, Germantown, MD), following manufacturer's instructions. After addition of 1 unit/μl DNAse I (Invitrogen), the cDNA was obtained using SuperScriptTM II Reverse Transcriptase (Invitrogen) in a final volume of 30 μl. Reaction cycles were performed on an Eppendorf Mastercycler gradient for 1 h (42° C for 60 min; 70° C for 15 min). PCR amplification was performed in an ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). Primers and TaqMan probe for TIMP-1 the GAPDH control reference gene were designed and synthesized according to Assay-by-Design (Applied Biosystems). Quantitative data was analyzed using the Sequence Detection System software (v1.0; Applied Biosystems). PCR reactions were carried out in a total volume of 25 ml, according the manufacturer's instructions. The standard curves of the target and reference genes showed similar results of efficacy (> 90%). The relative quantification was given by the ratio between the mean value of the target gene and the mean value of the reference gene (Gapdh) in each sample. The relative amount of PCR product generated from each primer set was determined on the basis of the Ct value. The relative quantification was calculated by 2-ΔΔCT (CT: fluorescence threshold value; ΔCT: CT of the target gene minus CT of the reference gene; ΔΔCT: tumor sample ΔCT minus reference sample ΔCT).
Twenty μg total RNA extracted each of the 4 control and 4 infected hearts were reverse transcribed into cDNA incorporating fluorescent Alexa Fluor®_647–aha-dUTP using SuperScriptTM Plus Direct cDNA Labeling System (Invitrogen, CA). Differently labeled biological replicas were co-hybridized overnight at 50°C with Duke MO30N mouse oligonucleotide arrays spotted with 30 k Operon 70-mer oligonucleotides V3.0.1 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL8938) using the “multiple yellow” strategy described in [10]. In this strategy, differently labeled biological replicas are co-hybridized with the array. Thus, we have hybridized two arrays with samples from 4 control hearts and other two arrays with samples from 4 infected hearts. After washing (0.1% SDS and 1% SSC) to remove the non-hybridized cDNAs, each array was scanned with an Axon 4000B dual-laser scanner (MDS, Toronto, Canada) and images were primarily analyzed with GenePixPro 6.0 (Axon Instruments, CA). Locally corrupted or saturated spots, as well as those for which the foreground median fluorescence did not exceed twice the median local background fluorescence in one sample were eliminated from the analysis in all samples. Microarray data were processed as described in our previous studies [10-12]. In brief, we used a normalization algorithm that alternates intra-chip and inter-chip normalization of the net fluorescence (i.e. background subtracted foreground) signals of the validated spots until the residual error is below 5% in subsequent steps. Intrachip normalization balances the averages of net fluorescence values in the two channels within each pin domain (subset of spots printed by the same pin), corrects the intensity-dependent bias (usually referred to as Lowess normalization), and forces the standard distribution (mean 0 and standard deviation 1) of log2 ratios (scale normalization) of net fluorescent values in the two channels for each array. Interchip normalization assigns a ratio between the corrected net fluorescence of each valid spot and the average net fluorescence of all valid spots in both control (C1, C2, C3, C4) and infected (I1, I2, I3, I4) samples. The spots probing the same gene were organized into redundancy groups and their background subtracted fluorescence replaced by a weighted average value. A gene was considered as significantly up- or down-regulated when comparing four infected with four control hearts if the absolute fold change was > 1.5× and the p-value of the Student's heteroscedastic t-test of equality of the means of the distributions with a Bonferroni-type adjustment for the redundancy groups was < 0.05. GenMapp [13] and MappFinder software (www.genmapp.org) and databases were used to identify the most affected GO (Gene Ontology) categories.
Morphometric, qRT-PCR, and cytokine data were analyzed using Student's t test. Differences were considered significant when P < 0.05.
Results
CCM caused by chronic infection with Colombian strain T. cruzi in C57Bl/6 mice
Upon infection with 1000 trypomastigote forms of Colombian strain T. cruzi, C57Bl/6 mice develop blood parasitemia peaking around 35 days after infection (Figure 1A). Mortality rate reached 28.5% during the first 100 days (Figure 1B), and about 31.4% after 8 months of infection. A progressive myocarditis accompanied by fibrosis occurs after the acute phase of infection. At eight months of infection, heart sections of chagasic mice revealed a multifocal inflammatory response composed mainly by mononuclear cells (Figures 1C and E) and exhibited areas of fibrosis (Figures 1D and F).
Figure 1.
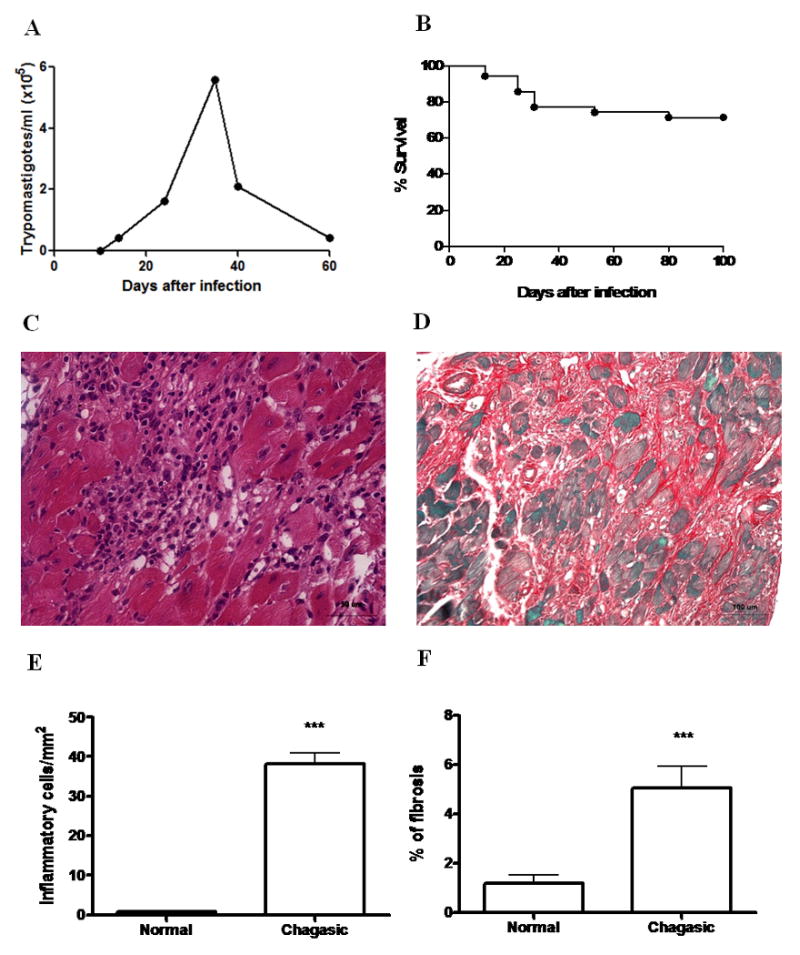
Infection of C57Bl/6 mice with Colombian strain T. cruzi. Mice (n=35) were infected with 1000 Colombian Strain trypomastigotes. A, Parasitemia and B, Mortality were evaluated during the acute phase of infection. Data in (A) represent the median of the individual parasitemia. C, Inflammation and D, fibrosis evidenced in heart sections of mice 8 months after infection, stained with H&E and Sirius red. E, Quantification of inflammatory cells and F, fibrosis area in heart sections of normal (n=4) and chagasic mice (n=9; 8 months after infection with T. cruzi) was done by morphometry. Bars represent the means±SEM. *** P<0.05.
