

Abstract
Skizzle (SkzL), secreted by Streptococcus agalactiae, has moderate sequence identity to streptokinase and staphylokinase, bacterial activators of human plasminogen (Pg). SkzL binds [Glu]Pg with low affinity (KD 3–16 μm) and [Lys]Pg and plasmin (Pm) with indistinguishable high affinity (KD 80 and 50 nm, respectively). Binding of SkzL to Pg and Pm is completely lysine-binding site-dependent, as shown by the effect of the lysine analog, 6-aminohexanoic acid. Deletion of the COOH-terminal SkzL Lys415 residue reduces affinity for [Lys]Pg and active site-blocked Pm 30-fold, implicating Lys415 in a lysine-binding site interaction with a Pg/Pm kringle. SkzL binding to active site fluorescein-labeled Pg/Pm analogs demonstrates distinct high and low affinity interactions. High affinity binding is mediated by Lys415, whereas the source of low affinity binding is unknown. SkzL enhances the activation of [Glu]Pg by urokinase (uPA) ∼20-fold, to a maximum rate indistinguishable from that for [Lys]Pg and [Glu]Pg activation in the presence of 6-aminohexanoic acid. SkzL binds preferentially to the partially extended β-conformation of [Glu]Pg, which is in unfavorable equilibrium with the compact α-conformation, thereby converting [Glu]Pg to the fully extended γ-conformation and accelerating the rate of its activation by uPA. SkzL enhances [Lys]Pg and [Glu]Pg activation by single-chain tissue-type Pg activator, ∼42- and ∼650-fold, respectively. SkzL increases the rate of plasma clot lysis by uPA and single-chain tissue-type Pg activator ∼2-fold, confirming its cofactor activity in a physiological model system. The results suggest a role for SkzL in S. agalactiae pathogenesis through fibrinolytic enhancement.
Keywords: Bacteria, Enzyme Kinetics, Enzyme Mechanisms, Fibrinolysis, Fluorescence, Plasmin, Plasminogen, Plasminogen Regulation, Protease, Protein-Protein Interactions
Introduction
Group B streptococci (GBS)4 cause severe diseases, including sepsis, meningitis, arthritis, and endocarditis in neonates and immune-compromised patients (1, 2). Streptococcus agalactiae serotype V is emerging as an invasive GBS pathogen with particular morbidity and mortality in neonates (3–5), but the mechanism of GBS pathogenesis is not fully understood. Unlike group A, C, and G streptococci, GBS does not express streptokinase (SK), a pathogenicity factor in group A streptococcal infection (6). Activation of the fibrinolytic system is a mechanism of bacterial pathogenesis employed by many bacteria through expression of cell-surface plasminogen (Pg)-binding proteins and the secretion of Pg activators. In this mechanism, aberrant up-regulation of the fibrinolytic system increases plasmin (Pm) generation, which degrades fibrin and extracellular matrix to disseminate the bacteria through soft tissue (7). Activation of Pg on the cell surface through Pg-binding proteins, such as Pg-binding group A streptococcal M-like protein (group A, C, and G streptococci), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; group A–C streptococci), and α-enolase (group A and B streptococci), results in coating of the cell surface with Pg or active Pm (8–12). A recent study indicated that Pg activation on the surface of S. agalactiae occurs through the GAPDH-bound Pg complex, facilitating GBS virulence and promoting bacterial dissemination in a murine model (10). Secreted Pg-activating proteins, such as SK and staphylokinase (SAK), are not enzymes themselves but form complexes with Pg or Pm to enable proteolytic Pm generation. This proteolysis can occur locally on bacterial cell surfaces through Pg-binding proteins or systemically by the free, secreted Pg activators in the vascular system. Although complete proteomic functional studies have not been performed, to date there are no known S. agalactiae secreted proteins with established human fibrinolytic activity.
Genomic sequencing of an S. agalactiae serotype V clinical isolate (2603 V/R) revealed 2175 predicted genes (13), including NP_688136.1, which corresponds to a novel protein of unknown function, subsequently named skizzle (SkzL). SkzL has 22% sequence identity to SK from Streptococcus equisimilis and pyogenes, 33% to SAK from Staphylococcus aureus, and 24% to PauB, a bovine Pg activator from Streptococcus uberis (14). In studies of genomic sequence diversity, SkzL (protein unnamed, gene name sk1A2) had weak similarity to SAK and SK (4). SAK, SK, and PauB facilitate the conversion of Pg into Pm, albeit through different mechanisms.
Native [Glu]Pg, the form circulating in blood, consists of an NH2-terminal PAN (Pg/apple/nematode) module, five kringle domains, and a serine proteinase catalytic domain. Kringles 1, 4, and 5 contain lysine-binding sites (LBS) with moderate to low affinity for l-lysine and higher affinity for the lysine analog 6-aminohexanoic acid (6-AHA) (15–20). Following cleavage by Pm, the NH2-terminal 77-residue PAN module of [Glu]Pg is released to yield [Lys]Pg (18). As a result of the NH2-terminal PAN module interaction with kringle 5, [Glu]Pg is stabilized in the compact α-conformation by physiological concentrations of chloride ion (21, 22). The α-conformation of [Glu]Pg, however, is in equilibrium with a low concentration of a partially extended β-conformation governed by the chloride ion concentration (17, 22–25). Lacking the PAN module, [Lys]Pg adopts the β-conformation in the absence or presence of chloride ion (22, 26). Upon low affinity binding of lysine analogs, such as 6-AHA to kringles 4 and 5, both [Glu]Pg and [Lys]Pg assume the fully extended γ-conformation (22, 23, 26–32).
Proteolytic Pg activation occurs through cleavage of the Arg561–Val562 peptide bond in the catalytic domain to form Pm. SK activates Pg through a conformational, nonproteolytic molecular sexuality mechanism (33–38). The SK NH2 terminus inserts into the NH2-terminal binding cleft in the Pg catalytic domain, inducing a conformational change to form a functional active site capable of proteolytically activating substrate Pg molecules (34–41). Binding of Pg as a substrate occurs through LBS interaction between kringle 5 and residues Arg235, Lys256, and Lys257 in the 250-loop of SK (42). SAK facilitates activation of Pg in the presence of catalytic concentrations of Pm. Binding of SAK to Pm changes the substrate specificity of Pm, allowing activation of Pg (43–45). PauB and its homolog PauA appear to follow a mechanism similar to SK, including formation of a nonproteolytically activated Pg complex, followed by proteolysis of substrate molecules of Pg but are specific for activation of bovine Pg (46–50).
Human Pg is proteolytically activated endogenously by either of two activators, urokinase and tissue-type plasminogen activator (tPA). High molecular weight single-chain urokinase (uPA) contains a catalytic domain in the COOH-terminal region and an epidermal growth factor domain and one kringle domain in the NH2-terminal region (51, 52). The epidermal growth factor and kringle domains are liberated by cleavage at Glu143–Leu144 by the metalloproteinase, pump-1, to form low molecular weight urokinase (52). Single-chain urokinase is converted to the more enzymatically active form, two-chain urokinase, through cleavage of Lys158–Ile159 in the catalytic domain by Pm or kallikrein (51–55). The low activity zymogen-like form of tPA, single-chain tPA (sctPA), is a 70-kDa protein that is cleaved by Pm at Arg275–Ile276 to form the more active two-chain tPA (56). One structural difference between tPA and uPA is the presence of a second kringle domain in tPA containing an LBS with high affinity for lysine, 6-AHA, and fibrin fragments (57, 58).
The modest sequence identity between SkzL and SK includes conservation of the NH2-terminal (Ile1–Ala2–Gly3) residues and the COOH-terminal Lys415 residue (Lys414 for SK) required for SK-induced Pg activation (36–39, 59). Despite the presence of these residues and predicted secondary structure similarity to SK, SkzL could not be shown to activate Pg to Pm, conformationally or proteolytically, in the absence or presence of catalytic concentrations of Pm. The conservation of the COOH-terminal Lys residue suggested a potential binding interaction of SkzL with the LBS of a Pg and Pm kringle. The present studies were undertaken to characterize the binding interactions of SkzL with Pg and Pm and to elucidate its function as a potential fibrinolytic cofactor of uPA- and sctPA-catalyzed Pg activation. The studies show that SkzL is secreted from S. agalactiae serotype V and binds both Pg and Pm in a LBS-dependent manner, primarily through Lys415. SkzL enhances uPA-catalyzed activation of [Glu]Pg by facilitating its conversion from the compact α-conformation to the fully extended γ-conformation, and it enhances uPA-mediated plasma clot lysis. SkzL also enhances [Glu]Pg and [Lys]Pg activation by sctPA and plasma clot lysis by sctPA. To our knowledge, SkzL is the first secreted protein from S. agalactiae to be identified that usurps the human fibrinolytic system, presumably to enable dissemination of infection.
EXPERIMENTAL PROCEDURES
Protein Purification and Characterization
[Glu]Pg, [Lys]Pg, and Pm (all carbohydrate form 2) were prepared as described previously (41, 60, 61). [5F]FFR-[Lys]Pg, [5F]FFR-[Glu]Pg, and [5F]FFR-Pm were generated by a thiol-specific reaction with 5-(iodoacetamido)fluorescein following inactivation of the SK-induced active site in [Lys]Pg and [Glu]Pg with Nα-[(acetylthio)acetyl]-d-Phe-Phe-Arg-CH2Cl essentially as described previously (39–41, 62). Native Pm activity was calculated based on known parameters for d-Val-Leu-Lys-p-nitroanilide (VLK-pNA) hydrolysis (41). Human high molecular weight uPA (Calbiochem) was reconstituted in ultra-pure water (yielding a solution containing 150 mm Cl− contributed by NaCl and Tris-Cl). Protein concentrations of uPA and sctPA were determined by the bicinchoninic acid protein assay (Pierce). sctPA (Molecular Innovations) was stored in 0.5 m HEPES, 0.5 m NaCl, pH 7.4, buffer to ensure solubility and was limited to one freeze-thaw cycle to maintain stable activity.
