

Abstract
The intra-S checkpoint response to 254 nm light (UVC)-induced DNA damage appears to have dual functions to slow the rate of DNA synthesis and stabilize replication forks that become stalled at sites of UVC-induced photoproducts in DNA. These functions should provide more time for repair of damaged DNA before its replication and thereby reduce the frequencies of mutations and chromosomal aberrations in surviving cells. This review tries to summarize the history of discovery of the checkpoint, the current state of understanding of the biological features of intra-S checkpoint signaling and its mechanisms of action with a focus primarily on intra-S checkpoint responses in human cells. The differences in the intra-S checkpoint responses to UVC and ionizing radiation-induced DNA damage are emphasized. Evidence that [6-4]pyrimidine-pyrimidone photoproducts in DNA trigger the response is discussed and the relationships between cellular responses to UVC and the molecular dose of UVC-induced DNA damage are briefly summarized. The role of the intra-S checkpoint response in protecting against solar radiation carcinogenesis remains to be determined.
“DNA is, in fact, so precious and so fragile that we now know that the cell has evolved a whole variety of repair mechanisms to protect its DNA from assaults by radiation, chemicals and other hazards. This is exactly the sort of thing that the process of evolution by natural selection would lead us to expect.” Francis Crick in What Mad Pursuit.
Introduction to DNA damage checkpoints
Cell cycle checkpoints represent positions of control that coordinate the temporal sequence of required events during the cell division cycle to achieve complete and accurate replication and segregation of the nuclear genome once per cycle. Just as a checkpoint may regulate the transit of travelers from one country to another, cell cycle checkpoints regulate progression from one phase of the cell division cycle to the next phase. The cell division cycle is for the most part composed of irreversible phase transitions. Once DNA replication is begun, the cell is committed to complete DNA replication before returning to G1. Similarly, once anaphase is initiated during mitosis, cells will not normally revert to metaphase but rather proceed to cytokinesis. Because cycle phase transitions are irreversible, it is important that progression from one cycle phase to the next will not occur until conditions are appropriate and opportune. It is inappropriate to begin mitosis before DNA replication is complete, and it is inopportune to replicate damaged DNA. Thus, checkpoints ensure the completion of dependent events during cell division and they provide more time for repair of DNA damage. Checkpoint signaling also can cause heavily damaged cells to die by apoptosis and stabilize replication forks that become stalled at sites of DNA damage. Checkpoint failure has serious consequences including generation of daughter cells with abnormalities of chromosome number (aneuploidy and polyploidy) and structure (allelic deletion, duplication or rearrangement). When these abnormalities are generated during development, various birth defects such as microcephalies can occur. When generated in differentiated somatic cells or tissue stem cells, cancer may develop. Cell cycle checkpoints, therefore, appear to play a vital role in maintaining cellular and organismal health.
The concept of a cell cycle checkpoint originated with studies by Weinert and Hartwell (1) showing that ionizing radiation (IR)-induced DNA damage caused yeast cells to delay growth in the G2 phase of the cell cycle. This delay was not simply a passive consequence of metabolic damage within cells, as certain gene mutations could prevent the delay. A checkpoint was seen as an active response to stress, dependent upon the gene product that, when mutated, reversed the delay. RAD9 was the first gene that was shown to enforce a checkpoint response to DNA damage. Treatment with IR or the alkylating agent methylmethanesulfonate caused cells with wild-type RAD9 to delay in G2, whereas cells with mutant RAD9 did not. This discovery solved an important conceptual problem in radiation biology and carcinogenesis, how to explain the attenuation or ablation of a DNA damage response.
The phenomenology of cell division delay during a DNA damage response had been described well before the concept of checkpoints appeared (2). It is possible to identify reports of IR-induced G2 delay in human cells dating back to 1959 (3) and drugs that reversed IR-induced G2 delay were recognized in 1969 (4). The ability of caffeine to reverse carcinogen-induced cell cycle delays was established in the 1970s and in 1980 Murnane et al. (5) showed that caffeine and other methylxanthines reversed an intra-S checkpoint response to the DNA cross-linking poison, nitrogen mustard. In this same year appeared three studies reporting that a cell cycle delay response to DNA damage was defective in patients with the inherited radiation hypersensitivity syndrome, ataxia telangiectasia (AT). Cells from AT patients displayed less inhibition of DNA replication than normal after treatment with IR (6–8). This radioresistant DNA synthesis included less inhibition of replicon initiation as well as less inhibition of DNA chain elongation (6). Zampetti-Bosseler and Scott (9) subsequently showed that the IR-induced G2 delay also was attenuated in cells from AT patients. The first models for the defect in AT cells proposed an alteration in chromatin structure that interfered with the DNA damage to prevent the cycle phase delays. We now know that AT cells have mutations that inactivate ATM (10), a phosphatidyl inositol kinase-like protein kinase that is activated by DNA double-strand breaks (dsb) and phosphorylates many proteins to slow or arrest progression through the cell cycle. Over the past 30 years, at least 50 genes have been shown to contribute to human cell cycle checkpoint responses (Table I). More genes are being added to the list yearly. In 2007, Matsuoka et al. used a high-throughput screen to identify >700 proteins that were phosphorylated by ATM or the ATM- and Rad3-related checkpoint kinase ATR in response to DNA damage. Of 37 proteins that had not previously been associated with DNA damage response and are not listed in Table I, 29 were found to affect the intra-S or G2 checkpoint responses to IR-induced DNA damage (56). ATM- and ATR-dependent DNA damage responses must occupy extensive cellular signaling networks.
Table I.
Human cell cycle checkpoint genes
| 53BP1 (11), BRCT repeat-containing protein that mediates responses to DNA dsb | Chk2 (12), non-essential checkpoint kinase that phosphorylates Cdc25 and p53 | Plk1 (13), mitotic protein kinase that mediates recovery from G2 checkpoint |
| ATM (10), non-essential checkpoint kinase that is activated by DNA dsb and changes in chromatin | Claspin (14), Chk1- and Tim-associated checkpoint mediator | PP2A (15), phosphatase that regulates checkpoint kinase activity |
| Abraxas (16), chaperone that with Rap80 brings BRCA1 to sites of DNA dsb | DNA-PK (17), non-essential protein kinase that is required for repair of DNA dsb by non-homologous end joining | PPM1D (18), p53-transactivated phosphatase that regulates checkpoint kinase activity |
| ATF2 (19), transcription factor in AP1 heterodimers that mediates ATM-dependent intra-S checkpoint | FAAP24 (20), with FANCM regulates FA complex loading on chromatin and mediates ATR/Chk1 signaling | Rad1 (21), member of 9-1-1 checkpoint clamp |
| ATR (22), essential checkpoint kinase that is activated by stalled replication forks | FANCM (20), DNA translocase that mediates ATR/Chk1 signaling | Rad17 (23), in complex with RFC2–5, loads 9-1-1 checkpoint clamp around DNA |
| ATRIP (24), ATR-interacting protein that binds RPA to bring ATR to stalled forks | H2AX (25), minor histone variant that organizes repair of DNA dsb | Rad18 (26), with Rad6 forms ubiquitin ligase that modifies PCNA |
| BACH1 (27), BRCA1-associated factor that mediates intra-S checkpoint response to IR | HCLK2 (28), circadian clock gene that regulates ATR expression | Rad50 (29), member of MRN complex that processes DNA dsb for repair and checkpoint activation |
| BARD1 (30), cofactor for BRCA1-associated ubiquitin ligase | HDAC4 (31), histone deacetylase that mediates checkpoint response | Rad9 (32), member of 9-1-1 checkpoint clamp that recruits the ATR cofactor TopBP1 |
| BRCA1 (33), checkpoint mediator required for repair of DNA dsb by homologous recombination | HNF8 (34), ubiquitin ligase that mediates responses at DNA dsb | Rap80 (16), ubiquitin-binding protein that interacts with abraxas to bring BRCA1 to DNA dsb |
| BRIT1/MCPH1 (35), mediator of checkpoint signaling | Hus1 (36), member of 9-1-1 checkpoint clamp | RB (37,38), regulates E2F transcription factors to control cell division |
| Bub1 (39), required for spindle assembly checkpoint | Mad1 (40), required for spindle assembly checkpoint | SMC1 (41), member of cohesin complex that mediates intra-S checkpoint |
| Bub1R (42), required for spindle assembly checkpoint | Mad2 (40), required for spindle assembly checkpoint | SMC3 (43), member of cohesin complex that mediates intra-S checkpoint |
| Cdc25A (44), phosphatase that activates cyclin-dependent kinases | MDC1 (45), BRCT repeat containing mediator of DNA damage checkpoints | Tim (46), member of RFPC that mediates intra-S checkpoint response to UVC |
| Cdc25B (47), phosphatase that activates cyclin-dependent kinases | MRE11 (29), member of MRN complex that processes DNA dsb for repair and checkpoint activation | Tipin (48), member of RFPC that mediates intra-S checkpoint response to UVC |
| Cdc25C (49), phosphatase that activates cyclin-dependent kinases | Myt1 (50), kinase that phosphorylates and inactivates cyclin-dependent kinases | TopBP1 (51), cofactor for essential ATR checkpoint kinase |
| Cep164 (52), mediator of checkpoint signaling | Nbs1 (29), member of MRN complex, mediates ATM activation | Wee1 (50), protein kinase that inhibits cyclin-dependent kinases |
| Chk1 (53), essential checkpoint kinase required for UVC-induced intra-S checkpoint | P53 (54), transcriptional activator and repressor that regulates checkpoint responses in trans | XPA (55), NER protein that mediates Chk1 activation |
Genes that have been shown to regulate or influence cell cycle checkpoint responses in human cells.
BRCT, BRCA1 carboxy terminus; MRN, MRE11/Rad50/NBS1 complex.
The importance of DNA damage checkpoint responses is revealed in cells that are unable to mount the response. AT cells are radiosensitive and display significantly increased frequencies of chromosomal aberrations after treatment with IR (57,58). Although AT cells do not display hypersensitivity to inactivation of colony formation by UVC, they do display increased frequencies of UVC-induced chromosomal aberrations (59,60), perhaps due to defective response to UVC-induced DNA dsb. AT patients also are radiosensitive and cancer prone, developing leukemias and lymphomas with 100-fold increased incidence rates (61). Because repair of DNA damage can ameliorate the genotoxicity of these radiations, the more time that cells have for DNA repair before DNA replication and mitosis the less induction of mutations and chromosomal aberrations as potentially heritable changes in the genome.
Checkpoint signaling not only provides more time for repair but also may enhance DNA repair. The checkpoint kinases ATM and ATR can activate p53, as the guardian of the genome, not only to induce expression of p21Waf1 for G1 arrest and BAX and PUMA for apoptosis but also XPC and XPE to enhance nucleotide excision repair (NER) of certain forms of DNA damage (62). ATR may phosphorylate XPA (56) to enhance NER in S phase cells (63). When activated by IR-induced DNA damage, ATM phosphorylates KAP-1 to enhance the repair of DNA dsb in heterochromatin (64). Thus, checkpoint signaling not only provides more time for DNA repair but also enhances or stimulates repair.
A full discussion of the field of cell cycle checkpoints is beyond the scope of this review and the reader is referred to other excellent reviews for primary references (62,65–68). Here the focus will be on a selected subtype of DNA damage checkpoint, the intra-S checkpoint response to UVC-induced DNA damage, that includes cyclobutane-type pyrimidine dimers (CPD) and [6-4]pyrimidine-pyrimidone (6-4PP) photoproducts in DNA as the principal forms of DNA damage.