Global gene expression analysis
Microarray data from this experiment have been deposited in Genebank (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17363). When comparing control hearts from C57Bl/6 mice to those from age and sex-matched mice chronically infected with the Colombian strain of T. cruzi, genes differentially expressed were detected. Spots corresponding to 14,356 unigenes satisfied the criteria of adequate quantitation on all eight microarrays. Of these, 1,221 (8.5%) were significantly upregulated in the chagasic hearts and 494 (3.4%) were significantly downregulated (>50% difference and p<0.05). A list of all genes that were found to be differentially expressed is presented in Table S1 (Supplemental Material), and subsets of the genes showing higher fold-change in expression ratio are considered below.
Pathways of proteins encoded by genes that were significantly affected by parasitic infection were determined using GenMapp (Gladstone Insititute), where significance is assessed by whether regulated genes are disproportionately represented within a gene ontogeny (GO) term. Pathways of genes significantly upregulated in infected hearts (p<0.05) are listed in Table 1 and prominently include immune response and related terms (eg, inflammatory response, intracellular signaling cascade, chemokine and cytokine receptor activity). The GenMapp illustrations of these altered genes are shown in Supplemental Figure 1A. In addition, upregulated pathways include phosphate transport, cell proliferation, actin binding (eg, Arp2/3 protein complex and actin filament organization, cytoskeleton, membrane ruffling). These genes related to the actin cytoskeleton are illustrated in Supplemental Figure 1B. In addition to these well-represented pathways, other smaller pathways showed prominent perturbation, including genes involved in cardiac differentiation (Tgfb2, Itgb1) and regulation of action potential (Gnaq, Hexa, Hab1 and Cd9).
Table 1. Up and downregulated GO categories in T. cruzi-infected heart.
| UPREGULATED | ||||||
|---|---|---|---|---|---|---|
| GOID | GO Name | Type | Measured | %Change | Z Score | PermuteP |
| 1726 | Ruffle | C | 21 | 38.10 | 5.039 | 0 |
| 6955 | immune response | P | 108 | 19.44 | 4.817 | 0 |
| 8009 | chemokine activity | F | 19 | 36.84 | 4.592 | 0 |
| 5764 | Lysosome | C | 44 | 31.82 | 4.487 | 0 |
| 5783 | endoplasmic reticulum | C | 305 | 14.43 | 3.806 | 0 |
| 7242 | Intracellular signaling cascade | P | 169 | 12.43 | 3.302 | 0 |
| 6935 | Chemotaxis | P | 31 | 25.81 | 3.254 | 0.004 |
| 30036 | actin cytoskeleton organization & biogenesis | P | 32 | 12.50 | 3.232 | 0.002 |
| 4180 | carboxypeptidase activity | F | 11 | 27.27 | 3.22 | 0.007 |
| 16798 | hydrolase activity, acting on glicosyl bonds | F | 25 | 28.00 | 3.172 | 0.004 |
| 15629 | actin cytoskeleton | C | 40 | 10.00 | 3.09 | 0.007 |
| 5938 | cell cortex | C | 10 | 20.00 | 3.035 | 0.01 |
| 7015 | actin filament organization | P | 15 | 26.67 | 2.914 | 0.015 |
| 5279 | amino acid-polyamine transporter activity | F | 11 | 18.18 | 2.91 | 0.016 |
| 42552 | Myelination | P | 13 | 23.08 | 2.806 | 0.024 |
| 8285 | negative regulation of cell proliferation | P | 45 | 15.56 | 2.67 | 0.015 |
| 8284 | positive regulation of cell proliferation | P | 56 | 16.07 | 2.669 | 0.014 |
| 45596 | negative regulation on cell differentiation | P | 13 | 7.69 | 2.603 | 0.014 |
| 8283 | cell proliferation | P | 55 | 16.36 | 2.588 | 0.012 |
| 8201 | heparin binding | F | 28 | 21.43 | 2.585 | 0.019 |
| 6954 | inflammatory response | P | 67 | 11.94 | 2.582 | 0.008 |
| 9986 | cell surface | C | 43 | 18.60 | 2.347 | 0.026 |
| 6817 | phosphate transport | P | 40 | 17.50 | 2.18 | 0.039 |
| 4896 | Hematopoietin/interferon class (D200 Domain) cytokine receptor activity | F | 16 | 25.00 | 2.18 | 0.048 |
| 48754 | branching morphogenesis of a tube | P | 12 | 33.33 | 2.123 | 0.048 |
| 45165 | cell fate commitment | P | 16 | 12.50 | 2.113 | 0.038 |
| 42127 | regulation of cell proliferation | P | 17 | 0.00 | 2.055 | 0.045 |
| 7264 | small GTPase mediated signal transduction | P | 83 | 10.84 | 2.055 | 0.05 |
| DOWNREGULATED | ||||||
|---|---|---|---|---|---|---|
| GOID | GO Name | Type | Measured | %Change | Z Score | PermuteP |
| 5739 | Mitochondrion | C | 351 | 17.38 | 13.839 | 0 |
| 5737 | Cytoplasm | C | 613 | 2.61 | 5.628 | 0 |
| 3824 | catalytic activity | F | 104 | 5.77 | 5.241 | 0 |
| 16491 | oxidoreductase activity | F | 208 | 6.73 | 4.835 | 0 |
| 3954 | NADH dehydrogenase activity | F | 10 | 30.00 | 4.599 | 0.005 |
| 50660 | FAD binding | F | 34 | 17.65 | 4.541 | 0 |
| 30170 | pyridoxal phosphate binding | F | 31 | 16.13 | 3.871 | 0.002 |
| 5777 | Peroxisome | C | 51 | 13.73 | 3.839 | 0.002 |
| 6118 | electron transport | P | 189 | 7.41 | 3.727 | 0 |
| 9055 | electron carrier activity | F | 60 | 8.33 | 3.349 | 0.003 |
| 5975 | carbohydrate metabolic process | p | 81 | 6.17 | 3.169 | 0.001 |
| 16874 | ligase activity | F | 121 | 8.26 | 3.123 | 0.005 |
| 8483 | transaminase activity | F | 10 | 20.00 | 2.947 | 0.017 |
| 7169 | transmembrane receptor protein tyrosine kinase signaling pathway | P | 30 | 13.33 | 2.801 | 0.012 |
| 6631 | fatty acid metabolic process | P | 33 | 12.12 | 2.735 | 0.013 |
| 166 | nucleotide binding | F | 715 | 3.78 | 2.698 | 0.007 |
| 9058 | Biosynthetic process | P | 31 | 12.90 | 2.511 | 0.009 |
| 7050 | cell cycle arrest | P | 24 | 12.50 | 2.43 | 0.034 |
| 8152 | metabolic process | P | 288 | 8.68 | 2.319 | 0.025 |
| 6629 | lipid metabolic process | P | 87 | 8.05 | 2.041 | 0.043 |
| 16740 | transferase activity | F | 558 | 4.84 | 1.995 | 0.044 |
Note. C = cellular location, F = molecular function, P = biological process.