The gene for SkzL (NP_688136.1) was cloned from S. agalactiae 2603 V/R genomic DNA (ATCC) into a modified pET30b(+) (Novagen) vector (59) containing a tobacco etch virus protease-cleavable NH2-terminal His6 tag. SkzL was expressed by Rosetta 2 (DE3) pLysS Escherichia coli (Novagen) in 0.1 mg/ml kanamycin following 0.5 mm isopropyl β-d-thiogalactopyranoside induction for 4 h at 30 °C. Following bacterial lysis by sonication, protein in the clarified supernatant was purified by Ni2+-iminodiacetic acid-Sepharose (5 ml) chromatography in 50 mm HEPES, 325 mm NaCl, 20 mm imidazole, pH 7.4, eluted with a 50-ml gradient of 20–500 mm imidazole in the equilibration buffer. Trace protein contaminants, including dimeric SkzL, were removed by anion exchange chromatography on Q-Sepharose (Amersham Biosciences) equilibrated with 50 mm HEPES, 50 mm NaCl, 1 mm EDTA, pH 7.4, eluted with a gradient of 50–500 mm NaCl over 30 column volumes. A 1:10 molar equivalent of recombinant tobacco etch virus protease was added to SkzL, and the mixture was incubated for 12 h to liberate the His6 tag, followed by repeated Ni2+-iminodiacetic acid-Sepharose chromatography to separate noncleaved protein and free His6 tag from cleaved protein (59). To preserve disulfide bond integrity, cleavage was performed in the absence of dithiothreitol. Tobacco etch virus protease cleavage resulted in the proper SkzL NH2-terminal sequence as confirmed by NH2-terminal sequencing (Ile1–Ala2–Gly3–Pro4–Ser5). Cysteine-to-serine substitution mutants and the COOH-terminal lysine deletion mutant (SkzLΔK415) were generated by QuikChange site-directed mutagenesis (Stratagene) and purified as described. [5F]-SkzL was generated by reaction of free thiol groups with 5-(iodoacetamido)fluorescein followed by chromatography on Sephadex G-25 and dialysis to remove excess probe. Probe incorporation was determined to be 1.0 mol of probe/mol of SkzL by absorbance measurements at 280 and 498 nm with an absorption coefficient of 84,000 m−1 cm−1 for fluorescein and an A280/A498 nm ratio of 0.19 in 6 m guanidine, 100 mm Tris-HCl, 1 mm EDTA, pH 8.5, buffer (63).
All proteins were snap-frozen and stored in 50 mm HEPES, 125 mm NaCl, pH 7.4, at −80 °C. Protein concentrations were determined from the 280-nm absorbance using the following absorption coefficients ((mg/ml)−1cm−1) and molecular masses (Da): [Glu]Pg, 1.69 and 92,000; [Lys]Pg and Pm, 1.69 and 84,000 (60, 64). The SkzL absorption coefficient at 280 nm of 1.23 was calculated from the amino acid sequence and confirmed by absorbance measurements (65). The molecular mass of 47,400 Da for SkzL was determined in excellent agreement with that calculated from the amino acid sequence by mass spectrometry using a Waters Acquity Ultra PerformanceTM LC/Thermo Finnigan LTQ with a lysozyme standard for optimization and calibration.
S. agalactiae Protein Expression
S. agalactiae serotype V was cultured in Todd-Hewitt yeast broth at 37 °C. Bacterial supernatant was collected at various time points by centrifugation at 39,000 × g for 15 min. Secreted proteins were obtained by precipitation with trichloroacetic acid (7.5%) and washed with acetone for use in Western blotting. A polyclonal rabbit anti-SkzL antibody was generated and purified by affinity chromatography on SkzL-coupled CNBr-activated Sepharose 4B (ProSci Inc.). Proteins were subjected to SDS-PAGE on 4–15% gradient gels, transferred onto polyvinylidine fluoride membranes, and developed with a BM chemiluminescence Western blotting kit (Roche).
Fluorescence Equilibrium Binding
Titrations of 50 nm [5F]FFR-[Lys]Pg, [5F]FFR-[Glu]Pg, and [5F]FFR-Pm with wtSkzL and SkzLΔK415 were performed in 50 mm HEPES, 125 mm NaCl, 1 mm EDTA, 1 mg/ml PEG 8,000, 10 μm FFR-CH2Cl, and 1 mg/ml bovine serum albumin, pH 7.4, at 25 °C using PEG 20,000-coated acyclic cuvettes. Titrations of active site-blocked fluorescent Pg/Pm analogs with wtSkzL and SkzLΔK415 performed without the use of polarizers revealed a high affinity decrease in fluorescence intensity and a low affinity decrease in fluorescence anisotropy. Consequently, identical titrations were performed with vertical excitation and emission at the magic angle to eliminate anisotropy effects. Excitation was at 500 nm, and emission was measured at 516 nm with 4-nm bandpasses.
Fluorescence titrations of native [Glu]Pg, [Lys]Pg, and FFR-Pm binding to 100 and 500 nm [5F]-SkzL were performed using identical buffer conditions. Fluorescence was measured with excitation at 492 nm and emission at 510 nm with 8-nm bandpasses without the use of fluorescence polarization. Competitive binding experiments with 100 nm [5F]-SkzL in the absence and presence of fixed concentrations of wtSkzL or SkzLΔK415 to [Lys]Pg and FFR-Pm were performed under identical conditions. In these titrations, mixtures of [5F]-SkzL and either wtSkzL or SkzLΔK415 were preincubated for 10 min at 25 °C prior to titration with [Lys]Pg or FFR-Pm. Following correction for buffer blank and probe dilution (≤10%), data were expressed as the fractional change in the initial fluorescence, (Fobs − Fo)/Fo = ΔF/Fo. Titrations were analyzed by nonlinear least squares fitting of the quadratic binding Equation 1,
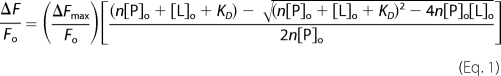 |
where ΔFmax/Fo is the maximum fluorescence change; KD is the dissociation constant; n is the stoichiometric factor; [P]o is the total concentration of probe-labeled protein, and [L]o is the total ligand concentration (39–41, 62). Competitive titrations were analyzed by nonlinear least squares fitting of the cubic competitive binding equation for tight binding interactions to give dissociation constants for both ligand and competitor, with the stoichiometry fixed at 1 for the competitors (66).
Direct titrations of wtSkzL binding to [5F]FFR-[Lys]Pg and [5F]FFR-Pm performed without polarizers revealed an apparent anisotropy decrease, representing a second distinct binding interaction. These data were analyzed by nonlinear least squares fitting of the sum of the solution to the quadratic equation (Equation 1) as ΔF1/Fo, with n = 1 for the high affinity interaction, and a hyperbola for the weak interaction with the assumption that [L]free ≈ [L]o for this interaction shown in Equation 2,
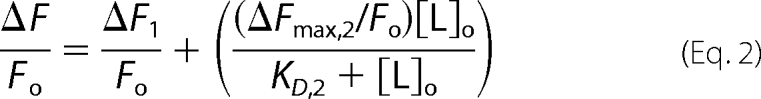 |
where KD,1 obtained from the quadratic equation and KD,2 are the dissociation constants for the high and low affinity interactions, respectively; ΔFmax,1/Fo from the quadratic equation and ΔFmax,2/Fo are the corresponding maximum fluorescence changes, and [L]o is the total ligand concentration. Experimental error in the fitted parameters represents ± 2 S.D.
Chromogenic Substrate Hydrolysis
The initial rates of hydrolysis of 200 μm pyro-Glu-Pro-Arg-pNA (pyro-EPR-pNA) by 10 nm Pm in PEG 20,000-coated microtiter plates was measured continuously at 405 nm as a function of SkzL concentration. kcat and Km were determined by fitting of the substrate dependence in the absence and presence of SkzL by the Michaelis-Menten equation. Rates of substrate hydrolysis were normalized to a 1-cm path length by a correction factor of 1.66 for reaction volumes of 300 μl. Titrations as a function of SkzL were normalized to the rate of pyro-EPR-pNA hydrolysis in the absence of SkzL and analyzed by Equation 2 (wtSkzL) or Equation 1 (SkzLΔK415) to determine dissociation constants.
Plasminogen Activation Kinetics
The kinetics of activation of 100 nm [Glu]Pg or 100 nm [Lys]Pg by uPA (0.32 or 0.11 nm, respectively) or sctPA (3 nm) as a function of SkzL concentration were performed by continuous measurement of hydrolysis of 200 μm VLK-pNA at 405 nm and 25 °C, as described previously (35, 42). Assays were performed in chloride-containing buffer (100 mm HEPES, 100 mm NaCl, 1 mm EDTA, 1 mg/ml PEG 8,000, pH 7.4) or no chloride buffer (100 mm HEPES, 100 mm sodium acetate, 1 mm EDTA, 1 mg/ml PEG 8,000, pH 7.4) using PEG 20,000-coated polystyrene cuvettes. In assays containing SkzL, uPA or sctPA and SkzL diluted in buffer were preincubated in the cuvette at 25 °C for 10 min prior to addition of Pg and substrate. Progress curves at varying SkzL concentrations were fit by the parabolic rate equation and truncated to include only data linear when plotted as absorbance against time2 and less than 10% substrate consumption.
Plasma Clot Lysis
Plasma clot lysis was measured by incubation of 160 μl of normal human plasma at 37 °C with final concentrations of 6 nm thrombin, 10 mm CaCl2, and 7 nm uPA or 1.5 nm sctPA, in a final volume of 200 μl. All proteins were diluted in 50 mm HEPES, 125 mm NaCl, pH 7.4, prior to addition. Increasing concentrations of SkzL were preincubated for 10 min at 37 °C with 10 mm CaCl2 and either uPA or sctPA in the above buffer prior to addition of thrombin and plasma to initiate the assay. Turbidity was measured at 650 nm in a microtiter plate reader. Results are reported as time to half-clot lysis as a function of SkzL concentration and analyzed by Equation 1, where [P]o was 1.6 μm, representing the dilution-corrected plasma Pg concentration of 2.0 μm (67).