Why study the effects of ultraviolet radiation on DNA replication?
Analysis of the mechanisms of chemical carcinogenesis in rat liver indicated that the combination of DNA damage and cell proliferation was essential; induction of DNA damage in quiescent hepatic tissue was without carcinogenic effect and hepatocyte proliferation alone did not induce liver cancer (69). In contrast, the combination of a single treatment with a chemical carcinogen during the phase of active cell division after a two-thirds partial hepatectomy produced liver cancer in a significant number of treated animals (70–72). The timing of the treatment with carcinogen was also important as hepatocytes appeared to display greatest risk of carcinogenesis at the time of DNA synthesis. Proliferating hepatocytes before or after the peak of DNA synthesis appeared to be less susceptible to carcinogenesis than hepatocytes during the peak of DNA synthesis. As the carcinogens that were used were direct acting and not influenced by variation in metabolic function after partial hepatectomy, it was concluded that replication of carcinogen-damaged DNA was required for initiation of carcinogenesis, and DNA repair before DNA replication reduced hepatocyte risk of initiation (Figure 1).
Fig. 1.

Cell cycle-dependent initiation of carcinogenesis. Replication of damaged DNA is required to induce mutations and chromosomal aberrations as permanent heritable changes in the genome that can initiate carcinogenesis. Repair of DNA damage before DNA replication reduces the frequencies of mutations and chromosomal aberrations and thereby reduces the risk of initiation of carcinogenesis.
Analysis of carcinogenesis using human skin fibroblast cultures reached similar conclusions concerning the sensitivity of S phase cells (73). For example, treatment with UVC at the time of entry of synchronized fibroblasts into S induced more mutations (74), chromosomal aberrations (60) and cellular transformation (75) than treatment early in the pre-replicative phase of growth or after completion of DNA synthesis. It appears that the S phase cell is the target of UVC carcinogenesis. Thus, it is important to understand how S phase cells respond to UVC-induced DNA damage.
UVC treatment inhibits DNA replication in S phase cells and delays progression through S phase (76). The inhibition of DNA replication is dose dependent, with increasing doses of DNA damage producing greater inhibitions of DNA synthesis (77). The recovery of DNA replication after UVC irradiation is also dose dependent. The greater the dose of DNA damage the longer that DNA replication is inhibited. UVC inhibits DNA synthesis by effects on two components of DNA replication, the rate of replicon initiation to generate active replication forks and the rate of DNA chain elongation in active replicons (78,79). The responses of human fibroblasts to UVC-induced DNA damage are not unique but rather are quite similar to responses produced by the chemical carcinogen, benzo[a]pyrene diolepoxide I (BPDE). Treatment with UVC or BPDE produced similar effects on DNA replication, with low doses inhibiting replicon initiation and higher doses inhibiting DNA chain elongation in active replicons (80,81). As the responses to UVC are similar to the responses to the chemical carcinogen, it appears that human fibroblasts display a stereotypic response to DNA damage. Thus, analysis of the specific effects of UVC on DNA replication in diploid skin fibroblasts may reveal details that are generally true for proliferating human cells in many tissues after exposure to a variety of environmental carcinogens. As replication of damaged DNA appears to be required to initiate carcinogenesis, the response of S phase cells to UVC-damaged DNA may provide clues to how environmental carcinogens cause cancer.
The intra-S checkpoint response to UVC-induced DNA damage
The phenomenon of intra-S checkpoint response to UVC was first recognized 30 years ago when the method of velocity sedimentation was applied to analysis of DNA replication in UVC-damaged diploid human fibroblasts and the HeLa cervical carcinoma cell line (79). The method involves separation of nascent DNA molecules according to size in alkaline sucrose gradients. A brief pulse of labeling with tritiated (3H)-thymidine added a radioactive tag to the growing ends of nascent DNA molecules in S phase cells. After separation by sedimentation in alkaline sucrose gradients, the labeled DNA molecules displayed a broad distribution of sizes, with a peak at ∼2 × 107 Da and a shoulder extending up to 2 × 108 Da, the limit of resolution of the gradients. Pulse-chase experiments showed that the sizes of the nascent molecules corresponded to their age. The smaller molecules originated in replicons that initiated DNA synthesis during the pulse with 3H-thymidine, and these small molecules grew with time to larger sizes by chain elongation and merging between adjacent replicons (78). A precursor–product relationship was evident with the smaller molecules of about half the average replicon size being precursors to larger multi-replicon-size intermediates. Although increasing times of incubation with 3H-thymidine produced increasing amounts of radioactivity in DNA, there was a period of incubation of 3–15 min when the size distribution of the labeled nascent DNA molecules did not change. Thus, a 10–15 min pulse revealed a steady-state distribution of nascent DNA molecules. By varying the dose of UVC and the time between UV irradiation and the pulse with 3H-thymidine, it was possible to describe changes in the steady-state distribution of nascent DNA as induced by UVC irradiation (78).
When HeLa cells or diploid fibroblasts were exposed to a graded series of fluences of UVC ranging from one that was sublethal without effect on colony formation efficiency to one that was twice the mean lethal dose and then incubated for 30 min before the pulse with 3H-thymidine, several changes in the steady-state distribution of nascent DNA molecules were evident (Figure 2). Low sublethal doses of UVC produced a selective inhibition of DNA synthesis in low-molecular weight intermediates that initiated DNA synthesis during or soon before the pulse labeling. Whereas synthesis of molecules of half the average replicon size was reduced by ∼50% after 1 J/m2 UVC, synthesis of multi-replicon-size intermediates was not inhibited after this low dose of UVC. However, after fluences >1 J/m2, there occurred a dose-dependent inhibition of DNA synthesis in the multi-replicon-size intermediates that had initiated DNA synthesis before the treatment with UVC. With higher doses of UVC that produced a severe inhibition of synthesis in the large multi-replicon-size intermediates, there appeared a new class of abnormally small nascent intermediates between the sizes of Okazaki fragments and newly initiated replicons. The sedimentation of these abnormally small intermediates overlapped with the position of sedimentation of newly initiated replicons, partially obscuring the inhibition of the newly initiated replicons seen at lower doses. UVC-induced DNA damage, therefore, was found to produce in normal fibroblasts and the HeLa cancer cell line a complex but reproducible pattern of inhibition of DNA synthesis that varied considerably according to dose (78,79).
Fig. 2.

Velocity sedimentation analysis of intermediates of DNA replication in UV-damaged human fibroblasts. The velocity sedimentation method involves separating 3H-thymidine-labeled nascent DNA molecules according to size by sedimentation in alkaline sucrose gradients. Incorporation of 3H-thymidine is normalized to DNA content, so profiles represent the specific activities of DNA synthesis in various size classes of nascent DNA. DNA size is a measure in part of replicon age. Cells were incubated with 3H-thymidine for 15 min beginning 30 min after sham or UV treatment. Sedimentation was from right to left. The peak of 3H radioactivity at fractions 16 and 17 in the no-UV control corresponds to nascent molecules of about half the average replicon size (2 × 107 Da) that initiated synthesis after the sham treatment. The selective inhibition of DNA synthesis in half-replicon-size intermediates seen 30 min after 0.5 or 1 J/m2 reflects the inhibition of replicon initiation. The inhibition of DNA synthesis in multi-replicon-size intermediates (fractions 5–12) reflects the UV dose-dependent inhibition of DNA chain elongation in replicons that had initiated synthesis before UV treatment. Note the abnormally small nascent intermediates that appear in fractions 18–21 after 5 and 10 J/m2. These probably arise by DNA synthesis between sites of UV-induced DNA damage.
As fibroblasts were examined further, the pattern of response to UVC also varied considerably according to the time after irradiation when the pulse with 3H-thymidine was applied (78). When cells were incubated for varying times after 0.8 J/m2, it was found that the inhibition of DNA synthesis spread between 30 and 90 min to include multi-replicon-size intermediates. This phenomenology was identical to that reported previously for HeLa cells that had been damaged with IR (82), and this selective inhibition of DNA synthesis in low-molecular weight intermediates that spread with time to include larger intermediates was attributed to an inhibition of replicon initiation.
The mechanism of inhibition of replicon initiation was unknown in the 1980s. It had initially been hypothesized that DNA strand breaks generated during NER triggered the response to UVC by relaxing DNA supercoils (83). However, NER-defective xeroderma pigmentosum (XP) cells also displayed the inhibition of replicon initiation, indicating that excision repair was not responsible (78). Indeed, XP cells displayed a longer lasting inhibition of replicon initiation. Whereas normal cells had recovered their rate of replicon initiation to 80% of control by 90 min after UVC irradiation, NER-defective XP cells did not recover replicon initiation even after 240 min. Because the dose of UVC that selectively inhibited replicon initiation by 50% produced ∼1 CPD per replicon, it was conceivable that encounter of replication forks with such dimers might be responsible for the effect. As will be discussed below, it appears that the encounter of replication forks with the 6-4PP photoproduct triggers the intra-S checkpoint response to inhibit replicon initiation.
As mentioned earlier, incrementally higher fluencies of UVC that inactivate colony formation by 20–70% produced additional changes in the sedimentation profiles of nascent DNA. There was a dose-dependent inhibition of DNA synthesis in multi-replicon-size intermediates and development of a new species of nascent DNA with abnormally small size. Studies at the time had shown that replicative DNA polymerases were blocked at sites in template strands of the most abundant UVC-induced photoproduct, the CPD (84). The velocity sedimentation phenomenology was interpreted to indicate that UVC-induced DNA damage blocked DNA chain elongation by DNA polymerase at active DNA replication forks and that initiation of DNA synthesis beyond the sites of blockage generated nascent molecules of abnormally small size. NER-defective XP cells displayed greater than normal sensitivity to inhibition of DNA chain elongation by UVC, indicating that repair of DNA damage before DNA replication was protective.
NER-proficient XP variant cells displayed even greater hypersensitivity to inhibition of DNA chain elongation by UVC than NER-defective XP cells and excessive production of the abnormally small nascent intermediates. XP variant cells were known to display a defect in replication of UVC-damaged DNA templates (85). We concluded at the time that an abundant UVC-induced DNA photoproduct inhibited DNA synthesis in XP variant cells but not in normal fibroblasts (78). We now know that XP variant cells have mutations that inactivate DNA pol eta (86,87) and this DNA polymerase is capable of holding a template thymine–thymine CPD in its active site and incorporating adenosine monophosphate into nascent DNA strands opposite the CPD by translesion synthesis (TLS) (88). Rad6- and Rad18-dependent ubiquitylation of PCNA appears to recruit the translesion DNA polymerases pol eta and pol iota to sites of DNA replication to permit rapid bypass of CPDs (89,90). XP variant cells lacking expression of DNA pol eta must rely upon some other mechanism for bypass of UVC-induced CPDs. This alternative bypass mechanism is more mutagenic than TLS by DNA pol eta, and so XP variant cells display enhanced mutagenesis after treatment with UVC (91–93). A recent study suggests that DNA pol iota may contribute to mutagenic TLS in XP variant cells (94). Failure to bypass the thymine–thymine CPDs in the XP variant cells also was associated with severely delayed recovery of replicon initiation after treatment with low UVC fluencies (76,78). Thus, blockage of DNA chain elongation by thymine–thymine CPDs produced in XP variant cells a long-lived signal to inhibit the initiation of DNA synthesis at origins of replication.