Pathways containing an over-representation of down-regulated genes (Table 1) included mitochondrion, enzymatic activity of several types and tyrosine kinase signaling. Genes downregulated in less extensive pathways included negative regulation of N+BMP signaling (Htra1, Twsg1) and regulation of vascular endothelial growth factor receptor signaling (Flt1).
Mice chronically infected with the Colombian strain of T. cruzi have intense myocarditis (Figure 1E). The inflammatory infiltrate is mainly composed by mononuclear cells, including CD4+ and CD8+ T lymphocytes (Figures 2A and B) and macrophages (Figure 2C). The analysis of genes that were upregulated ≥ 5-fold in the arrays showed alterations in a number of genes related to inflammation and immune responses. Genes coding for the macrophage cell surface marker CD68 and the lymphocyte antigens CD38 and CD52 had their expression increased in chronic chagasic hearts (Table 2), compatible with the presence of these cells in the inflammatory infiltrate.
Figure 2.
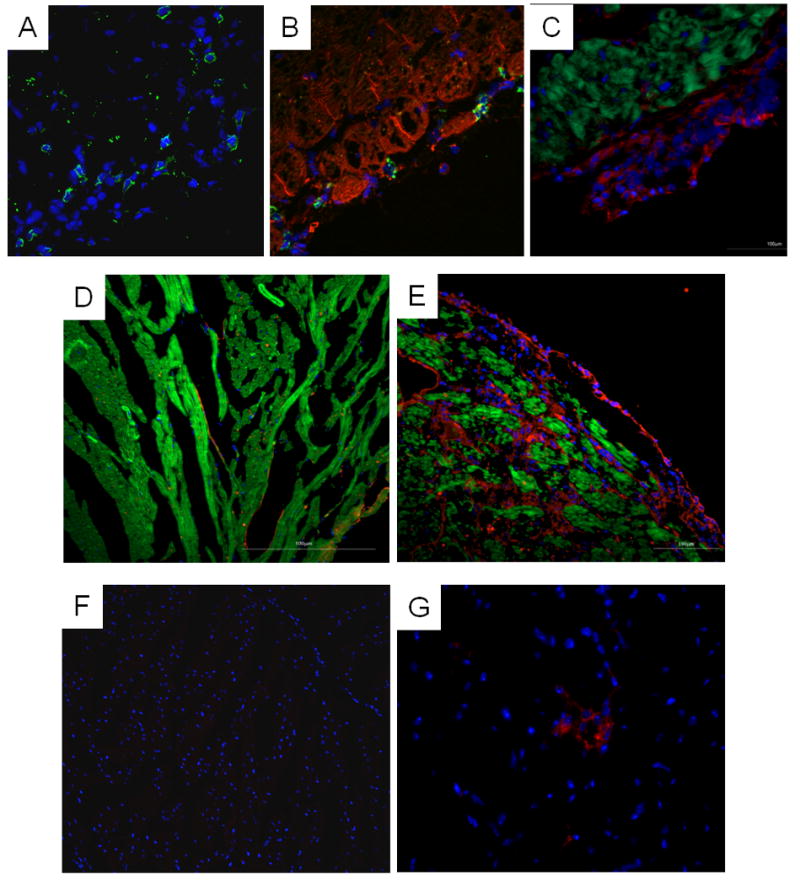
Analysis of heart sections of T. cruzi-infected mice. Hearts from non-infected controls (D and F) and chronic chagasic mice (A-C, E and G) were compared. A, CD4+ cells (green) were present in infected myocardium. B, Section stained with anti-CD8 antibody (green) and phalloidin (green). C, Presence of CD11b+ cells (red) in the inflammatory infiltrate, and phalloidin staining (green) reveals proximity of macrophages to cardiac myocytes. D (control) and E (infected), Sections stained with an anti-ICAM-1 antibody (red) and phalloidin (green) reveals upregulation of this protein in the chagasic heart. F (control) and G (infected), Sections stained with an anti-MHC-II (Ia/Ie) antibody (red), showing the presence of MHC-II expressing cells in the inflammatory infiltrate of chagasic hearts. All sections were stained with DAPI for nuclear visualization (blue).