RESULTS
Structural Characterization of SkzL
SkzL was cloned, expressed, and purified as a 47,400-Da monomer. The molecular mass was determined by mass spectrometry in excellent agreement with the molecular mass calculated from the amino acid sequence. SkzL exhibits 22% sequence identity to SK, including conservation of the NH2- and COOH-terminal residues of SK required for Pg activation. Primary sequence alignment and secondary structure prediction show the relatively weak structural similarity of SkzL to SK (Fig. 1) (42, 68, 69). One distinguishing primary structural feature of SkzL that is absent in SK is the presence of three Cys residues as follows: Cys120, Cys393, and Cys401. During the expression and purification of monomeric SkzL, trace amounts of a disulfide-bonded dimeric form were generated. Dimeric and monomeric SkzL were separated by anion exchange chromatography as described under “Experimental Procedures” to yield pure monomeric SkzL and a 1:1 mixture of monomeric and dimeric SkzL (Fig. 2A, lanes 2, 3, 6, and 7). Results of cysteine-to-serine mutagenesis studies indicated that Cys120 was buried within the protein and that Cys393 and Cys401 occurred partially as free thiols and partially as an internal disulfide bond (supplemental Table S1). The results also indicated that trace levels of disulfide-mediated dimeric SkzL may occur through Cys393 or Cys401. These results were confirmed by mass spectrometry analysis of a thiol-specific 5-fluorescein-labeled analog of SkzL monomer (Fig. 2A, lanes 1 and 5). Labeled and unmodified SkzL were subjected to chymotrypsin cleavage followed by liquid chromatography-tandem mass spectrometry. A mass shift of 387 Da, corresponding to the probe modification, was found in peptides containing either Cys393 or Cys401 (389PQAACYP395 and 399FACENTDSKYN409), indicating modification of both Cys residues under nondenaturing conditions. Peptides with unmodified Cys393 and Cys401 residues were also found. No modification was seen for Cys120 (117PVTCT121), supporting the results of the thiol quantification assays.
FIGURE 1.

Sequence alignment and secondary structure prediction for SkzL and SK. Sequence alignment of SkzL and SK from S. equisimilis was performed using the Clustal 2.0.10 multiple sequence alignment algorithm (69). Identical residue pairs are highlighted in blue with conserved residues in yellow. Secondary structure prediction was performed by PSIPRED for SkzL in green and SK in violet (68). SkzL Cys120, Cys 393, and Cys401 are highlighted in red. The pairs of Lys and Glu residues representing part (Lys256 and Lys257) of the SK motif involved in kringle 5-mediated Pg substrate recognition and the pairs of Lys and Glu residues in SkzL that may be involved in the low affinity LBS-dependent interaction of SkzL with Pg are also highlighted in red (42).
FIGURE 2.

Secretion of SkzL by S. agalactiae. A, protein-stained (top) and fluorescence (bottom) SDS-PAGE of nonreduced (lanes 1–3) and reduced (lanes 5–7) recombinant SkzL. [5F]-SkzL monomer (lanes 1 and 5), purified SkzL monomer (lanes 2 and 6), a 1:1 mixture of SkzL monomer and SkzL dimer (lanes 3 and 7), with the migration positions of molecular mass markers indicated (kDa). B, Western blotting of a mixture of recombinant SkzL monomer and dimer (lane 1) and proteins secreted from the lag, mid-logarithmic, late-logarithmic, early stationary, and late stationary growth phases (lanes 2–6), with molecular mass markers (kDa) from the corresponding Ponceau S-stained membrane. SDS-PAGE and blotting were performed, and membranes were probed with a polyclonal anti-SkzL antibody as described under “Experimental Procedures.”
Secretion of SkzL by S. agalactiae
As shown by Western blotting with a polyclonal anti-SkzL antibody, SkzL was secreted in monomeric form by S. agalactiae serotype V in all growth phases (Fig. 2B). The subsequent studies were restricted to monomeric SkzL as this was the secreted and putative physiological form of the protein. Mass spectrometry of triplicate bacterial supernatant samples confirmed that SkzL was present in both the mid-logarithmic and stationary phases of S. agalactiae growth (supplemental Table S2). Nineteen proteins were consistently observed in the S. agalactiae secretome during at least one growth phase, including the Pg-binding proteins α-enolase and GAPDH. All peptides identified distinct proteins with no cross-identifications. NP_687847.1, a putative staphylocoagulase homolog, was also identified in the proteomic analysis (71).
Binding of Pg and FFR-Pm to [5F]-SkzL
Fluorescence equilibrium binding studies were performed to quantitate binding of native [Glu]Pg, [Lys]Pg, and active site-blocked FFR-Pm to SkzL labeled at Cys393 and Cys401 with 5-(iodoacetamido)fluorescein ([5F]-SkzL). Titrations were performed in the absence and presence of 6-AHA to disrupt potential LBS interactions mediated by the kringle domains of Pg/Pm (Fig. 3). Binding of [Glu]Pg to [5F]-SkzL was weak, with a dissociation constant of ∼3 μm (Fig. 3A; Table 1). Titrations of [Lys]Pg and FFR-Pm were performed at two [5F]-SkzL concentrations to determine stoichiometry (Fig. 3, B and C). Binding of [Lys]Pg and FFR-Pm to [5F]-SkzL was characterized by similar dissociation constants of 15 ± 2 and 23 ± 8 nm, respectively, and unit stoichiometries (Table 1). Binding of all three proteins was eliminated by 10 mm 6-AHA, indicating completely LBS-dependent interactions.
FIGURE 3.

Fluorescence titrations of [5F]-SkzL with [Glu]Pg, [Lys]Pg, and FFR-Pm. A, titrations of the fractional change in fluorescence (ΔF/Fo) of 100 nm [5F]-SkzL as a function of total [Glu]Pg concentration ([[Glu]Pg]o) in the absence (●) and presence (○) of 10 mm 6-AHA. B, titrations of 100 nm (●) and 500 nm (Δ) [5F]-SkzL as a function of total [Lys]Pg ([[Lys]Pg]o) concentration in the absence of 6-AHA, and titration of 100 nm [5F]-SkzL with [Lys]Pg in the presence (○) of 10 mm 6-AHA. C, titrations as in B of 100 nm (●) and 500 nm (Δ) [5F]-SkzL, as a function of total FFR-Pm concentration ([FFR-Pm]o) in the absence of 6-AHA, and titration as in B of 100 nm [5F]-SkzL with FFR-Pm in the presence (○) of 10 mm 6-AHA. Solid lines represent the least squares fits of the data by the quadratic binding equation (Equation 1) with the parameters listed in Table 1. Experiments were performed and the data analyzed as described under “Experimental Procedures.”
TABLE 1.
Parameters for binding of [Glu]Pg, [Lys]Pg, and FFR-Pm to [5F]-SkzL and competitive binding of wtSkzL or SkzLΔK415
Dissociation constants (KD ligand), maximum fluorescence changes (ΔFmax/Fo), and stoichiometric factors (n) are listed from direct titrations of [5F]-SkzL with the indicated Pg/Pm ligands. Results of competitive binding studies for the indicated ligand and competitor were analyzed by simultaneous fitting of the titration in the absence and presence of one or two fixed concentrations of competitor to obtain the dissociation constant for the ligand (KD ligand) and competitor (KD competitor), with the stoichiometric factor for the ligand interaction fixed at its determined value, and fixed at 1 for the competitors. For analysis of the [Glu]Pg titrations, n was fixed at 1. ND represents not determined. Experimental error in parameters represents ± 2 S.D. Binding studies were performed and the data analyzed as described under “Experimental Procedures.”
| Probe | Ligand | Competitor | KD ligand | n | KD competitor | ΔFmax/Fo |
|---|---|---|---|---|---|---|
| nm | mol/mol | nm | % | |||
| [5F]-SkzL | [Glu]Pg | wtSkzL | 3000 ± 3000 | ND | −20 ± 3 | |
| [5F]-SkzL | [Lys]Pg | wtSkzL | 15 ± 2 | 0.92 ± 0.03 | 82 ± 13 | −35 ± 1 |
| [5F]-SkzL | [Lys]Pg | SkzLΔK415 | 14 ± 7 | 2500 ± 1200 | −34 ± 1 | |
| [5F]-SkzL | FFR-Pm | wtSkzL | 23 ± 8 | 0.97 ± 0.05 | 49 ± 23 | −30 ± 1 |
| [5F]-SkzL | FFR-Pm | SkzLΔK415 | 9 ± 3 | 1500 ± 600 | −34 ± 1 |
To determine the affinity of wtSkzL for native [Lys]Pg and Pm, competitive binding studies were performed by titration of [5F]-SkzL with [Lys]Pg and FFR-Pm in the absence and presence of two fixed concentrations of unlabeled wtSkzL (Fig. 4, A and C). Analysis of the competitive titrations for wtSkzL binding to [Lys]Pg and FFR-Pm gave KD values of 82 ± 13 and 49 ± 23 nm, respectively, 2–5-fold weaker than binding to [5F]-SkzL (Table 1). These KD values, unlike those for [5F]-SkzL, are independent of perturbations of the affinity due to the presence of the fluorescence probe, which accounts for the higher affinity of labeled SkzL for all of the proteins. It was not possible to carry out competitive binding experiments for [Glu]Pg because of the prohibitively high concentrations that would be required.
FIGURE 4.