Role of ATM, ATR and Chk1 in the intra-S checkpoint response to UVC
With the discovery that AT cells were defective in the inhibition of replicon initiation in response to IR-induced DNA damage, Painter (95) examined the response of an AT fibroblast line to UVC-induced DNA damage. The AT cells displayed less inhibition of replicon initiation after a low fluence of UVC than the normal control line, and so a conclusion was reached that AT cells also were unable to inhibit replicon initiation in response to UVC-induced DNA damage. This result did not at first glance seem unreasonable. Although AT cells do not display hypersensitivity to UVC-induced cytotoxicity, they do display severe hypersensitivity to UVC-induced chromosomal aberrations. In two separate studies, AT fibroblasts displayed ∼10 times more chromosomal aberrations after UVC treatment than normal fibroblasts (59,60). The cytotoxic lesion that is induced by UVC appears to be DNA photoproducts that block RNA transcription, as human cells in S phase are not noticeably more sensitive than cells in G1 or G2 (74). This is not true for induction of mutations and chromosomal aberrations, for which human cells are much more sensitive when in S than in G1 or G2. Thus, it seemed reasonable that AT cells were defective in the intra-S checkpoint response to UVC-induced DNA damage.
When ATM was cloned as the gene that is mutated in AT, analysis of the biochemistry of its gene product, the ATM protein, showed that it was activated as a protein kinase in cells with IR-induced DNA dsb, but not in cells with UVC-induced DNA damage (96). Then we found that two human fibroblast lines that were immortalized by transformation with SV40 virus or transduction of SV40 large T antigen displayed an inability to inhibit replicon initiation after treatment with UVC or IR (97). As Painter's prior analysis was done with an SV40-transformed AT fibroblast line, it was conceivable that the failure to inhibit replicon initiation after UVC was due to SV40 transformation and not derivation from an AT patient. A difficulty in working with AT fibroblasts in culture is their limited in vitro proliferative lifespan and low S phase fraction. This is one reason that the SV40-transformed AT fibroblast line was used. However, because SV40 transformation forces cells through a phase of crisis with severe chromosomal damage prior to establishment of the immortal line, the immortal lines that emerge from crisis may express idiosyncratic biology secondary to the chromosomal damage. With the discovery and cloning of the human telomerase reverse transcriptase (98,99), a much more efficient method for immortalization of human fibroblasts was available (100).
While human telomerase reverse transcriptase-expressing AT, AT-LD and NBS fibroblasts all were defective for the inhibition of replicon initiation after IR, as expected, these lines displayed an effective response after UVC (101). Thus, ATM, MRE-11 and NBS1 were not required for the inhibition of replicon initiation by UVC-induced DNA damage, casting serious doubt on the hypothesis that ATM enforced the response. ATR was found to be required. The U2OS human fibrosarcoma line that over-expressed kinase-active ATR displayed the selective inhibition of replicon initiation after a low dose of UVC; in contrast, U2OS cells that over-expressed a kinase-inactive ATR did not (101). Similarly, whereas over-expression of a kinase-active Chk1 allele in U2OS cells severely inhibited DNA replication, over-expression of a kinase-inactive Chk1 allele did not inhibit DNA replication and severely attenuated the UVC-induced inhibition of replicon initiation. Further evidence for the requirement for Chk1 in the response was its reversal in normal and AT fibroblasts by the Chk1 inhibitor UCN-01. UVC-induced DNA damage was shown to activate Chk1 through phosphorylation of ser317 and ser345 in Chk1 by ATR. Contemporaneous studies by Miao et al. (102) also concluded that Chk1 was required for the intra-S checkpoint response to UVC. Later studies using siRNA-mediated depletion of ATR and Chk1 in HeLa cells confirmed the requirements for ATR and Chk1 in the intra-S checkpoint response to UVC (103). These studies, therefore, defined an ATM-independent intra-S checkpoint response to UVC-induced DNA damage in human cells that appeared to operate through an ATR–Chk1 signaling pathway.
Downstream effectors of intra-S checkpoint signaling
The intra-S checkpoint response to IR-induced DNA dsb requires ATM, MRE-11 and NBS1 and appears to signal through apparently two separate branches to inhibit replicon initiation (41,104). One branch involves activation of Chk1 and Chk2 kinases that phosphorylate Cdc25A to induce its proteolytic degradation (44,105). The second branch involves signaling through the cohesin subunits, Smc1 and Smc3, by phosphorylation of serine's 957 and 966 in Smc1 and serine 1083 in Smc3 by ATM (41,43,104,106).
Cyclin E/Cdk2 appears to be required for replicon initiation (44,107,108). Phosphorylation of tyrosine 15 in Cdk2 blocks ATP binding and inhibits kinase activity. Human Cdk2 is regulated in part through phosphorylation at tyr15 by Wee1 (109,110). Cdc25A is a tyrosine phosphatase that removes the inhibitory phosphate on tyr15 in Cdk2. Because activation of Cdk2 requires dephosphorylation of tyr15 by Cdc25A, checkpoint-dependent proteolysis of Cdc25A represents an effective mechanism to inhibit Cdk2. Thus, ATM enforces two mechanisms to inhibit Cdk2 in response to IR. ATM-dependent phosphorylation and activation of p53 leads to induction of the cyclin-dependent kinase inhibitor p21Waf1 as a slow but principal effector of the G1 checkpoint response to IR (109). ATM-dependent, but p53-independent, phosphorylation and activation of Chk1 and Chk2 lead to phosphorylation and proteolysis of Cdc25A, as a rapid effector of the intra-S checkpoint response to IR.
A second arm of the ATM-dependent intra-S checkpoint response proceeds through the cohesin subunits, Smc1 and Smc3 (41,43,106). Smc1 forms a heterotetrameric complex with Smc3, Scc1 and SA1 or SA2 to form cohesin, a ring-like protein complex that can envelop two DNA duplexes and is loaded onto nascent DNA during DNA replication (111). Cohesion between daughter duplexes as enforced by cohesin appears to enhance recombinational repair of DNA dsb (112,113). It is thought that cohesion maintains daughter duplexes in spatial proximity and sequence alignment, so that when dsb occur in one chromatid the other undamaged chromatid is properly spaced and aligned to facilitate error-free recombinational repair of the double-strand break. Smc1- and Smc3-defective cells display an attenuation of the intra-S checkpoint response to IR (41,43,106), although the mechanism by which cohesin contributes to the intra-S checkpoint remains to be determined.
Although ATM did not appear to be required for the intra-S checkpoint response to UVC, it was conceivable that the signal from ATR passed directly to Smc1 and to Cdc25A through Chk1, and therefore the terminal phase of the UV checkpoint signaling pathway overlapped with the ATM-dependent intra-S checkpoint response to IR. A supralethal fluence of 50 J/m2 of UVC had been shown to induce the proteolysis of Cdc25A (44) and ATM-independent phosphorylation of Smc1 (106). Thus, it was conceivable that UVC produced the same effects on Smc1 and Cdc25A as IR when triggering the intra-S checkpoint. Timothy Heffernan set out to test a hypothesis that low fluences of UVC produce the selective inhibition of replicon initiation by proteolysis of Cdc25A and inhibition of cyclin E/Cdk2 (114). Dose–response studies showed that a 1 J/m2 fluence of UVC sufficient to inhibit replicon initiation by 50% had no effect on Cdc25A protein expression, and fluences >8 J/m2 were needed to produce a reduction in Cdc25A protein. Chemical inhibition of Cdc25A proteolysis reversed the IR-induced intra-S checkpoint response but not the UVC-induced response, supporting a conclusion that Cdc25A proteolysis is not required for the intra-S checkpoint response to UVC. Consistent with the biochemical analysis of Cdc25A, treatment with IR caused a 50% reduction in cyclin E/Cdk2 kinase activity, whereas treatment with UVC did not inhibit cyclin E/Cdk2. Finally, whereas 5 Gy of IR produced a clear signal of phosphorylation of Smc1, 1 J/m2 of UVC did not induce detectable phosphorylation of Smc1. These results indicate that the ATR–Chk1 signaling pathway that inhibits replicon initiation in UVC-damaged diploid human fibroblasts does not converge on the Cdc25A–Cdk2 and Smc1 regulatory circuits that are used by the ATM-dependent signaling pathway in response to IR-induced DNA damage (114).
The target of Chk1 signaling in the intra-S checkpoint response to UVC remains to be determined. Replicon initiation requires two protein kinases, cyclin E/Cdk2 and Dbf4/Cdc7, which may act sequentially to activate the replication complex at replicon origins (115–117). Whereas DNA damage caused the Dbf4–Cdc7 complex to dissociate in Xenopus egg extracts (118) and Abl-transformed leukemic cells (119), no evidence for dissociation of the complex was observed in UVC-damaged diploid human fibroblasts (114) or in etoposide- or hydroxyurea-treated HeLa cells (120). Some involvement of the Dbf4–Cdc7 complex in the response to UVC was suggested by the result that over-expression of Flag- or Myc-tagged Dbf4 reversed the intra-S checkpoint response to UVC, but not IR. This reversal of the checkpoint response was not associated with reversal of Chk1 activation by ATR, and so it was concluded that the over-expression of Dbf4 acted downstream of Chk1 to attenuate checkpoint signaling (114). As mentioned earlier, the chemical carcinogen BPDE triggers an intra-S checkpoint response that is similar to that provoked by UVC (80). Studies of the Chk1-dependent intra-S checkpoint response to BPDE indicated that Cdc45 binding to chromatin was reduced during the response (121). As Cdc45 and the GINS complex appears to be cofactors for the MCM helicase (122), reduction in Cdc45 association with the MCM helicase may reduce the efficiency of initiation of DNA replication at replicon origins. Dbf4/Cdc7 kinase phosphorylates MCM proteins and creates binding sites for GINS proteins and Cdc45 (116). The ATR–Chk1 checkpoint signaling pathway appears to act to reduce the content of Cdc45 on chromatin, and should this reduction affect the interaction Cdc45 with the MCM helicase complex, it could represent an effective mechanism for inhibiting replicon initiation. If the replicative helicase cannot denature template strands at origins of replication, replication forks are not generated and replicons are not initiated.
Two technologies have considerably enhanced efforts to elucidate the mechanisms of intra-S checkpoint response to UVC, siRNA-mediated protein depletion and fluorescence labeling of spread DNA fibers. siRNA-mediated protein depletion allows the selective removal of protein species by targeting mRNA stability and translation. It is possible now to deplete diploid human fibroblasts of most proteins of interest and determine the effect of protein depletion on the intra-S checkpoint response to UVC. Halogenated precursors for DNA synthesis that are incorporated into nascent DNA strands can be labeled with fluorescent antibodies after nuclear DNA is stripped of protein and spread on a glass slide. By incubating cells with different DNA precursors before and after treatment with UVC, fiber tracks are visualized that correspond to replicons that synthesized DNA before and after the UVC treatment. By counting the tracks that are labeled before, during or after the UVC treatment and measuring the lengths of the labeled tracks, it is possible to quantify replicon initiations and terminations, as well as rates of DNA chain elongation. These methods have led to the discoveries of new mediators of UVC-induced intra-S checkpoint response and the mechanisms of inhibition of DNA synthesis by the checkpoint.