Table 2. Selected upregulated (>5-fold) genes.
| Gene type | Gene name | Symbol | Fold regulation |
|---|---|---|---|
| Cytokines related genes | |||
| Chemokine (C-C motif) ligand 2 | Ccl2 | 26.5 | |
| Chemokine (C-C motif) ligand 7 | Ccl7/MCP3 | 16.2 | |
| Chemokine (C-C motif) ligand 8 | Ccl8 | 50.6 | |
| Chemokine (C-C motif) receptor 5 | Ccr5 | 12.1 | |
| Chemokine (C-X-C motif) ligand 12 | Cxcl12/SDF1 | 5.0 | |
| Interferon gamma induced GTPase | Igtp | 12.4 | |
| Interferon gamma inducible protein 30 | Ifi30 | 11.9 | |
| Interferon gamma inducible protein 47 | Ifi47 | 11.1 | |
| Interferon regulatory factor 1 | Irf1 | 7.7 | |
| Interferon regulatory factor 5 | Irf5 | 11.1 | |
| Interleukin 10 receptor, alpha | IL-10ra | 7.9 | |
| Interleukin 18 binding protein | IL-18bp | 6.6 | |
| Interleukin-4 receptor alpha chain precursor | IL-4R-alpha | 9.2 | |
| LPS-induced TN factor | Litaf | 9.0 | |
| Tumor necrosis factor, alpha-induced protein 2 | Tnfaip2 | 6.2 | |
| Tumor necrosis factor receptor superfamily, member 1b | Tnfrsf1b | 9.4 | |
| Tumor necrosis factor, alpha induced protein 8 like | Tnf p8l | 8.9 | |
| Immune responses-related genes | |||
| CD38 antigen | Cd38 | 7.0 | |
| CD52 antigen | Cd52/B7 | 21.6 | |
| CD68 antigen | Cd68 | 8.9 | |
| Complement component 4B | C4b | 6.0 | |
| Complement factor B | Cfb | 42.5 | |
| Fc receptor, IgE, high affinity I, gamma polypeptide | Fcer1g | 17.6 | |
| Fc receptor, IgG, low affinity III | Fcgr3 | 9.1 | |
| Histocompatibility 2, class II antigen A, alpha | H2-Aa | 38.5 | |
| Histocompatibility 2, class II, locus Mb1 | H2-DMb1 | 12.7 | |
| Histocompatibility 2, Q region locus 7 | H2-Q7 | 23.7 | |
| Histocompatibility 2, T region locus 10 | H2-T10 | 5.0 | |
| Semaphorin 4A | SemaA4 | 8.9 | |
| T-cell specific GTPase | Tgtp | 5.7 | |
| T-cell, immune regulator 1, ATPase, H+ transporting, lysosomal V0 protein A3 | Tcirg1, TIRC7 | 9.4 | |
| Toll-like receptor 2 | Tlr2 | 5.4 | |
| Cell adhesion | |||
| Galectin-3 | Gal3 | 36.9 | |
| Integrin beta 3 | Itgb3 | 6.0 | |
| Integrin beta 1 binding proteins 3 | Itgbp3 | 36.2 | |
| Intercellular adhesion molecule | Icam-1/Mala2 | 6.7 | |
| Enzymes | |||
| Cathepsin C | Ctsc | 12.5 | |
| Cathepsin H | Ctsh | 8.1 | |
| Cathepsin S | Ctss | 47.5 | |
| Cathepsin Z | Ctsz | 8.3 | |
| Lysozyme1 | Lyz1 | 8.7 | |
| Lysoyme2 | Lys2 | 7.0 | |
| Lysyl oxidase | Lox | 5.3 | |
| Phospholipase A2, group VII (platelet-activating factor acetylhydrolase, plasma) | Pla2g7 | 47.6 | |
| Proteasome (prosome, macropain) subunit, beta type 10 | Psmb10 | 6.56 | |
| Proteasome (prosome, macropain) subunit, beta type 8 (large multifunctional peptidase 7) | Psmb8 | 8.1 | |
| Matrix metalloproteinase 14 | Mmp14 | 10.8 | |
| ECM-related genes | |||
| Alpha-3 type ix collagen | Col9a3 | 7.3 | |
| Extracellular matrix protein 1 | Ecm1 | 5,5 | |
| Microfibrillar associated protein 5 | Mfap5 | 8.0 | |
| Procollagen, type I, alpha 2 | Col1a2 | 6.0 | |
| Tissue inhibitor of metalloproteinase 1 | Timp-1 | 49.6 | |
| Transforming growth factor, beta induced | Tgfbi | 15.4 | |
Upregulation of genes coding for chemoattractant factors Ccl2, Ccl7, Ccl8 and Ccl12 was observed (Table 2). Immunocytochemistry confirmed that the levels of Ccl12 (SDF-1) in hearts of chronic chagasic mice were increased in comparison with those of normal mice (Figure 3A). In addition, the expression of phospholipase A2, group VII (platelet-activating factor acetylhydrolase) and complement factor B genes were highly increased (47.6 and 42.5 fold, respectively) by chronic infection (Table 2).
Figure 3.
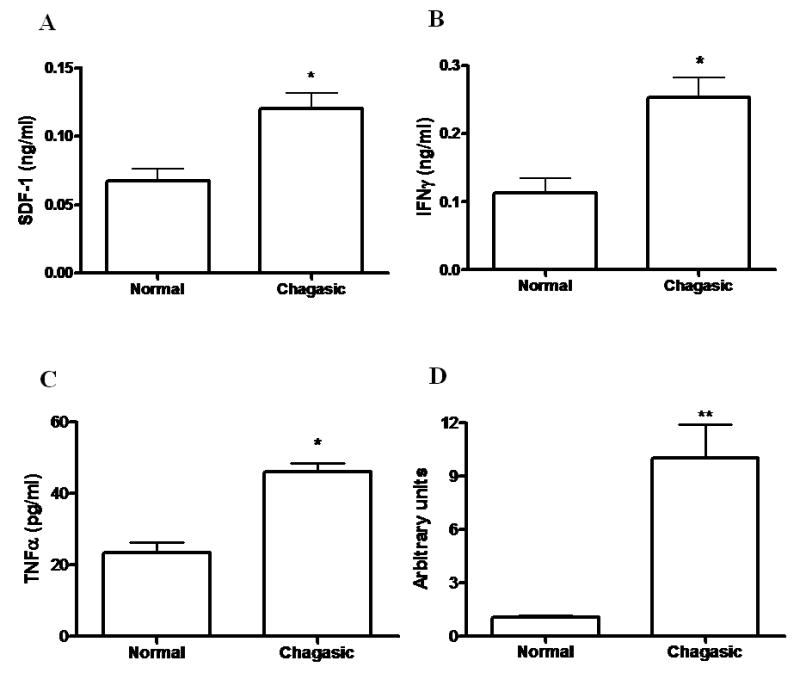
Increased production of SDF-1, IFNγ, and TNFα and transcript levels of Timp-1 in hearts of chronic chagasic mice. The levels of SDF-1 (A), IFNγ (B), and TNFα (C) were determined in heart homogenates of normal (n=4) and chagasic mice (n=9; 8 months after infection with T. cruzi), by ELISA. The analysis of Timp-1 (D) was done by quantitative real time RT-PCR using cDNA samples prepared from mRNA extracted from hearts of normal (n=5) or chronic chagasic mice (n=5). Values represent the means±SEM of values obtained from individual mice. * P<0.05.