Competitive binding of wtSkzL and SkzLΔK415 to Pg/Pm. A, titrations of the fractional change in fluorescence (ΔF/Fo) of 100 nm [5F]-SkzL as a function of total [Lys]Pg concentration ([[Lys]Pg]o) in the absence (●) and presence of 2000 (○) and 5000 nm (▴) wtSkzL. B, titrations as in A of 100 nm [5F]-SkzL with [Lys]Pg in the absence (●) and presence of 20 μm (○) SkzLΔK415. C, titrations as in A of 100 nm [5F]-SkzL as a function of total FFR-Pm concentration ([FFR-Pm]o) in the absence (●) and presence of 760 (○) and 2000 nm (▴) of wtSkzL. D, titrations as in A of 100 nm [5F]-SkzL with FFR-Pm in the absence (●) and presence of 20 μm (○) SkzLΔK415. Solid lines represent least squares fits of the data by the cubic competitive binding equation with the parameters listed in Table 1. Experiments were performed and the data analyzed as described under “Experimental Procedures.”
To elucidate the potential role of the COOH-terminal Lys415 residue in mediating LBS interactions, experiments were also performed with a SkzL mutant lacking this residue (SkzLΔK415). Competitive binding studies in the absence and presence of SkzLΔK415 (Fig. 4, B and D) gave probe-independent KD values for SkzLΔK415 binding to native [Lys]Pg and FFR-Pm of 2.5 ± 1.2 and 1.5 ± 0.6 μm, respectively (Table 1). Deletion of Lys415 resulted in a 30-fold decreased affinity for native [Lys]Pg and FFR-Pm compared with wtSkzL, indicating a major role of Lys415 in mediating the interactions.
LBS-mediated Binding of SkzL to Active Site-labeled Fluorescent Pg/Pm Analogs
To examine the interactions from a different perspective, equilibrium binding studies were performed using active site fluorescein-labeled analogs of [Glu]Pg, [Lys]Pg, and FFR-Pm in the absence and presence of 6-AHA (Fig. 5). Binding to [5F]FFR-[Glu]Pg was very weak (KD ∼10 μm), 3-fold weaker than observed with [5F]-SkzL, and was abolished by 10 mm 6-AHA (Fig. 5, A and B; Table 2). The source of the weaker KD values obtained for [5F]FFR-[Glu]Pg, -[Lys]Pg, and -Pm is explained by 2–3-fold decreases in affinity due to active site labeling of the proteins. Titrations of [5F]FFR-[Glu]Pg with SkzLΔK415 demonstrated no apparent fluorescence change, suggesting the absence of binding or an interaction occurring more weakly than could be detected under the experimental conditions used. Titrations of [5F]FFR-[Lys]Pg and [5F]FFR-Pm with wtSkzL revealed bimodal behavior, suggestive of two binding interactions. The high affinity interactions occurred with equivalent dissociation constants of 160–170 nm (Fig. 5, C and E; Table 2). The low affinity interactions were not well determined, with dissociation constants of ∼18 and ∼20 μm, respectively, and appeared to be due to a decrease in anisotropy. When vertically polarized light was used for excitation and emission, and polarizers were set to the magic angle to eliminate anisotropy, only the high affinity SkzL interaction was observed, with KD values for labeled [Glu]Pg, [Lys]Pg, and Pm indistinguishable from those obtained for the bimodal titrations (Fig. 5, D and F; Table 2). The values for the high affinity interactions were 2–3-fold weaker than the KD values from the competitive binding experiments (Table 1), due to fluorescence probe labeling of the active site.
FIGURE 5.

Binding of wtSkzL and SkzLΔK415 to [5F]FFR-[Glu]Pg, [5F]FFR-[Lys]Pg, and [5F]FFR-Pm. A and B, titrations of the fractional change in fluorescence (ΔF/Fo) of 50 nm [5F]FFR-[Glu]Pg as a function of total wtSkzL concentration ([SkzL]o) in the absence (●) and presence (○) of 10 mm 6-AHA and as a function of total SkzLΔK415 concentration ([SkzLΔK415]o) in the absence (▴) and presence (Δ) of 10 mm 6-AHA. C and D, titrations as in A and B of 50 nm [5F]FFR-[Lys]Pg as a function of total wtSkzL concentration in the absence (●) and presence (○) of 10 mm 6-AHA and as a function of total SkzLΔK415 concentration in the absence (▴) and presence (Δ) of 10 mm 6-AHA. E and F, titrations as in A and B of 50 nm [5F]FFR-Pm as a function of total wtSkzL concentration in the absence (●) and presence (○) of 10 mm 6-AHA, and as a function of total SkzLΔK415 concentration in the absence (▴) and presence (Δ) of 10 mm 6-AHA. Solid lines represent the least squares fits of the data by Equation 1 (A, B, D, and F) or Equation 2 (C and E) with the parameters listed in Table 2. The dashed line represents zero. Titrations were performed without polarizers (A, C and E), or with vertically polarized excitation and emission at the magic angle (B, D, and F). Titrations were performed and analyzed as described under “Experimental Procedures.”
TABLE 2.
Parameters for wtSkzL and SkzLΔK415 binding to active site fluorescein-labeled Pg/Pm analogs
Dissociation constants for the high affinity (KD,1) and low affinity (KD,2) interactions and corresponding maximum fluorescence changes (ΔFmax,1/Fo) and (ΔFmax,2/Fo) from titrations of [5F]FFR-[Glu]Pg, [5F]FFR-[Lys]Pg, and [5F]FFR-Pm (Probe-labeled protein) with wtSkzL or SkzLΔK415 (Ligand) as indicated. Titrations were performed without polarizers or with vertically polarized excitation and emission at the magic angle (Magic angle) to eliminate anisotropy. Experimental error in the parameters represents ± 2 S.D. Experiments were performed, and the results were analyzed as described under “Experimental Procedures.”
| Probe-labeled protein | Ligand | Magic angle | KD,1 | KD,2 | ΔFmax,1/Fo | ΔFmax,2/Fo |
|---|---|---|---|---|---|---|
| nm | nm | % | % | |||
| [5F]FFR-[Glu]Pg | wtSkzL | No | 10,000 ± 3200 | −18 ± 2 | ||
| [5F]FFR-[Glu]Pg | wtSkzL | Yes | 16,000 ± 9000 | −14 ± 4 | ||
| [5F]FFR-[Glu]Pg | SkzLΔK415 | No | No binding | No binding | ||
| [5F]FFR-[Glu]Pg | SkzLΔK415 | Yes | No binding | No binding | ||
| [5F]FFR-[Lys]Pg | wtSkzL | No | 170 ± 90 | ∼18,000 | −10 ± 2 | −22 ± 10 |
| [5F]FFR-[Lys]Pg | wtSkzL | Yes | 150 ± 40 | −9 ± 1 | ||
| [5F]FFR-[Lys]Pg | SkzLΔK415 | No | 12,000 ± 8000 | −15 ± 5 | ||
| [5F]FFR-[Lys]Pg | SkzLΔK415 | Yes | 2800 ± 1300 | −8 ± 1 | ||
| [5F]FFR-Pm | wtSkzL | No | 160 ± 80 | ∼20,000 | −11 ± 2 | −18 ± 12 |
| [5F]FFR-Pm | wtSkzL | Yes | 140 ± 14 | −14 ± 2 | ||
| [5F]FFR-Pm | SkzLΔK415 | No | 8000 ± 3000 | −18 ± 3 | ||
| [5F]FFR-Pm | SkzLΔK415 | Yes | 4400 ± 1600 | −16 ± 3 |
Titrations of [5F]FFR-[Lys]Pg and [5F]FFR-Pm with SkzLΔK415 under nonmagic angle (Fig. 5, C and E) and magic angle (Fig. 5, D and F) conditions gave weak KD values of 3–12 μm (Table 2). The low affinity binding of SkzLΔK415 was eliminated in the presence of 6-AHA, consistent with more than one LBS-dependent interaction between SkzL and Pg/Pm.
LBS-mediated Binding of SkzL to Active Pm
To detect any changes in Pm chromogenic substrate kinetics on binding to SkzL, assays were performed by continuous measurement of the initial rates of pyro-EPR-pNA hydrolysis by Pm as a function of SkzL concentration. Comparison of the Michaelis-Menten kinetic parameters in the absence and presence of 1 or 5 μm SkzL showed a 1.7-fold decrease in kcat compared with free Pm, no significant change in Km, and a 2-fold decrease in kcat/Km (Table 3). SkzL had no effect on the rate of hydrolysis of chromogenic substrates with Lys at the P15 position (d-VLK-pNA, pyro-Glu-Phe-Lys-pNA, and d-Val-Phe-Lys-pNA). However, similar effects on kcat were seen with other substrates with Arg at the P1 position (d-Ile-Pro-Arg-pNA and d-Val-Leu-Arg-pNA) (results not shown). Titration with SkzL at 200 μm pyro-EPR-pNA showed two distinct LBS-dependent binding interactions between SkzL and native Pm, with apparent dissociation constants of 170 ± 140 nm and ∼9 μm, where the latter value was poorly determined (Fig. 6). Titration with SkzLΔK415 demonstrated a loss of the high affinity interaction and an LBS-dependent low affinity interaction with a KD of 3.8 ± 1.2 μm (Fig. 6).
TABLE 3.
Parameters for the effect of SkzL on pyro-EPR-pNA hydrolysis by Pm
Kinetic parameters (kcat, Km, kcat/Km) were obtained from Michaelis-Menten analysis of the substrate dependence. Experiments were performed in the absence and presence of 1 and 5 μm total SkzL concentration ([SkzL]o). Experimental error in the parameters represents ± 2 S.D. Experiments were performed and the results analyzed as described under “Experimental Procedures.”
| [SkzL]o | kcat | Km | kcat/Km |
|---|---|---|---|
| μm | s−1 | μm | s−1 μm−1 |
| 0 | 78 ± 2 | 130 ± 2 | 0.60 |
| 1 | 46 ± 1 | 140 ± 11 | 0.33 |
| 5 | 47 ± 1 | 160 ± 22 | 0.30 |
FIGURE 6.