The DNA fiber-spreading method was first used to show that treatment with IR caused HeLa cells to slow their rate of replicon initiation. When treatment with IR was done between the time of labeling with iodo-deoxyuridine given first and chloro-deoxyuridine (CldU) given second, it was found that the treatment with IR reduced the relative proportion of tracks that were labeled exclusively with the post-IR DNA precursor CldU (123). As tracks that contained CldU only originated in replicons that initiated synthesis after treatment with IR, the result showed that IR caused an inhibition of replicon initiation. Treatment with the alkylating agent MMS, that had been shown previously to cause an inhibition of replicon initiation (124), also produced a reduction in CldU-only tracks (123). We compared fiber spreading and velocity sedimentation in UVC-damaged HeLa cells (103). In control cells that were treated with an siRNA to firefly luciferase, that does not appear to target a human protein, a low dose of UVC caused ∼50% reduction in the rate of replicon initiation as measured by velocity sedimentation analysis. The same treatment reduced the proportion of tracks that were labeled only with the post-UVC precursor by an average of 64%. Depletion of ATR and Chk1 protein expression by transduction of siRNA reversed the inhibition of replicon initiation as measured by both methods. Thus, the two methods of analysis of replicon initiation yielded comparable results and confirmed the requirement for ATR and Chk1 in the intra-S checkpoint response to UVC. The fiber-spreading method also revealed new information about the mechanisms of intra-S checkpoint function as will now be described.
A replication fork protection complex mediates the intra-S checkpoint response to UVC
As mentioned previously, new components of checkpoint signaling pathways continue to be discovered and recent studies have identified three proteins, claspin, Tim and Tipin, that mediate the activation of Chk1 by ATR at stalled DNA replication forks. Orthologues and analogs of these proteins in yeast species mediate DNA damage checkpoint signaling and were termed the replication fork protection complex (RFPC) (125). Note that in yeast cells, signaling from sites of stalled DNA replication forks not only inhibits initiation of late-firing replicons but also stabilizes DNA replication forks against demise or collapse (126). The essential function of the DNA damage checkpoint in yeast cells is to stabilize stalled replication forks (126), and it is possible that in humans a similar RFPC mediates the essential functions of ATR and Chk1 to stabilize stalled replication forks. Tim and Tipin form a self-stabilizing heterodimeric complex in vertebrate cells that can interact with claspin, Chk1, RPA, MCM2, ATR-interacting protein (ATRIP) and DNA pol delta (48,127–131). Based on studies of claspin, Tim and Tipin proteins in vertebrate cells, the term, RFPC, is appropriately descriptive of their function. The RFPC appears to be well placed to mediate activation of Chk1 at stalled DNA replication forks.
Claspin is a checkpoint protein that was first cloned based on its physical association with Chk1 (132). Depletion of claspin from human cells attenuated Chk1 activation in response to replication stress and DNA damage and attenuated the inhibition of DNA synthesis induced by UVC (14,133). We have found that depletion of claspin in diploid human fibroblasts attenuated Chk1 activation in response to a low dose of UVC by >50% (S.Smith-Roe, M.Cordeiro-Stone and W.K.Kaufmann, unpublished data). Thus, claspin appears to be one mediator of ATR phosphorylation of Chk1.
Timeless (Tim) was discovered as a fly gene that regulates the circadian clock (134,135). However, it also has homology to yeast proteins that participate in the DNA damage checkpoint response (128). Knockout of the Tim ortholog in mice was embryo lethal, suggesting that the Tim protein is essential (136). SiRNA-mediated depletion of Tim in mouse fibroblasts was associated with significantly increased frequencies of chromosomal aberrations and sister chromatid exchanges, suggesting that essential function of Tim is to stabilize the genome against lethal chromosomal damage (137). Tim was found to interact with Chk1 and ATRIP in human cells and this interaction was stimulated after treatment with UVC (46). Human Tim was also shown to interact with claspin, MCM2 and DNA pol delta (128), suggesting that Tim might couple the DNA growing point to the replicative helicase. Depletion of Tim expression in HeLa cells attenuated the intra-S checkpoint response to UVC and attenuated the activation of Chk1 in response to replication stress or UVC damage, indicating that Tim was a mediator of ATR phosphorylation of Chk1 and intra-S checkpoint response to UVC (46). Although studies with mammalian cells to date have not reported an effect of Tim depletion on sister chromatid cohesion, depletion of Tim in Caenorhabditis elegans caused defective loading of cohesin subunits on chromatin and premature centromeric separation (138). We have observed a similar phenotype of premature centromeric separation in Tim-depleted human fibroblasts (Smith-Roe, Cordeiro-Stone and Kaufmann, unpublished data).
A plausible mechanism whereby Tim might mediate Chk1 phosphorylation was identified with the discovery that Tim forms a self-stabilizing heterodimeric complex with Timeless-interacting protein (Tipin) (127). Depletion of Tim or Tipin by siRNA causes the partner in the complex also to be depleted (48,129,130), implying that each protein is required to stabilize its partner in the dimeric complex. It appears that the complex of Tim and Tipin is responsible for the mediation of Chk1 activation and intra-S checkpoint response to UVC, as depletion of either Tim or Tipin reversed the intra-S checkpoint response to UVC (48,128). Tipin also appears to be required to stabilize stalled DNA replication forks as depletion of Tipin in Xenopus egg extracts reduced the ability of replication forks to restart after they were stalled by incubation with the DNA polymerase inhibitor aphidicolin (131). Tipin was found to contain a domain that is homologous to a domain in the NER factor XPA that interacts with RPA. Tipin was shown to interact with RPA in HeLa cells and RPA promoted Tipin binding to DNA in an electrophoretic mobility shift assay in vitro (48). Tipin also promoted claspin loading on chromatin in Xenopus egg extracts (131). Tipin's role in the RFPC may be to bring Tim, claspin and Chk1 to stalled replication forks via its interaction with RPA. As activated Chk1 appears to diffuse freely through the nucleus (139), a RFPC composed of Tim, Tipin and claspin may increase the local concentration of Chk1 at stalled replication forks, thereby increasing the probability that Chk1 is phosphorylated and activated by ATR.
The intra-S checkpoint regulates replicon initiation and DNA chain elongation
Analysis of spread DNA fibers revealed an unexpected separation of Tim and Tipin function (48). Whereas depletion of either protein prevented the UVC-induced inhibition of new origin firing, the depletions had significantly different effects on DNA chain elongation in active replicons. Depletion of Tim inhibited DNA chain elongation by ∼50% in cells that had not been treated with UVC. Fiber tracks were shorter than in the control cells that had been transduced with non-targeting control siRNA. In contrast, the lengths of fiber tracks in Tipin-depleted cells were not significantly reduced relative to the non-targeted control. These results were interpreted to indicate that Tim protein is required for normal rates of DNA chain elongation, similar to recent studies showing that rates of DNA chain elongation are reduced by ∼50% in Chk1- and claspin-depleted cells (140,141). A low level of Chk1 signaling appears to be required to sustain normal rates of DNA chain elongation, and Tim and claspin may mediate this signaling. Because siRNA-mediated depletion of Tipin was associated with reduced expression of Tim but depletion of Tipin did not inhibit DNA chain elongation, it was proposed that residual Tipin may enforce the inhibition of DNA chain elongation in Tim-depleted cells (48). Given the interaction of Tipin with RPA, it is conceivable that residual Tipin outside of a complex with Tim could slow DNA synthesis by DNA polymerases.
As mentioned previously, UVC-induced DNA damage may block the replicative DNA polymerases producing a passive inhibition of DNA chain elongation. Analysis of the sizes of DNA fiber tracks that were labeled after a low dose of UVC revealed that DNA chain elongation was inhibited by UVC. Fiber tracks that were synthesized after treatment with 2.5 J/m2 were 50% of the size of tracks that were synthesized after a sham irradiation (no treatment with UVC but with the same experimental manipulations used for UVC treatment) (48). A previous velocity sedimentation analysis of UVC-treated HeLa cells indicated that 2.6 J/m2 inhibited DNA chain elongation by ∼30%, a reasonable correspondence between the two methods (79). However, in Tipin-depleted cells, the UVC treatment inhibited DNA chain elongation by only 10%, and this value was significantly less than that measured in the non-targeted control (48). Thus, it appeared that the inhibition of DNA chain elongation after treatment with a low dose of UVC was an active process that required the presence of the checkpoint mediator protein Tipin. Because depletion of Tipin's partner protein Tim reduced DNA chain elongation in un-irradiated cells, it was not possible to conclude that Tim was required for the UVC-induced inhibition of DNA chain elongation. This study revealed a second component of the intra-S checkpoint response to UVC, active inhibition of DNA chain elongation. Cells sense the presence of DNA damage in template strands to activate a signaling pathway that not only slows the rate of initiation of DNA synthesis at replicon origins but also slows the rate of DNA chain elongation in already active replicons.
Several other studies support that concept that the intra-S checkpoint has a dual function to inhibit both replicon initiation and DNA chain elongation. Painter and Young (6) first showed using velocity sedimentation analysis that IR produced an inhibition of DNA chain elongation and replicon initiation; AT cells displayed less inhibition of both arms of the response. Wang et al. (142) studied the effects on DNA replication of Hus1, a component of the Rad9-Rad1-Hus1 (9-1-1) checkpoint clamp. Because knockout of Hus1 caused a p53-dependent growth arrest, the effector of p53-dependent G1 checkpoint function, p21Waf1, also had to be deleted. Cells from p21Waf1 knockout mice displayed an effective intra-S checkpoint response to IR and the topoisomerase I inhibitor camptothecin, which induces replication-dependent DNA dsb. High doses of IR and camptothecin also produced a severe inhibition of DNA chain elongation in p21Waf1 null cells as measured using velocity sedimentation analysis. In contrast, the same treatments produced significantly less inhibition of DNA chain elongation in Hus1 null/p21Waf1 null cells. The results indicated that Hus1 was required for the inhibition of DNA chain elongation in IR- and camptothecin-treated cells. Note that Hus1 has also been shown to be required for the inhibition of DNA synthesis that follows treatment with a low dose of UVC or BPDE (36). Seiler et al. (143) studied DNA replication in camptothecin-damaged cells using the fiber-spreading method. Treatment with camptothecin inhibited DNA replication by inhibiting both replicon initiation and DNA chain elongation. Chemical inhibition of Chk1 or siRNA-mediated depletion of Chk1 reversed both inhibitions, indicating that Chk1 transduces checkpoint signals to inhibit replicon initiation and DNA chain elongation. These several reports now support a hypothesis that the intra-S checkpoint response to DNA damage includes not only active inhibition of replicon initiation but also active inhibition of DNA chain elongation. Not only is it valuable to delay the initiation of DNA replication to acquire more time for DNA repair before replication but there is also value in slowing the rate of DNA chain elongation in active replicons, which also provides more time for repair of DNA damage before it is encountered at replication forks by DNA polymerase.
The concept that cells may regulate the rate of DNA replication, changing the speed with which replication forks produce daughter duplexes, is appealing and, indeed, logical. Given the importance of DNA replication for the viability of cells and organisms, it is difficult to conceive of a system in which the rate of replication fork progression would have only one speed, fully on. A system with only one speed can replicate DNA with high efficiency in the absence of impediments along template strands, but once impediments are introduced into the system, the ability to regulate the speed of DNA replication gains considerable value. An appealing hypothesis is that the MCM–GINS–Cdc45 helicase complex represents the position of control of DNA chain elongation, with the helicase complex fixed in space by attachment to the nuclear matrix and daughter template strands actively extruded from the helicase at a variable rate depending upon the state of the template duplex. The rate that the helicase extrudes daughter template strands determines the rate with which DNA polymerase synthesizes daughter DNA. It may not be coincidental that the current data show similar ∼50% inhibitions of DNA chain elongation and replicon initiation after treatment with the low 1 J/m2 dose of UVC. Has the intra-S checkpoint system evolved to sense the presence of DNA damage and transmit a signal that acts to inhibit the replicative helicase complex thereby slowing the rate of replicon initiation as well as the rate of DNA chain elongation?