The expression of genes coding for adhesion molecules, such as galectin-3, P-selectin ligand (CD162), integrin beta 3 (CD61), and ICAM-1 (CD54), was also increased in hearts of chagasic mice (Table 2). Immunostaining revealed that ICAM-1 is virtually absent in control heart but in hearts of infected mice it is found mainly in inflammatory and endothelial cells (Figures 2D and E). The expression of genes coding for several cathepsins, which are proteases important in lysosomal degradation, were also up regulated (Table 2). Of special interest is cathepsin S, which mediates degradation of the invariant Ii chain in antigen presenting cells [13]. The expression of genes coding for MHC class II molecules IEb and IAa were highly altered. MHC class II molecules were observed to be highly expressed in cells of the inflammatory infiltrate in infected hearts (Figures 2F and G). In addition, the expression of genes encoding two proteasome subunits was also upregulated (Table 2).
Cytokine-associated genes were differentially expressed in hearts of chagasic mice (Table 2). Of special interest is up regulation of genes associated with two cytokines related to the severe form of chronic chagasic cardiomyopathy [14, 15], IFN-γ (Igtp, Ifi30, Ifi47, Irf1, and Irf5) and TNFα (Tnfaip2, Tnfrsf1b, and Litaf). Although regulation of genes encoding IFN-γ and TNFα could not be analyzed in this microarray dataset due to technical problems, the protein levels of both cytokines were increased in the hearts of chagasic mice compared to uninfected controls (Figures 3B and C). The expression of genes coding for surface receptors such as C3a receptor 1, Fc receptors for IgE (high affinity) and IgG (low affinity), and Toll like receptor 2 was also elevated in chagasic hearts (Table 2).
Fibrosis is characteristic of hearts from chronic chagasic mice (Figure 1F), and there was marked upregulation of genes related to synthesis of extracellular matrix components (Table 2). In addition, the gene expression of lysyl oxidase, an enzyme that promotes the cross-linking of collagen fibers, was increased (Table 2). Also, the tissue inhibitor of metalloproteinase 1 (Timp-1), an inhibitor of collagen degradation, was upregulated in chronic chagasic hearts (Table 2). Quantitative real time PCR analysis confirmed a significant overexpression in Timp-1 in hearts of chronic chagasic mice compared to normal controls (Figure 3D).
Discussion
The factors responsible for the establishment of the symptomatic form of chronic Chagas' heart disease are still not fully understood. However, it is likely that the damage sustained by the myocardium is derived from parasite as well as host factors [7]. Here we have identified, by microarray analysis, a number of genes up regulated in hearts of chronic chagasic mice that likely play a role in the processes of modulation of inflammation and fibrosis in this phase of infection, and thus may represent targets for therapeutic intervention in this disease.
A significant proportion of cells in the inflammatory infiltrate found in hearts of chronic chagasic mice are macrophages, as shown here by the expression of CD11b and upregulation of CD68 gene expression. These cells are found in close contact with myofibers and may directly contribute to their damage through the secretion of TNFα. In addition, macrophages are in close contact with T lymphocytes in the inflammatory foci presenting antigens by MHC II molecules to CD4+ T lymphocytes, which secrete IFNγ, increasing the cytotoxic potential of macrophages as well as of CD8+ T cells present in the inflammatory foci. Interestingly, we found expression of the toll-like receptor 2 (Tlr2) gene up regulated in hearts of chronic chagasic mice. T. cruzi molecules such as glycosylphosphatidylinositol (GPI) anchors and glycoinositolphospholipids (GIPLs) activate macrophages to produce IL-12, TNFα, and nitric oxide [16]. Thus, the residual parasitism found in the chronic infection likely contributes to the maintenance of TNFα levels directly and of IFNγ indirectly (through IL-12 production) in the hearts of chronic chagasic mice.
Although a growing body of evidence indicates that TNFα contributes to the pathogenesis of heart failure [17], other reports suggested beneficial effects of this cytokine in the heart [18]. Cardiac myocytes express both TNFα receptors, type 1 (TNFR1) and type 2 (TNFR2) [19], which mediate its functions. TNFR1 seems to mediate the majority of the deleterious effects of TNFα, such as TNFα-induced cell death [20]. In contrast, activation of TNFR2 appears to exert protective effects against cardiac myocyte damage and apoptosis [21-23]. The strong upregulation of the TNFR2 (Tnfrsf1b) gene in the hearts of chronic chagasic mice indicates that this receptor may contribute to the low number of apoptotic cardiac myocytes found during this phase of infection [24].
PLA2G7, another molecule secreted by monocytes and macrophages and found upregulated in our study, degrades platelet activating factor (PAF), a lipid mediator which activates various cell types and promotes inflammation. Mice lacking PAF receptor (PAFR) have increased inflammation and parasitism in the hearts during the acute infection [25]. This may be due to decreased parasite uptake and macrophage activation in absence of PAFR activation, since PAF has been shown to mediate nitric oxide production and resistance to T. cruzi infection in mice [26]. In our model of chronic chagasic myocarditis, the role of PAF degradation by PLA2G7 is unknown, but the reduction of PAF accumulation may be related to the progressive damage of the myocardium, since PAF was shown to have a cardioprotective effect in isolated hearts [27].
A number of molecules involved in the recruitment of inflammatory cells to the heart of chagasic mice, including adhesion molecules and chemoattractant factors, were found upregulated in our study. Intercellular adhesion molecule 1 (ICAM-1) expression in heart and endothelial cells was also increased in chagasic hearts, as described previously [28,29]. TNFα increases the adhesiveness of endothelium for leukocytes and induces ICAM-1 expression [30]. Thus, the pro-inflammatory cytokines produced at the inflamed heart may be promoting the maintenance of inflammation by increasing the expression of ICAM-1. In agreement with the present study, overexpression of galectin-3 has been reported in T. cruzi-infected mice [31]. Galectin-3 binds to extracellular matrix components, and was shown to participate in the adhesion of the parasite to coronary artery smooth muscle cells [32].