Effect of SkzL on pyro-EPR-pNA hydrolysis by Pm. Titrations of the fractional change in initial velocity (Δv/vo) of 10 nm Pm as a function of total wtSkzL concentration ([SkzL]o) in the absence (●) and presence (○) of 10 mm 6-AHA, and as a function of total SkzLΔK415 concentration ([SkzLΔK415]o) in the absence (▴) and presence (Δ) of 10 mm 6-AHA. Solid lines represent the least squares fits of the data by Equation 1 (▴) or Equation 2 (●), with the parameters given under “Results.” The dashed line represents zero. Titrations were performed and analyzed as described under “Experimental Procedures.”
Effect of SkzL on Plasminogen Activation by uPA
Chromogenic substrate kinetic assays of Pm formation were performed to determine the effect of SkzL on activation of [Glu]Pg and [Lys]Pg by uPA. To investigate the possible effects of SkzL on Pg conformation, assays were done in the absence and presence of 10 mm 6-AHA and in low chloride ion buffer, with the uPA preparation as the only source of Cl− (0.45 mm in the reactions) and in buffer containing 100 mm Cl−. In the presence of 100 mm Cl−, saturating SkzL concentrations resulted in an ∼2-fold decreased rate of [Lys]Pg activation with minimum rates of Pm generation equivalent to those in the presence of 10 mm 6-AHA (Fig. 7A). Fitting of the SkzL dependence in the absence of 6-AHA yielded an apparent KD value for SkzL-[Lys]Pg binding of 300 ± 120 nm (Fig. 7A; Table 4), which was ∼4-fold weaker than the KD of 80 nm obtained for SkzL binding to [Lys]Pg in competitive binding studies. An identical phenomenon was seen at low Cl− concentration, with an indistinguishable apparent KD of 340 ± 150 nm (Fig. 7B; Table 4), consistent with the lack of effect of Cl− on the [Lys]Pg β- to γ-conformational change.
FIGURE 7.

Effect of SkzL on Pg activation by uPA. Assays were performed in buffer containing 100 mm chloride (A) or 0.45 mm chloride (B). Initial rates of Pm generation ((nm Pm/s)/(nm uPA)) for activation of 100 nm [Lys]Pg by 0.11 nm uPA in the absence (●) and presence (○) of 10 mm 6-AHA as a function of total SkzL concentration ([SkzL]o) are shown. Initial rates of Pm generation ((nm Pm/s)/(nm uPA)) for activation of 100 nm [Glu]Pg by 0.32 nm uPA in the absence (▴) and presence (Δ) of 10 mm 6-AHA as a function of total SkzL concentration ([SkzL]o) are shown. Solid lines represent the least squares fits of the data by the quadratic binding equation with the parameters listed in Table 4. Experiments were performed and the data analyzed as described under “Experimental Procedures.”
TABLE 4.
Parameters for the effect of SkzL on Pg activation by uPA
Dissociation constants (KD) for the SkzL-dependent change in the rate of Pm generation for activation of 100 nm [Glu]Pg or [Lys]Pg by 0.11 nm uPA. Experiments were performed in the presence of low and high total chloride ion concentration ([chloride ion]o). Experimental error in the parameters represents ± 2 S.D. Experiments were performed and the results analyzed as described under “Experimental Procedures.”
| Substrate | [chloride ion]o | KD |
|---|---|---|
| mm | nm | |
| [Lys]Pg | 100 | 300 ± 120 |
| [Lys]Pg | 0.45 | 340 ± 150 |
| [Glu]Pg | 100 | 13,000 ± 12,000 |
| [Glu]Pg | 0.45 | 1900 ± 900 |
The rate of Pm generation from [Glu]Pg by uPA in the presence of chloride ion was greatly enhanced, ∼20-fold to a rate indistinguishable from that for [Lys]Pg at saturating concentrations of SkzL or 6-AHA (Fig. 7A; Table 4). Fitting of the SkzL dependence gave an apparent KD value for SkzL-[Glu]Pg binding of 13 ± 12 μm, in good agreement with the estimates of 10–16 μm for SkzL binding to [5F]FFR-[Glu]Pg (Table 2). The rate of [Glu]Pg activation by uPA in the presence of 10 mm 6-AHA was essentially the same as the rate in the presence of saturating concentrations of SkzL. This indicated that, similar to the effect of 6-AHA, the α-conformation of [Glu]Pg was being extended by SkzL to resemble the γ-conformation of [Lys]Pg. The basal rate of [Glu]Pg activation by uPA was enhanced ∼11-fold at low Cl− concentration compared with high Cl− concentration (Fig. 7B; Table 4). Saturating SkzL concentrations resulted in a slight, ∼2-fold further enhancement of [Glu]Pg activation. This small enhancement resulted in rates of Pm generation equivalent to those in the presence of 10 mm 6-AHA (Fig. 7B; Table 4).
Effect of SkzL on Plasminogen Activation by sctPA
Chromogenic substrate kinetic assays of Pm formation were performed to determine the effect of SkzL on activation of [Lys]Pg and [Glu]Pg by sctPA. The rate of Pm generation from [Lys]Pg and [Glu]Pg by sctPA was greatly enhanced ∼42- and ∼650-fold, respectively, in a totally LBS-dependent manner (Fig. 8), although the maximum rate at saturating SkzL was 3.4-fold higher for [Lys]Pg compared with [Glu]Pg. Fitting of the SkzL dependence gave KD values for [Lys]Pg and [Glu]Pg of 12 ± 7 and 14 ± 13 μm, respectively. Although the apparent KD value of SkzL for [Glu]Pg was consistent with those obtained in the binding studies, the KD value for [Lys]Pg was ∼60–140-fold weaker than the affinity determined in the binding studies for the high affinity interaction (Table 2). This very large difference suggested that a different mechanism was involved for sctPA.
FIGURE 8.

Effect of SkzL on Pg activation by sctPA. Initial rates of Pm generation ((nm Pm/s)/(nm sctPA)) for activation of 100 nm [Lys]Pg by 3 nm sctPA in the absence (●) and presence (○) of 10 mm 6-AHA as a function of total SkzL concentration ([SkzL]o) are shown. Initial rates of Pm generation ((nm Pm/s)/(nm sctPA)) for activation of 100 nm [Glu]Pg by 3 nm sctPA in the absence (▴) and presence (Δ) of 10 mm 6-AHA as a function of total SkzL concentration ([SkzL]o) are shown. Solid lines represent the least squares fits by the quadratic binding equation with parameters given under “Results”, and the dashed line represents zero. Experiments were performed and data analyzed as described under “Experimental Procedures.”
SkzL Enhances uPA- and sctPA-mediated Plasma Clot Lysis
Plasma clot lysis experiments were performed to assess the role of SkzL as an effector of uPA- and sctPA-mediated Pg activation in a physiological model system of fibrinolysis. Human plasma and 6 nm human thrombin were reacted with a preincubated mixture of either 7 nm uPA or 1.5 nm sctPA and increasing concentrations of SkzL. The results were expressed as time to half-clot lysis as a function of SkzL concentration (72). SkzL enhanced clot lysis by uPA and sctPA, decreasing the time to half-clot lysis by ∼2-fold (Fig. 9). No clot lysis was observed in the absence of uPA or sctPA.
FIGURE 9.

Effect of SkzL on uPA- and sctPA-mediated plasma clot lysis. Clot lysis is represented by the time to half-lysis (time to half-lysis) (●) as a function of total SkzL concentration ([SkzL]o) for reactions containing uPA (A) or sctPA (B). Following dilution of all proteins in 50 mm HEPES, 125 mm NaCl, pH 7.4, SkzL was preincubated with 10 mm CaCl2, 7 nm uPA, or 1.5 nm sctPA for 10 min at 37 °C prior to addition of 6 nm thrombin and 160 μl of plasma (200-μl assay volume). Clot lysis was measured by turbidity as described under “Experimental Procedures.” Analysis of clot lysis by uPA and sctPA gave apparent KD values of 1.9 ± 0.9 and 2.5 ± 2.5 μm, respectively.
DISCUSSION
SkzL from S. agalactiae has modest homology to the bacterial Pg activators SAK, SK, and PauB and is secreted as the monomeric 47,400-Da form by S. agalactiae throughout its growth cycle. Nineteen proteins were identified in the S. agalactiae secretome, including α-enolase and GAPDH, which are known to bind Pg. In contrast to SK, SAK, and PauB, SkzL does not conformationally or proteolytically activate Pg to Pm (results not shown). SkzL, however, binds Pg and Pm in an LBS-dependent manner, resulting in enhanced [Glu]Pg activation by the endogenous human Pg activators, uPA and sctPA. To our knowledge, SkzL is the first S. agalactiae-secreted protein that targets uPA, sctPA, and Pg to facilitate activation of the human fibrinolytic system.
Equilibrium binding studies show that SkzL binds to [Glu]Pg with low affinity, likely due to low accessibility of the LBS in the kringle domains of the compact [Glu]Pg α-conformation. Nevertheless, the low affinity interaction of SkzL with [Glu]Pg is eliminated by 6-AHA, indicating that LBS interactions are involved. Competitive binding studies indicate that wtSkzL binds to unlabeled, native [Lys]Pg and FFR-Pm with near-equivalent KD values of 80 and 50 nm, respectively. The large increase in affinity compared with [Glu]Pg is likely due to the increased LBS accessibility in the partially extended [Lys]Pg/Pm β-conformation. Binding of SkzL to [Lys]Pg and FFR-Pm is blocked by 6-AHA, indicating that binding is entirely mediated by LBS interactions.