Which form of UV-induced DNA damage activates the intra-S checkpoint?
A low dose of UVC (1 J/m2) that produces a 50% inhibition of replicon initiation yields ∼1 CPD per 60 kb replicon in diploid human fibroblasts (78,144). Conceivably, the presence of a CPD in DNA could be sensed to activate ATR and initiate the signaling pathway. It was shown that ATR kinase activity was stimulated by UVC-damaged DNA although the degree of stimulation was only ∼2-fold even after a high dose of UVC (145). Studies of the effect of replication stress on ATR activation have suggested that single-stranded DNA at stalled replication forks represents a strong stimulus for ATR phosphorylation of Chk1 (146,147). Soon after discovery of ATR as an essential checkpoint kinase (knockout mice display embryo lethality), ATR was shown to exist in a complex with an associated protein, ATRIP (148). ATRIP appears to be a chaperone helping to bring the kinase to its substrates. ATRIP has affinity for the single-stranded DNA binding protein RPA (24) that is required for DNA replication and repair. Accordingly, when DNA polymerases are inhibited by depletion of DNA precursors, uncoupling of the helicase and polymerase components at the replication fork may generate extended regions of single-stranded template DNA that become coated with RPA. RPA-coated DNA is a binding site for ATRIP, thereby bringing ATR to stalled replication forks. Junctions of duplex and single-stranded DNA appear to recruit an alternative form of the RFC clamp-loading complex, in which RFC1 is replaced by Rad17 (149). The Rad17–RFC2–5 complex loads the 9-1-1 checkpoint clamp at the stalled replication fork. Phosphorylation of Rad9 creates a binding site for TopBP1, an activating cofactor for ATR (32,150). TopBP1 was shown in a Xenopus cell-free system to stimulate ATR kinase activity by ∼100-fold (150), and studies with purified proteins from human cells show that TopBP1 stimulates ATR phosphorylation of Chk1 (151).
When a leading-strand DNA polymerase is blocked at a UVC-induced DNA photoproduct, uncoupling of polymerase and helicase activities may generate an extended region of single-stranded template DNA. This single-stranded DNA was first recognized as S1 nuclease-sensitive sites in nascent DNA in UVC-treated human fibroblasts (152). It was also visualized in replicating episomes with a site-specific CPD (153). The single-stranded DNA and dsDNA–ssDNA junctions at stalled replication forks attract RPA, Rad17/RFC2–5, 9-1-1, ATR/ATRIP and TopBP1 with the purpose to phosphorylate and activate Chk1, which then transduces the checkpoint signal throughout the nucleus. The RFPC of claspin, Tim and Tipin serves as a mediator of Chk1 phosphorylation. Depletion of any one of the RFPC proteins can significantly squelch the signal of activated Chk1 in response to stalling of DNA replication forks. Reinforcing interactions among RPA, claspin, Chk1, Tim and Tipin have the effect of recruiting Chk1 to regions of RPA-coated DNA at stalled replication forks, bringing the transducer checkpoint kinase Chk1 to the activator checkpoint kinase ATR. Indeed, Tim interaction with ATRIP and Chk1 appeared to be enhanced in UVC-damaged cells (46). Once phosphorylated by ATR, Chk1 rapidly diffuses away from the stalled fork and throughout the nucleus transmitting the signals to delay replicon initiation and inhibit DNA chain elongation in active replicons. This model recognizes two contributions to the inhibition of DNA chain elongation by UVC-induced DNA damage, an active checkpoint-mediated response to stalled replication forks and a passive inhibition by photoproducts in DNA template strands that cannot be incorporated into the active sites of replicative DNA polymerases. By blocking DNA polymerization, UV-induced photoproducts produce discontinuities in nascent DNA strands that later are removed either by translesion DNA synthesis or a recombinational bypass mechanism (154).
Analysis of the kinetics of Chk1 phosphorylation in UVC-damaged human fibroblasts suggested that CPDs were not the lesion that activates the intra-S checkpoint (155). After treatment with the moderate fluence, 6 J/m2, that inhibits DNA chain elongation by 50–70% and colony formation by ∼40%, there occurred a burst of Chk1 phosphorylation that was maximal 45–120 min after irradiation. By 6 h after irradiation, the signal of Chk1 activation had declined to near the basal levels seen in untreated controls. Quantification of UVC-induced CPDs and 6-4PPs showed that 80% of the 6-4PPs were removed from DNA within 3 h after irradiation with 12 J/m2, whereas >75% of CPDs remained in the DNA. Thus, rapid removal of 6-4PPs by NER was associated with substantial attenuation of the phospho-Chk1 signal of active intra-S checkpoint signaling. At a time (6 h post-UVC) when most of the UVC-induced CPDs still remained in DNA, the signal of active Chk1 was extinguished. As even more cells are in S phase 6 h after treatment with 6 J/m2 than in the sham-treated control, the reduced signal of phospho-Chk1 cannot be attributed to a reduced fraction of S phase cells. The result implies that the CPDs that remain in S phase cells do not activate the intra-S checkpoint. It is conceivable that the switching of the replicative polymerases alpha, delta and epsilon with polymerase eta at sites of template CPDs occurs so rapidly that insufficient uncoupling of helicase and polymerase occurs to activate the intra-S checkpoint. The 6-4PP photoproduct cannot be bypassed by polymerase eta alone, bypass requires a more complicated process of polymerase switching (156–158), and consequently the growing point of nascent DNA may be blocked longer at a 6-4PP lesion than at a CPD. Polymerase blockage at the 6-4PP lesions appears to generate sufficient uncoupling from the helicase to generate the single-stranded DNA that binds RPA and the many other intra-S checkpoint signaling factors.
The substantial decay in the phospho-Chk1 checkpoint signal seen 6 h after UVC may also define an upper limit for the timing of bypass of 6-4PPs by translesion DNA polymerases or recombinational mechanisms (154). Growing points that were blocked by 6-4PPs soon after UVC irradiation probably are bypassed by 6 h after irradiation in order for the signal of phospho-Chk1 to decay to near basal levels. Alternatively, a phenomenon of checkpoint adaptation (13,159) may extinguish the phospho-Chk1 signal before bypass of the 6-4PP has been accomplished. We found that the signal of activated Chk1 in BPDE-treated human fibroblasts also decayed to baseline within 6 h after carcinogen challenge (155), consistent with a report showing rapid repair of BPDE–DNA adducts in human fibroblasts (160).
A second independent analysis is consistent with a model that 6-4PPs represent the principal fork stalling UV-induced DNA photoproducts. As XP variant cells lack pol eta-dependent TLS across TT CPDs (and possibly across other CPDs), they display the consequences of fork stalling at both CPDs and 6-4PPs. Quantification of the dose kinetics for inhibition of DNA chain elongation demonstrated that XP variant fibroblasts were three times more sensitive to UVC irradiation than normal human fibroblasts (144). The implication of this result is that replication forks stall in normal fibroblasts at one-third of the template lesions that stall forks in XP variant cells. As 6-4PPs represent a third of the UV-induced photoproducts, it is possible that only the 6-4PPs produce a measurable inhibition of DNA chain elongation (fork stalling) in diploid human fibroblasts.
The conclusion that much of the intra-S checkpoint signaling in response to UVC-induced damage derives from uncoupling of helicase and polymerase activities at template 6-4PPs is at variance with a study that used photolyase to reverse CPDs in murine dermal fibroblasts (161). One output of checkpoint activation is phosphorylation of H2AX as a sensitive marker not only of DNA dsb (162) but also stalled DNA replication forks without formation of dsb (163). The intra-S checkpoint kinase ATR phosphorylates H2AX within the first 2 h after UV irradiation of S phase HeLa cells (164,165) and diploid human skin fibroblasts and melanocytes (W.K.Kaufmann, D.A.Simpson and M.Cordeiro-Stone, unpublished data). In murine diploid skin fibroblasts, phosphorylation of H2AX was slowly induced by UVC treatment being barely detectable at 2 h and not reaching a maximum until 48 h after irradiation (161). This signal was reduced by 50% by photoreactivation to remove CPDs from DNA. Thus, persisting CPDs accounted for about half of the phosphorylated H2AX, with the other half presumably due to some other long-lived cellular damage. Studies of the kinetics of checkpoint activation and photoproduct repair in human and murine skin fibroblasts yield highly divergent results. It is conceivable and indeed likely that mouse fibroblasts respond to radiation-induced DNA damage with sensitivities and kinetics that differ from human fibroblasts. For example, we found that a dose of IR (0.5 Gy) sufficient to arrest 80% of G1 human skin fibroblasts behind the ATM- and p53-dependent G1 checkpoint was unable to arrest mouse skin fibroblasts and 8 Gy of IR was needed to arrest 60% of G1 mouse skin fibroblasts (166). While murine models have proven to be of great value in understanding mechanisms of carcinogenesis, they do not fully replicate human DNA metabolism nor cell biology.
Checkpoint activation and translesion DNA synthesis.
The checkpoint activation and polymerase-switching mechanisms at stalled replication forks appear to be connected. Rad6 and Rad18 form a ubiquitin ligase complex that mono-ubiquitylates PCNA in response to UVC-induced DNA damage, and this ubiquitylation of PCNA appears to be required for pol eta-dependent bypass of CPDs (167,168). Rad18-dependent PCNA mono-ubiquitylation also may be required for switching of DNA pol kappa with replicative DNA polymerases for efficient bypass of BPDE–DNA adducts (26,169). Inactivation of ATR and Chk1 was found to attenuate mono-ubiquitylation of PCNA by ∼50% in H1299 carcinoma cells showing a possible connection between intra-S checkpoint response to polymerase blockage and ubiquitin-mediated polymerase switch for efficient bypass of BPDE–DNA adducts (26). However, ATR was not required for PCNA ubiquitylation in UVC-treated HeLa cells or SV40-transformed human fibroblasts (170,171). Chk1 protein, but not catalytic activity, was required for PCNA ubiquitylation in HeLa cells (170). This physical requirement for Chk1 protein may be related to its interaction with claspin as depletion of Chk1 caused depletion of claspin in HeLa cells (170,172). Because depletion of claspin also reduced PCNA ubiquitylation (170), it is possible that the requirement for Chk1 is to stabilize claspin. Whereas Chk1 depletion appeared to reduce PCNA ubiquitylation through its effect on claspin stability, Tim depletion also reduced PCNA ubiquitylation independent of an effect on claspin stability (170). Tim was found to physically interact with PCNA and claspin (170). Thus, it appears that claspin and Tim both must be physically present for the Rad6–Rad18 complex to mono-ubiquitylate PCNA at stalled replication forks. These studies suggest a tight association between mediators of intra-S checkpoint response to UVC-induced DNA damage and the Rad6–Rad18 pathway of polymerase switch for translesion DNA synthesis.