In the present study we found that chemokine genes encoding for CCL2, CCL8, and CCL7 (MCP-1, 2, and 3, respectively) are upregulated in hearts of chronic chagasic mice. A number of studies have shown that T. cruzi infection stimulates the production of chemokines by macrophages as well as by cardiomyocytes [33-35]. These chemokines are known to recruit monocytes and T lymphocytes. Other studies have demonstrated an association between the expression of MCP-1 and 2 in the heart with myocarditis and heart dysfunction [36, 37], suggesting a role of these cytokines in the maintenance of chagasic myocarditis.
CCR5 is a receptor for several chemokines of the CC family, including CCL3, CCL4, and CCL5, known to be up regulated by infection with the Colombian strain of T. cruzi [38], and also for CCL8. Hearts of CCR5-deficient mice infected with T. cruzi have reduced migration of T cells. Since this receptor is predominantly expressed on the surface of Th1 cells [39], and a type 1 response with production of IFNγ is associated with severity of CCM, CCR5 may play an important role in the pathogenesis of chronic chagasic myocarditis, as described in a model of autoimmune myocarditis [40]. In fact, treatment of chagasic mice with a selective CCR1 and CCR5 antagonist (Met-RANTES) decreased heart inflammation and fibrosis [41]. A positive correlation between severity of cardiomyopathy and the presence of CCR5+ IFNγ+ T cells was found in chronic Chagas' disease patients [14].
We describe here that CXCL12 (SDF-1) expression is increased in hearts of chronic chagasic mice. SDF-1 is a potent chemoattractant factor for lymphocytes [42], and therefore its expression may be relevant for the maintenance of immune-mediated heart destruction during the chronic phase of infection. Conversely, since this chemokine also is a stem cell recruitment factor, its increased expression may contribute to tissue regeneration of the damaged myocardium, as reported previously in a model of myocardial ischemia [43]. In addition, MCP-3 (CCL7) was recently shown to be a mesenchymal stem cell homing factor to the myocardium [44]. Thus, the increased expression of these chemokines indicates that migration of stem cells can be promoted in chronic chagasic myocarditis by the presence of stem cell chemoattractant factors such as SDF-1 and MCP-3. In fact, we have shown that intravenously injected bone marrow cells migrate to the hearts of chronic chagasic mice [45].
The myocardial interstitial collagen matrix surrounds and supports cardiac myocytes and the coronary microcirculation, and its integrity is critical for the proper function of the heart. Thus, alterations in the collagen matrix will result in the disruption of myocardial mechanical properties and ventricular function [46]. In Chagas' disease, as a consequence of the sustained inflammatory process found in the myocardium during the chronic phase of infection, fibrosis is evident and contributes to cardiac remodeling. We also observed alterations in genes related to extracellular matrix (ECM) deposition, such as ECM components and Timp-1. Plasma concentrations of TIMP-1 are significantly elevated in patients with terminal heart failure compared to healthy controls [52], suggesting that this metalloproteinase inhibitor may also play a role in the evolution of heart failure in chronic Chagas' disease. In addition, lysyl oxidase was increased in chagasic hearts. This enzyme promotes the cross-linking of collagen fibers, irreversibly altering the structure and function of the ECM proteins, causing dysfunction of the cardiomyocytes and, consequently, of the heart [46], and therefore is likely playing an important role in the evolution of fibrosis in the chronic chagasic hearts.
To understand the delicate balance of multiple factors involved in the pathogenesis of Chagas' disease is a complex task. Microarray approaches have been used before in mouse models of T. cruzi infection (C3H/HeN mice infected with T. cruzi Sylvio X10/4 strain [47] and C57Bl/6·129sv mice infected with T. cruzi Brazil strain [48, 49] as well as in hearts of chronic chagasic patients [50]. Using the model of infection with Colombian T. cruzi strain, we have identified new potentially important genes that may serve as a basis for the development of therapeutic interventions in chronic Chagas' heart disease.
Supplementary Material
Acknowledgments
The authors thank Carine Machado and Fabiola Encinas Rosa for technical assistance.
Financial support: Financial support for these studies was provided by the National Institutes of Health (HL-73732, HD-32573, AI-076248, AI-052739), CAPES, CNPq, FAPERJ and FAPESB. RCSG and LR were supported by a Fogarty International Training Grant D43TW007129. This study was presented in III International Symposium of advanced therapies and stem cell, September, 2008, Curitiba, Paraná, Brazil.
Footnotes
Potential conflicts of interest: None to declare.