Because of the use of multiple experimental approaches to quantitate SkzL binding to different forms of Pg and Pm, it is important to address some apparent discrepancies among the dissociation constants obtained. [5F]-SkzL binds with 5- and 2-fold enhanced affinity for native [Lys]Pg and FFR-Pm, respectively, compared with the affinity of unlabeled wtSkzL determined by competitive binding. The dissociation constants determined by competitive binding are independent of the effects that fluorescence probe labeling may have on binding affinity. The enhanced binding of [5F]-SkzL may be a result of the location of probe incorporation. The [5F]-labeled SkzL has a stoichiometry of labeling of 1.0 mol of [5F]/mol of SkzL, which reflects probe incorporation on Cys393 and Cys401, only 14 residues from Lys415 (see Fig. 1). On this basis, higher affinity binding to [5F]-SkzL may reflect a favorable direct interaction of Pg with the probes. Conversely, wtSkzL binds with 2- and 3-fold reduced affinity to active site-labeled [5F]FFR-[Lys]Pg and [5F]FFR-Pm, respectively. The small decrease in affinity associated with Pg/Pm active site labeling may result from distortion of the conformation of the catalytic domain of Pg/Pm due to the presence of the probe and linking tripeptide inhibitor. These opposing effects of probe labeling on [Glu]Pg binding are compounded in comparison of [Glu]Pg binding with [5F]-SkzL and SkzL binding with [5F]FFR-[Glu]Pg, which differ by 3–5-fold.
SkzL binding to active site-labeled [5F]FFR-[Lys]Pg and [5F]FFR-Pm revealed a high affinity interaction (KD ∼140–170 nm) represented by a decrease in fluorescence intensity and a low affinity interaction (KD ∼20 μm) represented by a decrease in anisotropy. The latter values were not well determined because little saturation was obtained at the highest SkzL concentrations achievable (20 μm). Titrations performed with vertically polarized excitation and emission at the magic angle resulted in near-elimination of the anisotropy change and observation of the high affinity interaction (KD 140–150 nm) only, indicating that the low affinity interaction is associated with a decrease in anisotropy. Considering the 2–3-fold increase in the KD of SkzL binding due to probe labeling, the KD values of 140–150 nm for the labeled proteins were close enough (≤3-fold) to the values of 50 and 80 nm for high affinity SkzL binding to unlabeled FFR-Pm and native [Lys]Pg to support the conclusion that they represent the same high affinity interaction. It should be noted that 2-fold differences in dissociation constants are generally not considered significant.
Because the low affinity interactions had some of the hallmarks of nonspecific binding, the presence of two distinct interactions between SkzL and Pm was confirmed through analysis of the effect of SkzL on the rates of pyro-EPR-pNA hydrolysis by Pm. These studies demonstrated mediation of the high affinity LBS-dependent interaction by Lys415 and the presence of a second LBS-dependent ∼9 μm KD interaction, which was poorly determined.
Deletion of SkzL Lys415 results in undetectable binding of [Glu]Pg, suggesting that binding is mediated by Lys415 exclusively. Binding of both native [Lys]Pg and FFR-Pm to wtSkzL is weakened 30-fold by Lys415 deletion. The results indicate that Lys415 plays a major role in the LBS-dependent binding of all forms of Pg and Pm. Loss of Lys415 decreases the affinity of SkzL for native [Lys]Pg and FFR-Pm to KD values of 2.5 and 1.5 μm, respectively (Table 1), demonstrating that Lys415 is responsible for the high affinity interaction. The remaining interaction is also eliminated by 6-AHA, indicating that there are two sites on SkzL that mediate LBS-dependent binding to labeled [Lys]Pg/Pm. A sequence containing pairs of Lys and nearby Glu residues in SkzL (Fig. 1; Lys218, Lys219, Glu227, Glu228) is similar to that of the 250-loop of SK, with the exception of Arg253 (Lys256, Lys257, Glu262, Glu263) and a motif in SAK (Lys97-Lys-Glu-Glu100), certain residues of which are implicated in mediating LBS-dependent Pg substrate interactions with SK·Pg* and SAK·Pm catalytic complexes (42, 73). The similar motif in SkzL may be responsible for the low affinity interaction. Future mutagenesis studies may be used to establish the SkzL residues involved in mediating this weak interaction.
Pg can adopt three distinct conformations that affect its potential for activation by tPA and uPA. The compact nature of the α-conformation of [Glu]Pg, stabilized by physiological chloride concentrations, makes it a poor substrate for uPA, tPA, and SK, compared with the partially extended, chloride-independent β-conformation of [Lys]Pg (17, 22–25, 49, 66). Upon occupation of [Glu]Pg and [Lys]Pg kringles 1, 4, and 5 by 6-AHA, the transition to the fully extended γ-conformation also results in enhanced activation by uPA (21, 23–25).
Because of the highly LBS-dependent nature of SkzL binding to Pg, it was hypothesized that SkzL-Pg binding could mimic the conformational change induced by 6-AHA binding, resulting in the extension of [Glu]Pg and [Lys]Pg from the α- and β-conformations, respectively, to the γ-conformation. Activation of [Lys]Pg by uPA, however, was inhibited by SkzL to a rate equivalent to that in the presence of either 10 mm 6-AHA or saturating SkzL. In these experiments, the apparent dissociation constants for SkzL-[Lys]Pg binding were ∼4-fold weaker than the native affinity determined by competitive binding. This discrepancy may be due to the inability to discern both the high and low affinity interactions between SkzL and [Lys]Pg, resulting in a weaker apparent affinity representative of a mixture of binding interactions between SkzL and [Lys]Pg. Activation of [Lys]Pg by uPA is inhibited by SkzL, suggesting that although SkzL binding may alter the conformation of [Lys]Pg from the β- to the γ-conformation, similar to 6-AHA, it does not enhance [Lys]Pg activation by uPA.
In the presence of 100 mm chloride ion, activation of [Glu]Pg by uPA is very slow; however, binding of SkzL enhances the rate of Pm generation ∼20-fold to the same level as [Lys]Pg in the presence of saturating SkzL or 6-AHA. Rates of Pm generation in the presence of 6-AHA are identical to those in the presence of saturating SkzL, suggesting that occupation of the [Glu]Pg LBS and stabilization of the γ-conformation by SkzL is responsible for the observed SkzL-enhanced rate of [Glu]Pg activation. Together, the results show that SkzL is a specific effector of [Glu]Pg activation by uPA. Moreover, the mechanism of SkzL-enhanced [Glu]Pg activation by uPA is shown for the first time to be caused by SkzL-mediated transformation of [Glu]Pg from the activation-resistant α-conformation to the more rapidly activated γ-conformation.
This mechanism and a hypothetical interpretation of the results of the binding studies for [Lys]Pg and Pm are presented in Fig. 10. In this scheme, [Glu]Pg in the α-conformation is in unfavorable equilibrium (Reaction 1) with the β-conformation, which is governed by the Cl− concentration (17, 23–25). SkzL binds preferentially to the β-conformation, mediated by Lys415 binding to kringle 4, and is accompanied by transition of [Glu]Pg to the γ-conformation (Reactions 1 and 2), which is more susceptible to activation by uPA (Reaction 3). The proposal of kringle 4 as the Lys415-binding site is based on the preference of kringle 4 for COOH-terminal Lys residues (26, 31, 74), and preferential binding of SkzL to the [Glu]Pg β-conformation is supported by evidence that kringle 4 is masked in the α-conformation (26, 75–77). LBS-dependent monoclonal antibody binding specific for kringle 4 has been hypothesized to induce an activation-enhancing conformational change (23, 26, 30) distinct from the α- to β-conformational change caused by disruption of the PAN module-kringle 5 interaction (27, 78), suggesting a role for kringle 4 in modulating the β- to γ-conformational change (23, 30, 32). Reactions 4 and 5 illustrate that binding of SkzL to [Lys]Pg (or Pm) can occur through two distinct interactions as follows: high affinity Lys415-kringle 4 binding and a weak interaction between the internal SkzL motif and kringle 5, supported by the demonstrated specificity of kringle 5 for alkylamines and internal Lys residues (79, 80). All of the putative [Lys]Pg·Pm complexes are consistent with the present results, but the Pg kringles to which the two sites on SkzL bind and the compositions of the complexes have not been substantiated experimentally and therefore remain hypothetical.
FIGURE 10.

Diagram of the proposed mechanism of SkzL-mediated [Glu]Pg activation by uPA and postulated LBS-dependent complexes formed with [Lys]Pg and Pm mediated by two sites on SkzL. SkzL is shown in purple with Lys415 (K) on a flexible segment, and the postulated SkzL internal motif that mediates kringle binding is represented by the “nose.” [Glu]Pg in light orange is shown with the NH2-terminal PAN module in red, five numbered kringle domains, and the COOH-terminal catalytic domain. [Glu]Pg in the compact α-conformation ([Glu]Pg-α) is shown as a spiral structure that is maintained by the PAN domain interacting with kringle 5. [Glu]Pg is in equilibrium (Reaction 1) with a low concentration of the partially extended β-conformation ([Glu]Pg-β), in which the PAN module has been released by kringle 5. The proposed mechanism of SkzL-enhanced [Glu]Pg activation by uPA is shown in Reactions 1–3. SkzL binds preferentially to the β-conformation of [Glu]Pg, mediated by Lys415 binding to the LBS of kringle 4, which converts the SkzL·[Glu]Pg complex to the γ-conformation (SkzL·[Glu]Pg-γ), thereby accelerating the rate of [Glu]Pg activation to [Lys]Pm (active site in green) by uPA (Reaction 3). Conversion of the initial product [Glu]Pm into [Lys]Pm by the Pm generated is not shown. [Lys]Pg, lacking the PAN module, is represented in the partially extended β-conformation ([Lys]Pg-β). Hypothetical complexes formed by wtSkzL and [Lys]Pg or Pm mediated by its two binding sites are as follows: Reaction 4, formation of a ternary SkzL·[Lys]Pg2 complex in which one Pg molecule is bound through the Lys415-kringle 4 interaction in the γ-conformation and binding of the other Pg molecule mediated by the SkzL internal motif-kringle 5 interaction, with Pg in the β-conformation (SkzL·[Lys]Pg-γ·[Lys]Pg-β); and Reaction 5, formation of an SkzL·[Lys]Pg complex in the γ-conformation with both the internal motif and Lys415 engaged with their respective kringles. Reaction 6 illustrates the complex formed between SkzLΔK415 and [Lys]Pg or Pm mediated by the internal motif in SkzL.