New developments in S phase DNA damage responses
As technology improves, new tests are applied to old problems, revealing unexpected results. We reported in 1981 that a fibroblast strain from a patient with XP belonging to complementation group A displayed an effective intra-S checkpoint response of inhibition of replicon initiation after a low fluence of UVC (173). This result was interpreted to mean that NER was not required for the inhibition of replicon initiation. However, it was reported recently that XPA was required for the recruitment of ATRIP and ATR to nuclear foci and for rapid activation of Chk1 in response to UVC-induced DNA damage (55). XPA fibroblasts displayed reduced phosphorylation of Chk1 in comparison with isogenic XPA-complemented fibroblasts 1 and 2 h after UVC but not at 4 h. Thus, XPA was not required for Chk1 activation but appeared to stimulate the rapid activation of Chk1 after UVC. As sites of NER appear to be able to activate ATR (and, therefore, phosphorylation of Chk1) (163,174,175), it is likely that XPA contributes to activation of Chk1 in interphase cells. However, other NER factors were not required for rapid Chk1 activation, suggesting that XPA was required for the intra-S checkpoint response to UVC-induced DNA damage with a role that is outside its function in NER. It was suggested that XPA may bind UVC-induced DNA damage to enhance phosphorylation of Chk1 by ATR (55). As discussed above, it appears that 6-4PPs are the UV-induced template lesion that activates Chk1 and intra-S checkpoint signaling (155). Both XPA and RPA have greater affinity for 6-4PPs than for undamaged DNA (176), so it is possible that 6-4PPs in unwound template strands at stalled DNA replication forks may be bound by XPA and RPA. Moreover, given that XPA and RPA display affinity one for the other, cooperative binding of XPA to 6-4PPs and RPA, when coupled to RPA binding to single-stranded DNA, could further enhance accumulation of XPA at sites of 6-4PPs in unwound template strands (177). XPA cells displayed less ATRIP accumulation at sites of UVC-induced DNA damage implying that XPA enhanced the interaction of ATRIP with RPA at stalled replication forks. Therefore, XPA may have a physical requirement outside of NER to enhance the assembly of the intra-S checkpoint signaling complex at sites of blockage of DNA polymerases at template 6-4PPs. Given that XPA and Tipin can compete for binding to RPA (48), it is possible that XPA association with RPA at stalled replication forks might also attenuate Chk1 activation by blocking binding of the Tim–Tipin mediator complex.
The potential role of XPA in intra-S checkpoint response has gained additional support from studies of a newly discovered checkpoint mediator. Cep164, a protein that is required for primary cilia formation (178), was recently identified as a mediator of intra-S checkpoint responses to replication stress, UVC and IR (52). SiRNA-mediated depletion of Cep164 severely attenuated UVC-induced activation of Chk1 (52), reduced the rate of repair of CPDs and sensitized cells to killing by UVC (179). Cep164 was phosphorylated by ATR in response to UVC. Cep164 was also found to interact with XPA and this interaction was stimulated by UVC irradiation (179). The Cep164-interacting domain on XPA was required for full activation of Chk1 in UVC-damaged cells (179), supporting a hypothesis that a complex of XPA and Cep164 mediates phosphorylation of Chk1 by ATR.
The combination of a 6-4PP-specific antibody and flow cytometry was used to show that ATR was required for the rapid repair of 6-4PPs during S phase (63). Repair of 6-4PPs in G1 and G2 cells did not require ATR. The slower repair of CPDs also did not require ATR. It is tempting to speculate that this role for ATR in NER of 6-4PPs in S phase might somehow be connected to the role of XPA and Cep164 as mediators of intra-S checkpoint response.
Why would cells evolve to regulate NER of 6-4PPs differently in S phase than in G1 or G2? The answer to this question may lie in the special circumstances of S with DNA replication forks coursing through the nuclear chromatin. The presence of NER factors on template strands could interfere with bypass of CPDs and 6-4PPs by translesion DNA polymerases or could cause scission of single-stranded template strands to produce DNA dsb and chromosomal aberrations. UV-induced chromosomal aberrations are S dependent, suggesting that stalled replication forks turn into DNA dsb. Un-repaired DNA dsb are seen at mitosis as chromatid breaks and mis-repaired DNA dsb are seen as chromatid exchanges. NER-defective human and hamster cells display increased frequencies of UVC-induced chromosomal aberrations, indicating that repair of UVC-induced DNA damage protects against UV clastogenesis (180,181). The assembly and activity of the XPF/ERCC1 and XPG NER endonucleases on DNA at sites of UVC-induced photoproducts are strictly regulated by protein–protein interactions and DNA structure (176,177). A study with Chinese hamster ovary cells indicated that the ratio of UVC-induced chromatid exchanges and breaks was less in ERCC1- and XPF-defective cell lines than in repair-proficient cells or other NER-defective lines (181). This result suggested that ERCC1/XPF heterodimer might contribute to UVC-induced chromatid exchanges. A role for ERCC1/XPF in recombinational repair of DNA dsb has been identified (182,183), implying that UVC-induced chromatid exchanges may arise from ERCC1/XPF-dependent recombinational repair of DNA dsb. It remains to be determined the mechanism whereby blockage of DNA polymerase at sites of template CPDs or 6-4PPs leads to DNA dsb and chromosomal aberrations.
Given the high affinity of XPA and RPA for DNA containing 6-4PPs (176), it seems reasonable that S phase cells suppress the assembly of NER complexes on this lesion when it resides within a stalled DNA replication fork or a replicative gap. The assembly of NER complexes on template strands could interfere with the polymerase-switch mechanism of postreplication repair. Accumulation of ERCC1/XPF or XPG endonucleases at such sites could lead to strand scission and formation of a DNA dsb. The severe reduction in global 6-4PP repair in ATR-depleted S phase cells implies that ATR regulates a factor that regulates NER in S phase. Cep164 has the attributes of being phosphorylated by ATR and interacting with XPA. Future studies should determine whether Cep164 influences global repair of 6-4PPs in S phase cells.
Working model of intra-S checkpoint response to UV-induced DNA damage
Based upon the above-mentioned considerations, the following working model of intra-S checkpoint response to UVC-induced DNA damage can be proposed (Figure 3 and Supplementary data are available at Carcinogenesis Online). UVC-induced DNA damage including CPDs and 6-4PPs blocks DNA synthesis by replicative DNA polymerases (alpha, delta and epsilon) when encountered in single-stranded template strands. The junction of single-stranded DNA and double-stranded DNA at the site of polymerase blockage attracts the Rad17/RFC2–5 checkpoint clamp loader, which loads the 9-1-1 complex at the blocked growing point. Uncoupling of helicase and replicative DNA polymerases generates extended regions of RPA-coated single-stranded DNA beyond the blocked growing point and this RPA-coated single-stranded DNA attracts ATR–ATRIP complexes and Tim–Tipin complexes. Phosphorylation of Rad9 by ATR attracts TopBP1 to enhance ATR kinase activity. The Tim–Tipin complex attracts Chk1 and claspin to the site of replicative arrest, presenting Chk1 to ATR for its phosphorylation and activation. Diffusion of activated Chk1 to other replication complexes transduces the signal to inhibit replicon initiation and DNA chain elongation, possibly by transient disassembly of the MCM–GINS–Cdc45 helicase complex.
Fig. 3.
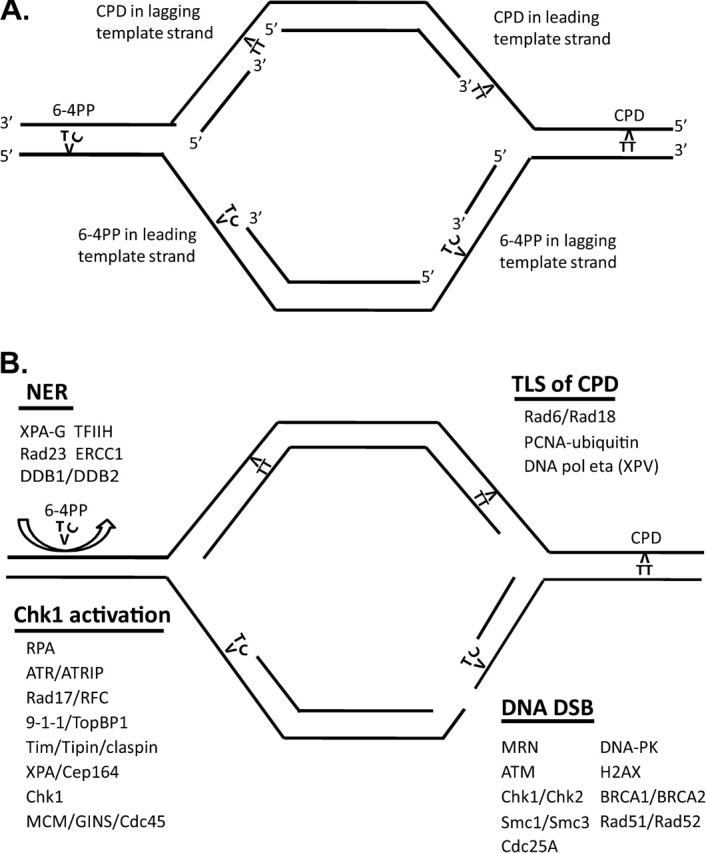
Working model of intra-S checkpoint response to UV-induced DNA damage. (A) Schematic structure of a replicon with UV-induced CPDs and 6-4PPs in template strands blocking leading-strand and lagging-strand DNA polymerases. (B) Rapid NER excision repair of 6-4PPs ahead of DNA growing points reduces the blocking of replicative polymerases and DNA growing points. Rapid TLS of TT CPDs by DNA pol eta suppresses intra-S checkpoint signaling. Sustained blockage at 6-4PPs and uncoupling of the replicative helicase and polymerases stimulate Rad17/RFC2–5-dependent loading of 9-1-1 complexes and generate RPA-coated single-stranded templates that attract ATR–ATRIP and Tim–Tipin–claspin complexes. Binding of TopBP1 to phospho-Rad9 and Chk1 to Tim and claspin completes the assembly of the RFPC to allow ATR to phosphorylate and activate Chk1. XPA and Cep164 appear to stimulate Chk1 phosphorylation. Activated Chk1 diffuses to dissociate MCM–GINS–Cdc45 complexes to inhibit replicon initiation and DNA chain elongation. Scission of the 6-4PP-containing template strand at a blocked DNA growing point produces a DNA dsb, thereby triggering the ATM-dependent checkpoint signaling pathway and recruiting DNA dsb repair pathways. See the Supplementary data at Carcinogenesis Online for an animated version of the working model.
Signals generated by ATR and Chk1 also appear to affect other elements of DNA metabolism. Polymerase switch for efficient TLS across CPDs by DNA pol eta and for less-efficient TLS across 6-4PPs may include regulation of Rad18-dependent mono-ubiquitylation of PCNA by ATR, claspin or Tim. Because TLS across TT CPDs by DNA pol eta is efficient, DNA growing points are not blocked for long at CPDs and uncoupling of helicase and polymerase activities appears to be insufficient to generate a measurable checkpoint signal. The blockage of replicative DNA polymerases at 6-4PPs appears to produce sufficient uncoupling of helicase and polymerase activities and sufficient RPA-coated DNA at the stalled replication fork to attract all the components of the intra-S checkpoint that are needed to activate Chk1. We do not yet know where to place the XPA–Cep164 complex in this model although given the affinities of XPA for 6-4PPs and RPA, it might properly be placed on the 6-4PP at the blocked growing point.
ATR, Tim and Chk1 are all required for the intra-S checkpoint response to UV. These proteins also are essential for organismal development, as nullizygous mice die early in embryogenesis. ATR-nullizygous embryos die with massive chromosomal damage (184), siRNA-mediated depletion of Tim in mouse fibroblasts induces chromosomal aberrations (137) and we have found that siRNA-mediated depletion of ATR, Tim and Chk1 in diploid human fibroblasts produced significant increases in S-dependent, chromatid-type chromosomal aberrations (S.Smith-Roe, M.Cordeiro-Stone and W.K.Kaufmann, unpublished data). Thus, it appears that the ATR–Chk1 signaling pathway that is mediated by Tim–Tipin complexes is required to prevent the formation of lethal chromosomal aberrations during DNA replication. This may reflect an essential function of the ATR-, Tim- and Chk1-dependent intra-S checkpoint to stabilize stalled DNA replication forks. The implication of this conclusion is that DNA replication forks stall naturally during the cell division cycle and the ATR–Chk1 signaling pathway evolved to stabilize these stalled replication forks.