References
- 1.World Health Organization. Chagas' disease: important advances in elimination of transmission in four countries in Latin America. Vol. 183. WHO Feature; 1995. pp. 1–3. [Google Scholar]
- 2.Dias JCP, Coura JR. Epidemiologia. In: Dias JCP, Coura JR, editors. Clínica e terapêutica da doença de Chagas: uma abordagem prática para o clínico geral. 2nd. Brazil: FIOCRUZ; 1997. pp. 36–66. [Google Scholar]
- 3.Rosenbaum MB. Chagasic myocardiopathy. Prog Cardiovasc Dis. 1964;7:199–225. doi: 10.1016/s0033-0620(64)80020-7. [DOI] [PubMed] [Google Scholar]
- 4.Chiale PA, Rosenbaum MB. In: Clinical and pharmacological characterization and treatment of potentially malignant arrythmias of chronic chagasic cardiomiopathy Handbook of Experimental Pharmacology. 5. Williams EMV, Campbell TJ, editors. Spring-Verlag; 1989. pp. 601–20. [Google Scholar]
- 5.Tanowitz HB, Kirchhoff LV, Simon D, Morris SA, Weiss LM, Wittner M. Chagas' disease. Clin Microbiol Rev. 1992;5:400–19. doi: 10.1128/cmr.5.4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petkova SB, Huang H, Factor SM, et al. The role of endothelin in the pathogenesis of Chagas' disease. Int J Parasitol. 2001;31:499–511. doi: 10.1016/s0020-7519(01)00168-0. [DOI] [PubMed] [Google Scholar]
- 7.Soares MB, Pontes-De-Carvalho L, Ribeiro-dos-Santos R. The pathogenesis of Chagas' disease: when autoimmune and parasite-specific immune responses meet. An Acad Bras Cienc. 2001;73:547–59. doi: 10.1590/s0001-37652001000400008. [DOI] [PubMed] [Google Scholar]
- 8.Garcia S, Ramos CO, Senra JF, et al. Treatment with benznidazole during the chronic phase of experimental Chagas' disease decreases cardiac alterations. Antimicrob Agents Chemother. 2005;49:1521–28. doi: 10.1128/AAC.49.4.1521-1528.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Federici EE, Abelmann WN, Neva FA. Chronic and progressive myocarditis in C3H mice infected with Trypanosoma cruzi. Am J Trop Med Hyg. 1964;13:272–80. doi: 10.4269/ajtmh.1964.13.272. [DOI] [PubMed] [Google Scholar]
- 10.Iacobas DA, Fan C, Iacobas S, Spray DC, Haddad GG. Transcriptomic changes in developing kidney exposed to chronic hypoxia. Biochem Biophys Res Comm. 2006;349(1):329–38. doi: 10.1016/j.bbrc.2006.08.056. [DOI] [PubMed] [Google Scholar]
- 11.Iacobas DA, Iacobas S, Li WE, Zoidl G, Dermietzel R, Spray DC. Genes controlling multiple functional pathways are transcriptionally regulated in connexin43 null mouse heart. Physiol Genomics. 2005b;20:211–23. doi: 10.1152/physiolgenomics.00229.2003. [DOI] [PubMed] [Google Scholar]
- 12.Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- 13.Cunha-Neto E, Rizzo LV, Albuquerque F, et al. Cytokine production profile of heart-infiltrating T cells in Chagas' disease cardiomyopathy. Braz J Med Biol Res. 1998;31:133–7. doi: 10.1590/s0100-879x1998000100018. [DOI] [PubMed] [Google Scholar]
- 14.Gomes JA, Bahia-Oliveira LM, Rocha MO, Busek SC, Teixeira MM, Silva JS, Correa-Oliveira R. Type 1 chemokine receptor expression in Chagas' disease correlates with morbidity in cardiac patients. Infect Immun. 2005;12:7960–6. doi: 10.1128/IAI.73.12.7960-7966.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campos MA, Almeida IC, Takeuchi O, et al. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J Immunol. 2001;167:416–23. doi: 10.4049/jimmunol.167.1.416. [DOI] [PubMed] [Google Scholar]
- 16.Anker SD, Coats AJ. How to RECOVER from RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int J Cardiol. 2002;86:123–30. doi: 10.1016/s0167-5273(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 17.Kurrelmeyer KM, Michael LH, Baumgarten G, et al. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci USA. 2000;97:5456–61. doi: 10.1073/pnas.070036297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torre-Amione G, Kapadia S, Lee J, Bies RD, Lebovitz R, Mann DL. Expression and functional significance of tumor necrosis factor receptors in human myocardium. Circulation. 1995;92:1487–93. doi: 10.1161/01.cir.92.6.1487. [DOI] [PubMed] [Google Scholar]
- 19.Shen HM, Pervaiz S. TNF receptor superfamily-induced cell death: redox-dependent execution. Faseb J. 2006;20:1589–98. doi: 10.1096/fj.05-5603rev. [DOI] [PubMed] [Google Scholar]
- 20.Higuchi Y, McTiernan CF, Frye CB, McGowan BS, Chan TO, Feldman AM. Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor-alpha-induced cardiomyopathy. Circulation. 2004;109:1892–7. doi: 10.1161/01.CIR.0000124227.00670.AB. [DOI] [PubMed] [Google Scholar]
- 21.Ramani R, Mathier M, Wang P, et al. Inhibition of tumor necrosis factor receptor-1-mediated pathways has beneficial effects in a murine model of postischemic remodeling. Am J Physiol Heart Circ Physiol. 2004;287:H1369–77. doi: 10.1152/ajpheart.00641.2003. [DOI] [PubMed] [Google Scholar]
- 22.Defer N, Azroyan A, Pecker F, Pavoine C. TNFR1 and TNFR2 signaling interplay in cardiac myocytes. J Biol Chem. 2007;282:35564–73. doi: 10.1074/jbc.M704003200. [DOI] [PubMed] [Google Scholar]
- 23.Rossi MA, Souza AC. Is apoptosis a mechanism of cell death of cardiomyocytes in chronic chagasic myocarditis? Int J Cardiol. 1999;68:325–31. doi: 10.1016/s0167-5273(98)00375-1. [DOI] [PubMed] [Google Scholar]
- 24.da Silva RP, Gordon S. Phagocytosis stimulates alternative glycolysation of macrosialin (mouse CD68), a macrophage-specific endosomal protein. Biochem J. 1999;338:687–94. [PMC free article] [PubMed] [Google Scholar]
- 25.Aliberti JC, Machado FS, Gazzinelli RT, Teixeira MM, Silva JS. Platelet-activating factor induces nitric oxide synthesis in Trypanosoma cruzi-infected macrophages and mediates resistance to parasite infection in mice. Infect Immun. 1999a;67:2810–4. doi: 10.1128/iai.67.6.2810-2814.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penna C, Alloatti G, Cappello S, et al. Platelet-activating factor induces cardioprotection in isolated rat heart akin to ischemic preconditioning: role of phosphoinositide 3-kinase and protein kinase C activation. Am J Physiol Heart Circ Physiol. 2005;288:H2512–20. doi: 10.1152/ajpheart.00599.2004. [DOI] [PubMed] [Google Scholar]
- 27.Laucella S, Salcedo R, Castaños-Velez E, et al. Increased expression and secretion of ICAM-1 during experimental infection with Trypanosoma cruzi. Parasite Immunol. 1996;18:227–39. doi: 10.1046/j.1365-3024.1996.d01-95.x. [DOI] [PubMed] [Google Scholar]