The enhanced rate of [Glu]Pg activation caused by SkzL is not specific to uPA, as kinetic studies demonstrate that SkzL also enhances Pg activation by sctPA. In contrast to the results with uPA, however, increased rates of Pm generation are seen for sctPA activation of both [Glu]Pg and [Lys]Pg. The effect of SkzL on [Glu]Pg activation by sctPA may be due to SkzL-mediated conformational changes in [Glu]Pg similar to those seen with uPA. However, the apparent dissociation constant for SkzL-enhanced [Lys]Pg activation by sctPA appears to be ∼12 μm, ∼60–140-fold weaker than the KD value for SkzL-[Lys]Pg binding determined by fluorescence. This suggests that the mechanism of enhanced [Lys]Pg activation by sctPA is different and cannot be explained by SkzL-[Lys]Pg binding interactions alone. Although further studies are needed to evaluate this hypothesis, preliminary results suggest that SkzL interacts with sctPA though the LBS on kringle 2 of sctPA, which is not present in uPA. Binding of wtSkzL and SkzLΔK415 to a fluorescent sctPA analog was characterized by KD values of 14 and 15 μm, respectively, in an LBS-dependent manner.6 This result suggests that weak SkzL binding to sctPA is due to an interaction of kringle 2 of sctPA with a site distinct from Lys415 on SkzL. As SkzL binding to [Lys]Pg is largely mediated by Lys415, such an interaction with sctPA may result in higher order complex formation, which will require further experimentation to evaluate.
The present studies show that SkzL is a novel protein that is modestly homologous to SK, secreted from S. agalactiae, and acts as a cofactor of sctPA and uPA in accelerating Pg activation. Recent studies have established that an interaction of cell surface-bound GAPDH with Pg contributes to the virulence of GBS invasive infection in an in vivo murine model (10). The present studies reveal SkzL as an additional component in the interactions between GBS and the proteins of the human fibrinolytic system. As a secreted protein, SkzL may play a role as a Pg-binding protein different from the cell surface-bound Pg-binding proteins, α-enolase and GAPDH, in the context of GBS infection, or it could act in cooperation with these proteins through as yet unknown interactions. SkzL has high affinity for Pg and Pm relative to the plasma Pg concentration of 2 μm (67), and it binds through a strictly LBS-dependent mechanism involving the kringle domains of Pg and Pm. SkzL is a novel effector of uPA-mediated [Glu]Pg activation through an LBS-dependent mechanism similar to 6-AHA (22, 29). The studies suggest that the role of SkzL as an effector of Pg activation by uPA is specific for the circulating form of Pg in blood, [Glu]Pg. SkzL also enhances sctPA-mediated [Glu]Pg and [Lys]Pg activation through a different mechanism that will be examined in further studies. The finding that SkzL enhances clot lysis by both physiological Pg activators in plasma provides strong support for a role of SkzL in the interaction of GBS with the human fibrinolytic system. SkzL has the potential to be a new virulence factor in life-threatening S. agalactiae infections of neonates and immune-compromised individuals.
Acknowledgments
We thank Dr. Lisa Zimmerman of the Vanderbilt University Mass Spectrometry Research Center Proteomics Core for work on the S. agalactiae secreted protein profile. We also thank Drs. Jonathan Creamer and Anthony Tharp for assistance in protein purification and data analysis, respectively.
This work was supported in whole or in part by National Institutes of Health Grant RO1 HL056181 from the NHLBI (to P. E. B.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental “Experimental Procedures,” “Results and Discussion,” Tables S1 and S2, and an additional reference.
The Schechter and Berger (70) notation refers to the residues of a substrate (from the NH2-terminal end) as …P4-P3-P2-P1-P1′-P2′… with the scissile bond at P1-P1′.
K. G. Wiles, P. Panizzi, H. K. Kroh, and P. E. Bock, unpublished results.
- GBS
- group B streptococci
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- SkzL
- skizzle
- wtSkzL
- recombinant wild-type SkzL
- SK
- streptokinase
- SAK
- staphylokinase
- Pg
- plasminogen
- [Glu]Pg
- native plasminogen
- [Lys]Pg
- plasminogen lacking the amino-terminal 77-residue PAN module
- Pm
- [Lys]plasmin
- uPA
- high molecular weight urokinase
- sctPA
- single-chain tissue-type plasminogen activator
- FFR-CH2Cl
- d-Phe-Phe-Arg-CH2Cl
- [5F]FFR-[Glu]Pg
- [5F]FFR-[Lys]Pg, and [5F]FFR-Pm, [Glu]Pg, [Lys]Pg, or Pm labeled at the active site with 5-(iodoacetamido)fluorescein attached to the thiol generated on Nα-[(acetylthio)acetyl]-d-Phe-Phe-Arg-CH2Cl
- 6-AHA
- 6-aminohexanoic acid
- pNA
- para-nitroaniline
- VLK-pNA
- d-Val-Leu-Lys-p-nitroanilide
- pyro-EPR-pNA
- pyro-Glu-Pro-Arg-pNA
- LBS
- lysine-binding sites
- [5F]-SkzL
- SkzL labeled at Cys393 and Cys401 with 5-(iodoacetamido)fluorescein
- SkzLΔK415
- skizzle lacking the COOH-terminal Lys415 residue
- tPA
- tissue-type plasminogen activator
- PAN
- Pg/apple/nematode.
REFERENCES
- 1.Schuchat A. (1999) Lancet 353, 51–56 [DOI] [PubMed] [Google Scholar]
- 2.Pietrocola G., Schubert A., Visai L., Torti M., Fitzgerald J. R., Foster T. J., Reinscheid D. J., Speziale P. (2005) Blood 105, 1052–1059 [DOI] [PubMed] [Google Scholar]
- 3.Blumberg H. M., Stephens D. S., Modansky M., Erwin M., Elliot J., Facklam R. R., Schuchat A., Baughman W., Farley M. M. (1996) J. Infect. Dis. 173, 365–373 [DOI] [PubMed] [Google Scholar]
- 4.Brochet M., Couve E., Zouine M., Vallaeys T., Rusniok C., Lamy M. C., Buchrieser C., Trieu-Cuot P., Kunst F., Poyart C., Glaser P. (2006) Microbes Infect. 8, 1227–1243 [DOI] [PubMed] [Google Scholar]
- 5.Fluegge K., Schweier O., Schiltz E., Batsford S., Berner R. (2004) Eur. J. Clin. Microbiol. Infect. Dis. 23, 818–824 [DOI] [PubMed] [Google Scholar]
- 6.Sun H., Ringdahl U., Homeister J. W., Fay W. P., Engleberg N. C., Yang A. Y., Rozek L. S., Wang X., Sjöbring U., Ginsburg D. (2004) Science 305, 1283–1286 [DOI] [PubMed] [Google Scholar]
- 7.Henkin J., Marcotte P., Yang H. C. (1991) Prog. Cardiovasc. Dis. 34, 135–164 [DOI] [PubMed] [Google Scholar]
- 8.Bergmann S., Wild D., Diekmann O., Frank R., Bracht D., Chhatwal G. S., Hammerschmidt S. (2003) Mol. Microbiol. 49, 411–423 [DOI] [PubMed] [Google Scholar]
- 9.Lähteenmäki K., Kuusela P., Korhonen T. K. (2001) FEMS Microbiol. Rev. 25, 531–552 [DOI] [PubMed] [Google Scholar]
- 10.Magalhães V., Veiga-Malta I., Almeida M. R., Baptista M., Ribeiro A., Trieu-Cuot P., Ferreira P. (2007) Microbes Infect. 9, 1276–1284 [DOI] [PubMed] [Google Scholar]
- 11.Whiting G. C., Evans J. T., Patel S., Gillespie S. H. (2002) J. Med. Microbiol. 51, 837–843 [DOI] [PubMed] [Google Scholar]
- 12.Cork A. J., Jergic S., Hammerschmidt S., Kobe B., Pancholi V., Benesch J. L., Robinson C. V., Dixon N. E., Aquilina J. A., Walker M. J. (2009) J. Biol. Chem. 284, 17129–17137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tettelin H., Masignani V., Cieslewicz M. J., Eisen J. A., Peterson S., Wessels M. R., Paulsen I. T., Nelson K. E., Margarit I., Read T. D., Madoff L. C., Wolf A. M., Beanan M. J., Brinkac L. M., Daugherty S. C., DeBoy R. T., Durkin A. S., Kolonay J. F., Madupu R., Lewis M. R., Radune D., Fedorova N. B., Scanlan D., Khouri H., Mulligan S., Carty H. A., Cline R. T., Van Aken S. E., Gill J., Scarselli M., Mora M., Iacobini E. T., Brettoni C., Galli G., Mariani M., Vegni F., Maione D., Rinaudo D., Rappuoli R., Telford J. L., Kasper D. L., Grandi G., Fraser C. M. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 12391–12396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 15.Brockway W. J., Castellino F. J. (1972) Arch. Biochem. Biophys. 151, 194–199 [DOI] [PubMed] [Google Scholar]