What types of DNA or chromatin structure might stall DNA replication forks? Detailed analysis of DNA replication in eukaryotic cells has defined natural replication slow zones, pause sites and barriers (68,185). DNA–protein interactions, RNA synthesis, unusual DNA structures such as G4 quadruplexes and DNA damage all may slow or arrest DNA replication. A study in yeast cells suggested that heterochromatin may represent a natural replication barrier that requires the claspin analog, MRC1, for stable replication (186). The question now is not whether DNA replication forks stall during S thereby activating the ATR–Chk1 signaling pathway but rather how does the ATR–Chk1 signaling pathway stabilize the stalled forks?
Stalled forks may degrade to produce a single-end DNA dsb when single-stranded template strands at the fork break by spontaneous, chemically induced and/or enzymatic scission of the deoxyribose-phosphodiester backbone of DNA. UVC irradiation was shown to induce DNA dsb in human skin fibroblast lines (187), supporting the hypothesis that DNA replication forks that are stalled at 6-4PPs may collapse to form DNA dsb. One model of fork collapse has the replication fork regressing with renaturation of newly synthesized daughter strands to produce a four-stranded ‘chicken foot’-like structure that resembles the Holliday junctions that are produced during homologous recombination. This fork regression model provides one mechanism for bypass of UVC-induced 6-4PPs in template strands (188). Completion of the blocked daughter strand using the unblocked daughter strand as the template would allow bypass of the blocking lesion upon de-regression of the DNA replication fork. However, resolution of the chicken foot by Holliday junction-specific endonucleases (189), before de-regression can be accomplished, generates a DNA dsb. We do not know the mechanisms whereby stalled DNA replication forks collapse or degrade to produce DNA dsb. The ATR–Chk1 signaling pathway may be required to prevent fork collapse and/or for repair of the DNA dsb to restart DNA replication by re-establishment of the replication fork. Chk1 signaling is required for efficient recombinational repair of DNA dsb that arise at DNA replication forks (190). However, the essential function of Chk1 in maintaining cell viability in the absence of experimentally induced DNA damage could be separated from its requirements for regulation of DNA replication and it was suggested that the essential function of Chk1 is in mitotic progression (191). A resolution of the essential functions of ATR, Tim and Chk1 and their roles in stabilizing DNA replication forks that are stalled at sites of UVC-induced DNA damage awaits further experimentation.
Are there other high-dose intra-S checkpoint signals?
This discussion has focused on the intra-S checkpoint response to UVC-induced DNA damage as it occurs in human cells. It is a remarkable fact that the activity of this checkpoint reduces the rate of replicon initiation in S phase cells by 50% within 30 min after a 1 J/m2 dose of UVC that is without detectable mutagenic or cytotoxic effect. This checkpoint response has evolved to respond to very low levels of DNA damage that are below the threshold for induced mutagenesis and in the shoulder region of survival curves. It is instructive to consider the levels of DNA damage that are associated with the various cellular responses to UVC.
Many studies of UVC-induced DNA damage have quantified the yields of CPDs and 6-4PPs that are produced in human cells in culture. A consensus value of 1 CPD/J/m2/60 kb DNA has been determined for 254 nm UVC (144). Treatment with a UVC fluence of 0.8–1 J/m2 to induce ∼1 CPD per replicon was shown to produce a 50% inhibition of replicon initiation measured in the first hour after irradiation (70,114). A fluence of 2.5 J/m2 was also shown to inhibit DNA chain elongation by ∼50% in the first hour after irradiation (70). Thus, very significant effects on DNA replication can be observed after very low doses of UVC. In survival studies, it is common to see little or no effect on colony formation after UVC fluences up to 3 J/m2 (91,192–195). Similarly, analysis of UVC-induced mutations often reveals a threshold dose below which UVC does not produce an increase above the sham-treated control, and this threshold is also around 3 J/m2 (193,194). Analysis of the rates of reparative strand incision by NER in diploid human fibroblasts indicated a Km of 3 J/m2 (196), i.e. this fluence activated NER to half its maximal rate. Human skin fibroblasts have evolved to respond rapidly and effectively to UV-induced DNA damage to prevent cell death and mutations in survivors. As many as three CPDs per replicon can be tolerated without genotoxic effect in normal human fibroblasts.
The maximal velocity of reparative strand incision (Vmax) determined for diploid human fibroblasts was achieved after 7 J/m2 and with higher fluences NER is saturated (196). This means that after irradiation with fluences >7 J/m2, UVC-induced photoproducts may persist in DNA for an extended interval of time before being repaired by the limited supply of NER factors. The studies of UV-induced cytotoxicity in diploid human fibroblasts that were referenced above reported 70–90% inactivation of cell colony formation by UV fluences in the range of 8–10 J/m2. It is instructive to recognize this biological fact when examining human cell responses to still higher UVC fluences.
There is second realm of cellular response to UVC irradiation that is little expressed in diploid human fibroblasts after fluences below 10 J/m2. This realm appears to depend upon UVC photons interacting with aromatic bonds in proteins or with UVC-induced reactive oxygen species (ROS) oxidizing sulfhydryl groups in proteins (197). The result of protein modification by UV is activation of several intracellular signaling pathways. UV-induced protein modification has been shown to activate the mitogen-activated protein kinase (MAPK) signaling cascade, NF-κB and AKT, with substantial biological effects (197). Whereas studies of UVC-induced intra-S checkpoint signaling that use fluences <3 J/m2 can be expected to reveal responses to comparatively low levels of DNA damage, studies using fluences of >10 J/m2 will add UVC-induced cytoplasmic protein responses to the high levels of DNA damage.
One example of a high-dose UVC-induced checkpoint response is the G2 checkpoint response that delays progression of G2 cells into mitosis. Treatment with the 1 J/m2 fluence that inhibited replicon initiation in diploid human fibroblast by 50% had no apparent effect on the G2 to M transition, although the higher fluence of 10 J/m2 was found to reduce the mitotic index by 30–50% by inhibiting the G2 to M transition (198). This UVC-induced G2 checkpoint has been extensively investigated by Bulavin et al. (47,199) and shown to require signaling by the MAPK terminal kinase p38 to Cdc25B. Inactivation of p38 by chemical inhibition or genetic deletion restored the G2 to M progression in UVC-treated cells. Subsequent studies have shown that one substrate of p38 is MAPKAP kinase-2, a serine/threonine kinase (200). Phosphorylation by p38-activated MAPKAP kinase-2, which is capable of phosphorylation of Cdc25B and C. Thus, high fluences of UVC that activate the MAPK signaling pathway trigger a p38- and MAPKAP kinase-2-dependent G2 delay. Treatment of U2OS fibrosarcoma cells with 10 J/m2 was also shown to inhibit DNA synthesis severely 12 h after irradiation. Cells with S phase DNA content did not appear to incorporate bromo-deoxyuridine into DNA. Depletion of MAPKAP kinase-2 in U2OS cells reversed this inhibition of DNA synthesis, suggesting that MAPKAP kinase-2 might regulate an intra-S checkpoint response to UVC.
Although the low dose of 1 J/m2 that inhibited replicon initiation by 50% activated Chk1, it did not activate Chk2, induce proteolysis of Cdc25A or inhibit cyclin E/Cdk2 (114). High doses of UVC can activate Chk1 and Chk2 to phosphorylate Cdc25A, leading to proteolysis of Cdc25A and inhibition of cyclin E–Cdk2 complexes (44). As discussed previously, this is one arm of the ATM-dependent intra-S checkpoint response to IR-induced DNA dsb. It may be that activation of p38 and MAPKAP kinase-2 in lethally irradiated human cells can produce the same signals to degrade Cdc25A for generation of an intra-S checkpoint response and phosphorylate Cdc25B/C for generation of a G2 checkpoint response. It remains to be determined whether these pathways of biochemical signaling in response to UVC are expressed in the targets of UV carcinogenesis, proliferative keratinocytes and melanocytes, under conditions of human solar radiation exposure.
What about damage produced by UVB and UVA?
This discussion has focused on an intra-S checkpoint response to UVC-induced DNA damage, which our recent analyses suggest develops in response to stalling of replicative DNA polymerases at 6-4PP photoproducts in DNA template strands. UV wavelengths that induce this form of DNA damage should trigger this response. A recent study quantified the yields of CPDs and 6-4PPs in human skin fibroblasts and keratinocytes after irradiation with 312 nm UVB radiation (201). Isolated fibroblasts incurred ∼60% more photoproducts in their DNA than keratinocytes. The 6-4PPs represented 17% of photoproducts in fibroblasts and 24% in keratinocytes. Repair of the 6-4PPs was very rapid in both fibroblasts and keratinocytes, with >90% removal of 6-4PPs from keratinocyte DNA within 4 h after irradiation. We have observed 80% removal of 6-4PPs within 3 h of irradiation of human skin melanocytes and fibroblasts with 12 J/m2 UVC (W.K.Kaufmann, D.L.Mitchell and M.Cordeiro-Stone, unpublished data). In whole-skin samples incubated in culture medium, >60% of 6-4PPs that were induced by UVB irradiation were removed from DNA within 4 h after irradiation (201). These results confirm that wavelengths of solar radiation that penetrate the UVC-protective ozone layer produce the same forms of damage in DNA that are produced in the laboratory using 254 nm UVC radiation and the primary cellular targets for solar radiation carcinogenesis in human skin, keratinocytes and melanocytes express a functional NER system. The 2000-fold increase in incidence rates of melanoma, squamous cell carcinoma and basal cell carcinoma in XP patients indicates that NER and TLS of UV-induced DNA damage protect against human carcinogenesis (202).
Solar UVA also penetrates the ozone layer and human skin to induce DNA damage. Remarkably, although CPDs are induced by UVA, they appear to arise by a different mechanism than those induced by UVB, but probably involving ROS (203). Moreover, while irradiation of skin keratinocytes with UVA inhibited DNA replication in association with activation of ATM, ATR and p38, phosphorylation of Chk1 and Chk2 and degradation of Cdc25A, the inhibition of DNA replication induced by UVA could not be reversed by inhibition of the ATM, ATR or p38 signaling pathways (204). This result suggested that the inhibition of DNA replication by UVA in cultured keratinocytes was mediated by ROS-induced damage to protein, not DNA. The challenge for scientists who wish to understand the mechanisms of solar radiation carcinogenesis in humans now is to determine the interplay between cellular responses to ROS-induced protein and DNA damage and UV photon-induced DNA damage.
Summary
Solar radiation is ubiquitous to terrestrial existence and humans have evolved within an environment with alternating high and low solar radiation exposures. The circadian clock cycle that is entrained by day/night cycles is one example of cellular adaptation to this environment. Human fibroblasts appear to have evolved a mechanism for response to low levels of UVC-induced DNA damage in S phase cells that differs from the response to IR-induced DNA dsb. The UVC-induced intra-S checkpoint requires signaling from ATR to Chk1 with mediation by claspin, Tim, Tipin, XPA and Cep164. The target of this signaling network is unknown but the MCM–GINS–Cdc45 helicase complex is an attractive possibility as it can influence both the rate of initiation of DNA synthesis at replicon origins as well as the rate of DNA chain elongation in active replicons. By slowing DNA synthesis, the intra-S checkpoint provides more time for repair of DNA damage before DNA replication. The intra-S checkpoint may also stabilize replication forks that are stalled at sites of UVC-induced DNA damage. The fact that knockout of ATR, Tim and Chk1 is embryo lethal in mice suggests that components of the intra-S checkpoint response to UVC may also have an essential function to stabilize replication forks that are stalled at natural replication barriers.