- 28.Huang H, Calderon TM, Berman JW, Braunstein VL, Weiss LM, Wittner M, Tanowitz HB. Infection of endothelial cells with Trypanosoma cruzi activates NF-kappaB and induces vascular adhesion molecule expression. Infect Immun. 1999;67:5434–40. doi: 10.1128/iai.67.10.5434-5440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ledebur HC, Parks TP. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. J Biol Chem. 1995;270:933–43. doi: 10.1074/jbc.270.2.933. [DOI] [PubMed] [Google Scholar]
- 30.Vray B, Camby I, Vercruysse V, et al. Up-regulation of galectin-3 and its ligands by Trypanosoma cruzi infection with modulation of adhesion and migration of murine dendritic cells. Glycobiology. 2004;14:647–57. doi: 10.1093/glycob/cwh068. [DOI] [PubMed] [Google Scholar]
- 31.Kleshchenko YY, Moody TN, Furtak VA, Ochieng J, Lima MF, Villalta F. Human galectin-3 promotes Trypanosoma cruzi adhesion to human coronary artery smooth muscle cells. Infect Immun. 2004;72:6717–21. doi: 10.1128/IAI.72.11.6717-6721.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villalta F, Zhang Y, Bibb KE, Kappes JC, Lima MF. The cysteine-cysteine family of chemokines RANTES, MIP-1alpha, and MIP-1beta induce trypanocidal activity in human macrophages via nitric oxide. Infect Immun. 1998;66:4690–5. doi: 10.1128/iai.66.10.4690-4695.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aliberti JC, Machado FS, Souto JT, Campanelli AP, Teixeira MM, Gazzinelli RT, Silva JS. beta-Chemokines enhance parasite uptake and promote nitric oxide-dependent microbiostatic activity in murine inflammatory macrophages infected with Trypanosoma cruzi. Infect Immun. 1999b;67:4819–26. doi: 10.1128/iai.67.9.4819-4826.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Machado FS, Martins GA, Aliberti JC, Mestriner FL, Cunha FQ, Silva JS. Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation. 2000;102:3003–8. doi: 10.1161/01.cir.102.24.3003. [DOI] [PubMed] [Google Scholar]
- 35.Kolattukudy PE, Quach T, Bergese S, et al. Myocarditis induced by targeted expression of the MCP--1 gene in murine cardiac muscle. Am J Pathol. 1998;152:101–1. [PMC free article] [PubMed] [Google Scholar]
- 36.Shen Y, Xu W, Chu YW, Wang Y, Liu QS, Xiong SD. Coxsackievirus group B type 3 infection upregulates expression of monocyte chemoattractant protein 1 in cardiac myocytes, which leads to enhanced migration of mononuclear cells in viral myocarditis. J Virol. 2004;78:12548–56. doi: 10.1128/JVI.78.22.12548-12556.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Talvani A, Ribeiro CS, Aliberti JC, et al. Kinetics of cytokine gene expression in experimental chagasic cardiomyopathy: tissue parasitism and endogenous IFN-gamma as important determinants of chemokine mRNA expression during infection with Trypanosoma cruzi. Microbes Infect. 2000;2:851–66. doi: 10.1016/s1286-4579(00)00388-9. [DOI] [PubMed] [Google Scholar]
- 38.Turner JE, Steinmetz OM, Stahl RA, Panzer U. Targeting of Th1-associated chemokine receptors CXCR3 and CCR5 as therapeutic strategy for inflammatory diseases. Mini Rev Med Chem. 2007;7:1089–96. doi: 10.2174/138955707782331768. [DOI] [PubMed] [Google Scholar]
- 39.Gong X, Feng H, Zhang S, Yu Y, Li J, Wang J, Guo B. Increased expression of CCR5 in experimental autoimmune myocarditis and reduced severity induced by anti-CCR5 monoclonal antibody. J Mol Cell Cardiol. 2007;42:781–91. doi: 10.1016/j.yjmcc.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 40.Marino AP, da Silva A, dos Santos P, Pinto LM, Gazzinelli RT, Teixeira MM, Lannes-Vieira J. Regulated on activation, normal T cell expressed and secreted (RANTES) antagonist (Met-RANTES) controls the early phase of Trypanosoma cruzi-elicited myocarditis. Circulation. 2004;110:1443–9. doi: 10.1161/01.CIR.0000141561.15939.EC. [DOI] [PubMed] [Google Scholar]
- 41.Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1) J Exp Med. 1996;184:1101–9. doi: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Askari AT, Unzek S, Popovic ZB, et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet. 2003;362:697–703. doi: 10.1016/S0140-6736(03)14232-8. [DOI] [PubMed] [Google Scholar]
- 43.Schenk S, Mal N, Finan A, et al. Monocyte chemotactic protein-3 is a myocardial mesenchymal stem cell homing factor. Stem Cells. 2007;25:245–51. doi: 10.1634/stemcells.2006-0293. [DOI] [PubMed] [Google Scholar]
- 44.Soares MB, Lima RS, Rocha LL, Takyia CM, Pontes-de-Carvalho L, de Carvalho AC, Ribeiro-dos-Santos R. Transplanted bone marrow cells repair heart tissue and reduce myocarditis in chronic chagasic mice. Am J Pathol. 2004;164:441–7. doi: 10.1016/s0002-9440(10)63134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Souza RR. Aging of myocardial collagen. Biogerontology. 2002;3:325–35. doi: 10.1023/a:1021312027486. [DOI] [PubMed] [Google Scholar]
- 46.Milting H, Ellinghaus P, Seewald M, et al. Plasma biomarkers of myocardial fibrosis and remodeling in terminal heart failure patients supported by mechanical circulatory support devices. J Heart Lung Transplant. 2008;27:589–96. doi: 10.1016/j.healun.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 47.Garg N, Popov VL, Papaconstantinou J. Profiling gene transcription reveals a deficiency of mitochondrial oxidative phosphorylation in Trypanosoma cruzi-infected murine hearts: implications in chagasic myocarditis development. Biochim Biophys Acta. 2003;1638:106–20. doi: 10.1016/s0925-4439(03)00060-7. [DOI] [PubMed] [Google Scholar]
- 48.Mukherjee S, Belbin TJ, Spray DC, et al. Microarray analysis of changes in gene expression in a murine model of chronic chagasic cardiomyopathy. Parasitol Res. 2003;3:187–96. doi: 10.1007/s00436-003-0937-z. [DOI] [PubMed] [Google Scholar]
- 49.Mukherjee S, Nagajyothi F, Mukhopadhyay A, et al. Alterations in myocardial gene expression associated with experimental Trypanosoma cruzi infection. Genomics. 2008;91:423–32. doi: 10.1016/j.ygeno.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cunha-Neto E, Dzau VJ, Allen PD, et al. Cardiac gene expression profiling provides evidence for cytokinopathy as a molecular mechanism in Chagas' disease cardiomyopathy. Am J Pathol. 2005;167:305–13. doi: 10.1016/S0002-9440(10)62976-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.