- 16.Tordai H., Bányai L., Patthy L. (1999) FEBS Lett. 461, 63–67 [DOI] [PubMed] [Google Scholar]
- 17.Markus G., DePasquale J. L., Wissler F. C. (1978) J. Biol. Chem. 253, 727–732 [PubMed] [Google Scholar]
- 18.Robbins K. C., Boreisha I. G., Arzadon L., Summaria L. (1975) J. Biol. Chem. 250, 4044–4047 [PubMed] [Google Scholar]
- 19.Sjöholm I. (1973) Eur. J. Biochem. 39, 471–479 [DOI] [PubMed] [Google Scholar]
- 20.Violand B. N., Byrne R., Castellino F. J. (1978) J. Biol. Chem. 253, 5395–5401 [PubMed] [Google Scholar]
- 21.Castellino F. J., Brockway W. J., Thomas J. K., Liano H. T., Rawitch A. B. (1973) Biochemistry 12, 2787–2791 [DOI] [PubMed] [Google Scholar]
- 22.Urano T., Chibber B. A., Castellino F. J. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 4031–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Markus G. (1996) Fibrinolysis 10, 75–85 [Google Scholar]
- 24.Markus G., Priore R. L., Wissler F. C. (1979) J. Biol. Chem. 254, 1211–1216 [PubMed] [Google Scholar]
- 25.Weisel J. W., Nagaswami C., Korsholm B., Petersen L. C., Suenson E. (1994) J. Mol. Biol. 235, 1117–1135 [DOI] [PubMed] [Google Scholar]
- 26.Ponting C. P., Marshall J. M., Cederholm-Williams S. A. (1992) Blood Coagul. Fibrinolysis 3, 605–614 [PubMed] [Google Scholar]
- 27.Marshall J. M., Brown A. J., Ponting C. P. (1994) Biochemistry 33, 3599–3606 [DOI] [PubMed] [Google Scholar]
- 28.McCance S. G., Castellino F. J. (1995) Biochemistry 34, 9581–9586 [DOI] [PubMed] [Google Scholar]
- 29.Urano T., Sator de Serrano V., Chibber B. A., Castellino F. J. (1987) J. Biol. Chem. 262, 15959–15964 [PubMed] [Google Scholar]
- 30.Cummings H. S., Castellino F. J. (1985) Arch. Biochem. Biophys. 236, 612–618 [DOI] [PubMed] [Google Scholar]
- 31.Lerch P. G., Rickli E. E., Lergier W., Gillessen D. (1980) Eur. J. Biochem. 107, 7–13 [DOI] [PubMed] [Google Scholar]
- 32.Ploplis V. A., Cummings H. S., Castellino F. J. (1982) Biochemistry 21, 5891–5897 [DOI] [PubMed] [Google Scholar]
- 33.Bode W., Huber R. (1976) FEBS Lett. 68, 231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boxrud P. D., Bock P. E. (2004) J. Biol. Chem. 279, 36642–36649 [DOI] [PubMed] [Google Scholar]
- 35.Boxrud P. D., Verhamme I. M., Bock P. E. (2004) J. Biol. Chem. 279, 36633–36641 [DOI] [PubMed] [Google Scholar]
- 36.Boxrud P. D., Verhamme I. M., Fay W. P., Bock P. E. (2001) J. Biol. Chem. 276, 26084–26089 [DOI] [PubMed] [Google Scholar]
- 37.Wang S., Reed G. L., Hedstrom L. (1999) Biochemistry 38, 5232–5240 [DOI] [PubMed] [Google Scholar]
- 38.Wang S., Reed G. L., Hedstrom L. (2000) Eur. J. Biochem. 267, 3994–4001 [DOI] [PubMed] [Google Scholar]
- 39.Bean R. R., Verhamme I. M., Bock P. E. (2005) J. Biol. Chem. 280, 7504–7510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boxrud P. D., Bock P. E. (2000) Biochemistry 39, 13974–13981 [DOI] [PubMed] [Google Scholar]
- 41.Boxrud P. D., Fay W. P., Bock P. E. (2000) J. Biol. Chem. 275, 14579–14589 [DOI] [PubMed] [Google Scholar]
- 42.Tharp A. C., Laha M., Panizzi P., Thompson M. W., Fuentes-Prior P., Bock P. E. (2009) J. Biol. Chem. 284, 19511–19521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Esmon C. T., Mather T. (1998) Nat. Struct. Biol. 5, 933–937 [DOI] [PubMed] [Google Scholar]
- 44.Grella D. K., Castellino F. J. (1997) Blood 89, 1585–1589 [PubMed] [Google Scholar]
- 45.Jespers L., Vanwetswinkel S., Lijnen H. R., Van Herzeele N., Van Hoef B., Demarsin E., Collen D., De Maeyer M. (1999) Thromb. Haemost. 81, 479–485 [PubMed] [Google Scholar]
- 46.Ward P. N., Field T. R., Rosey E. L., Abu-Median A. B., Lincoln R. A., Leigh J. A. (2004) J. Mol. Biol. 342, 1101–1114 [DOI] [PubMed] [Google Scholar]
- 47.Johnsen L. B., Rasmussen L. K., Petersen T. E., Etzerodt M., Fedosov S. N. (2000) Biochemistry 39, 6440–6448 [DOI] [PubMed] [Google Scholar]
- 48.Sazonova I. Y., Houng A. K., Chowdhry S. A., Robinson B. R., Hedstrom L., Reed G. L. (2001) J. Biol. Chem. 276, 12609–12613 [DOI] [PubMed] [Google Scholar]
- 49.Ward P. N., Leigh J. A. (2002) J. Bacteriol. 184, 119–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ward P. N., Leigh J. A. (2004) Indian J. Med. Res. 119, 136–140 [PubMed] [Google Scholar]
- 51.Lijnen H. R., De Cock F., Collen D. (1994) Eur. J. Biochem. 224, 567–574 [DOI] [PubMed] [Google Scholar]
- 52.de Munk G. A., Groeneveld E., Rijken D. C. (1993) Thromb. Haemost. 70, 481–485 [PubMed] [Google Scholar]
- 53.Lijnen H. R., Nelles L., Van Hoef B., Demarsin E., Collen D. (1988) Eur. J. Biochem. 177, 575–582 [DOI] [PubMed] [Google Scholar]
- 54.Lijnen H. R., Van Hoef B., Nelles L., Collen D. (1990) J. Biol. Chem. 265, 5232–5236 [PubMed] [Google Scholar]
- 55.Nelles L., Lijnen H. R., Collen D., Holmes W. E. (1987) J. Biol. Chem. 262, 5682–5689 [PubMed] [Google Scholar]
- 56.Urano T., Sator de Serrano V., Gaffney P. J., Castellino F. J. (1988) Biochemistry 27, 6522–6528 [DOI] [PubMed] [Google Scholar]
- 57.Novokhatny V., Medved L., Lijnen H. R., Ingham K. (1995) J. Biol. Chem. 270, 8680–8685 [DOI] [PubMed] [Google Scholar]
- 58.de Munk G. A., Caspers M. P., Chang G. T., Pouwels P. H., Enger-Valk B. E., Verheijen J. H. (1989) Biochemistry 28, 7318–7325 [DOI] [PubMed] [Google Scholar]
- 59.Panizzi P., Boxrud P. D., Verhamme I. M., Bock P. E. (2006) J. Biol. Chem. 281, 26774–26778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castellino F. J., Powell J. R. (1981) Methods Enzymol. 80, 365–378 [DOI] [PubMed] [Google Scholar]
- 61.Deutsch D. G., Mertz E. T. (1970) Science 170, 1095–1096 [DOI] [PubMed] [Google Scholar]
- 62.Bock P. E., Day D. E., Verhamme I. M., Bernardo M. M., Olson S. T., Shore J. D. (1996) J. Biol. Chem. 271, 1072–1080 [DOI] [PubMed] [Google Scholar]
- 63.Bock P. E. (1992) J. Biol. Chem. 267, 14963–14973 [PubMed] [Google Scholar]
- 64.Violand B. N., Castellino F. J. (1976) J. Biol. Chem. 251, 3906–3912 [PubMed] [Google Scholar]
- 65.Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. (1995) Protein Sci. 4, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bock P. E., Olson S. T., Björk I. (1997) J. Biol. Chem. 272, 19837–19845 [DOI] [PubMed] [Google Scholar]
- 67.Bachmann F. (1994) in Hemostasis and Thrombosis: Basic Principles and Clinical Practice (Colman R. W., Hirsh J., Marder V. J., Salzman E. W. eds) 3rd Ed., pp. 1592–1622, J. B. Lippincott, Philadelphia [Google Scholar]
- 68.Jones D. T. (1999) J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 69.Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 70.Schechter I., Berger A. (1967) Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 71.Panizzi P., Friedrich R., Fuentes-Prior P., Bode W., Bock P. E. (2004) Cell. Mol. Life Sci. 61, 2793–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fredenburgh J. C., Nesheim M. E. (1992) J. Biol. Chem. 267, 26150–26156 [PubMed] [Google Scholar]
- 73.Rajamohan G., Dahiya M., Mande S. C., Dikshit K. L. (2002) Biochem. J. 365, 379–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petros A. M., Ramesh V., Llinás M. (1989) Biochemistry 28, 1368–1376 [DOI] [PubMed] [Google Scholar]
- 75.Hochschwender S. M., Laursen R. A. (1981) J. Biol. Chem. 256, 11172–11176 [PubMed] [Google Scholar]
- 76.Plow E. F., Collen D. (1981) J. Biol. Chem. 256, 10864–10869 [PubMed] [Google Scholar]
- 77.Váli Z., Patthy L. (1982) J. Biol. Chem. 257, 2104–2110 [PubMed] [Google Scholar]
- 78.Cockell C. S., Marshall J. M., Dawson K. M., Cederholm-Williams S. A., Ponting C. P. (1998) Biochem. J. 333, 99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang Y., Mochalkin I., McCance S. G., Cheng B., Tulinsky A., Castellino F. J. (1998) Biochemistry 37, 3258–3271 [DOI] [PubMed] [Google Scholar]
- 80.Novokhatny V. V., Matsuka YuV., Kudinov S. A. (1989) Thromb. Res. 53, 243–252 [DOI] [PubMed] [Google Scholar]