Supplementary material
Supplementary material can be found at http://carcin.oxfordjournals.org/
Funding
US PHS (ES10126, ES015856, ES014635).
Acknowledgments
The author is indebted to the students, postdoctoral trainees, research assistants and investigators in the University of North Carolina Program in Environmental Melanoma for stimulating discussions of human solar radiation carcinogenesis.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- AT
ataxia telangiectasia
- ATRIP
ATR-interacting protein
- BPDE
benzo[a]pyrene diolepoxide I
- CldU
chloro-deoxyuridine
- CPD
cyclobutane-type pyrimidine dimers
- IR
ionizing radiation
- MAPK
mitogen-activated protein kinase
- NER
nucleotide excision repair
- 6-4PP
[6-4]pyrimidine-pyrimidone
- 9-1-1
Rad9-Rad1-Hus1
- RFPC
replication fork protection complex
- ROS
reactive oxygen species
- TLS
translesion synthesis
- XP
xeroderma pigmentosum
References
- 1.Weinert T, et al. Control of G2 delay by the rad9 gene of Saccharomyces cerevisiae. J. Cell Sci. 1989;12(suppl):145–148. doi: 10.1242/jcs.1989.supplement_12.12. [DOI] [PubMed] [Google Scholar]
- 2.Hartwell LH, et al. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 3.Painter RB, et al. Effect of irradiation and theory of role of mitotic delay on the time course of labeling of HeLa S3 cells with tritiated thymidine. Radiat. Res. 1959;11:206–217. [PubMed] [Google Scholar]
- 4.Scaife JF, et al. An investigation of factors influencing mitotic G2 delay in synchronous cultures of human kidney cells after x-irradiation. Can. J. Biochem. 1969;47:237–249. doi: 10.1139/o69-038. [DOI] [PubMed] [Google Scholar]
- 5.Murnane JP, et al. Effects of methylated xanthines on mammalian cells treated with bifunctional alkylating agents. Nature. 1980;285:326–329. doi: 10.1038/285326a0. [DOI] [PubMed] [Google Scholar]
- 6.Painter RB, et al. Radiosensitivity in ataxia-telangiectasia: a new explanation. Proc. Natl Acad. Sci. USA. 1980;77:7315–7317. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houldsworth J, et al. Effect of ionizing radiation on DNA synthesis in ataxia telangiectasia cells. Nucleic Acids Res. 1980;8:3709–3720. doi: 10.1093/nar/8.16.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edwards MJ, et al. Unusual levels of (ADP-ribose)n and DNA synthesis in ataxia telangiectasia cells following gamma-ray irradiation. Nature. 1980;287:745–747. doi: 10.1038/287745a0. [DOI] [PubMed] [Google Scholar]
- 9.Zampetti-Bosseler F, et al. Cell death, chromosome damage and mitotic delay in normal human, ataxia telangiectasia and retinoblastoma fibroblasts after x-irradiation. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1981;39:547–558. doi: 10.1080/09553008114550651. [DOI] [PubMed] [Google Scholar]
- 10.Savitsky K, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 11.Wang B, et al. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;3:3. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 12.Matsuoka S, et al. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 13.Syljuasen RG, et al. Adaptation to the ionizing radiation-induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer Res. 2006;66:10253–10257. doi: 10.1158/0008-5472.CAN-06-2144. [DOI] [PubMed] [Google Scholar]
- 14.Chini CC, et al. Human claspin is required for replication checkpoint control. J. Biol. Chem. 2003;278:30057–30062. doi: 10.1074/jbc.M301136200. [DOI] [PubMed] [Google Scholar]
- 15.Dozier C, et al. Regulation of Chk2 phosphorylation by interaction with protein phosphatase 2A via its B’ regulatory subunit. Biol. Cell. 2004;96:509–517. doi: 10.1016/j.biolcel.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Wang B, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arlander SJ, et al. DNA protein kinase-dependent G2 checkpoint revealed following knockdown of ataxia-telangiectasia mutated in human mammary epithelial cells. Cancer Res. 2008;68:89–97. doi: 10.1158/0008-5472.CAN-07-0675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu X, et al. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–1174. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhoumik A, et al. ATM-dependent phosphorylation of ATF2 is required for the DNA damage response. Mol. Cell. 2005;18:577–587. doi: 10.1016/j.molcel.2005.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collis SJ, et al. FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol. Cell. 2008;32:313–324. doi: 10.1016/j.molcel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 21.Bao S, et al. Disruption of the Rad9/Rad1/Hus1 (9-1-1) complex leads to checkpoint signaling and replication defects. Oncogene. 2004;23:5586–5593. doi: 10.1038/sj.onc.1207753. [DOI] [PubMed] [Google Scholar]
- 22.Cliby WA, et al. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bao S, et al. ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature. 2001;411:969–974. doi: 10.1038/35082110. [DOI] [PubMed] [Google Scholar]
- 24.Zou L, et al. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 25.Kang J, et al. Functional interaction of H2AX, NBS1, and p53 in ATM-dependent DNA damage responses and tumor suppression. Mol. Cell. Biol. 2005;25:661–670. doi: 10.1128/MCB.25.2.661-670.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bi X, et al. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol. Cell. Biol. 2006;26:3527–3540. doi: 10.1128/MCB.26.9.3527-3540.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu X, et al. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell. Biol. 2004;24:9478–9486. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collis SJ, et al. HCLK2 is essential for the mammalian S-phase checkpoint and impacts on Chk1 stability. Nat. Cell Biol. 2007;9:391–401. doi: 10.1038/ncb1555. [DOI] [PubMed] [Google Scholar]
- 29.Carney JP, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 30.Fabbro M, et al. BRCA1-BARD1 complexes are required for p53Ser-15 phosphorylation and a G1/S arrest following ionizing radiation-induced DNA damage. J. Biol. Chem. 2004;279:31251–31258. doi: 10.1074/jbc.M405372200. [DOI] [PubMed] [Google Scholar]
- 31.Kao GD, et al. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J. Cell Biol. 2003;160:1017–1027. doi: 10.1083/jcb.200209065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delacroix S, et al. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cortez D, et al. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. [In Process Citation] Science. 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 34.Wang B, et al. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl Acad. Sci. USA. 2007;104:20759–20763. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu X, et al. Microcephalin is a DNA damage response protein involved in regulation of CHK1 and BRCA1. J. Biol. Chem. 2004;279:34091–34094. doi: 10.1074/jbc.C400139200. [DOI] [PubMed] [Google Scholar]
- 36.Weiss RS, et al. Critical role for mouse Hus1 in an S-phase DNA damage cell cycle checkpoint. Mol. Cell. Biol. 2003;23:791–803. doi: 10.1128/MCB.23.3.791-803.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slebos RJ, et al. p53-dependent G1 arrest involves pRB-related proteins and is disrupted by the human papillomavirus 16 E7 oncoprotein. Proc. Natl Acad. Sci. USA. 1994;91:5320–5324. doi: 10.1073/pnas.91.12.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eguchi T, et al. RB silencing compromises the DNA damage-induced G2/M checkpoint and causes deregulated expression of the ECT2 oncogene. Oncogene. 2007;26:509–520. doi: 10.1038/sj.onc.1209810. [DOI] [PubMed] [Google Scholar]
- 39.Cahill DP, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 40.Luo X, et al. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol. Cell. 2002;9:59–71. doi: 10.1016/s1097-2765(01)00435-x. [DOI] [PubMed] [Google Scholar]
- 41.Yazdi PT, et al. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002;16:571–582. doi: 10.1101/gad.970702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan GK, et al. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J. Cell Biol. 1999;146:941–954. doi: 10.1083/jcb.146.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo H, et al. Regulation of intra-S phase checkpoint by ionizing radiation (IR)-dependent and IR-independent phosphorylation of SMC3. J. Biol. Chem. 2008;283:19176–19183. doi: 10.1074/jbc.M802299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falck J, et al. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 45.Goldberg M, et al. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- 46.Unsal-Kacmaz K, et al. Coupling of human circadian and cell cycles by the timeless protein. Mol. Cell. Biol. 2005;25:3109–3116. doi: 10.1128/MCB.25.8.3109-3116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bulavin DV, et al. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature. 2001;411:102–107. doi: 10.1038/35075107. [DOI] [PubMed] [Google Scholar]
- 48.Unsal-Kacmaz K, et al. The human Tim/Tipin complex coordinates an intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell. Biol. 2007;27:3131–3142. doi: 10.1128/MCB.02190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peng CY, et al. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. [see comments] Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, et al. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer Biol. Ther. 2004;3:305–313. doi: 10.4161/cbt.3.3.697. [DOI] [PubMed] [Google Scholar]
- 51.Kim JE, et al. Human TopBP1 ensures genome integrity during normal S phase. Mol. Cell. Biol. 2005;25:10907–10915. doi: 10.1128/MCB.25.24.10907-10915.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sivasubramaniam S, et al. Cep164 is a mediator protein required for the maintenance of genomic stability through modulation of MDC1, RPA, and CHK1. Genes Dev. 2008;22:587–600. doi: 10.1101/gad.1627708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Q, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 54.Kastan MB, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 55.Bomgarden RD, et al. Opposing effects of the UV lesion repair protein XPA and UV bypass polymerase eta on ATR checkpoint signaling. EMBO J. 2006;25:2605–2614. doi: 10.1038/sj.emboj.7601123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 57.Taylor AM, et al. Is chromatid-type damage in ataxia telangiectasia after irradiation at G0 a consequence of defective repair? Nature. 1976;260:441–443. doi: 10.1038/260441a0. [DOI] [PubMed] [Google Scholar]
- 58.Taylor AM, et al. Ataxia telangiectasia: a human mutation with abnormal radiation sensitivity. Nature. 1975;258:427–429. doi: 10.1038/258427a0. [DOI] [PubMed] [Google Scholar]
- 59.Ejima Y, et al. Enhanced expression of X-ray- and UV-induced chromosome aberrations by cytosine arabinoside in ataxia telangiectasia cells. Mutat. Res. 1986;159:117–123. doi: 10.1016/0027-5107(86)90120-x. [DOI] [PubMed] [Google Scholar]
- 60.Kaufmann WK, et al. G1 arrest and cell-cycle-dependent clastogenesis in UV-irradiated human fibroblasts. Mutat. Res. 1994;314:67–76. doi: 10.1016/0921-8777(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 61.Swift M, et al. Incidence of cancer in 161 families affected by ataxia-telangiectasia. [see comments] N. Engl. J. Med. 1991;325:1831–1836. doi: 10.1056/NEJM199112263252602. [DOI] [PubMed] [Google Scholar]
- 62.Harper JW, et al. The DNA damage response: ten years after. Mol. Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 63.Auclair Y, et al. ATR kinase is required for global genomic nucleotide excision repair exclusively during S phase in human cells. Proc. Natl Acad. Sci. USA. 2008;105:17896–17901. doi: 10.1073/pnas.0801585105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goodarzi AA, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 65.Kastan MB, et al. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 66.Sancar A, et al. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 67.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 68.Lambert S, et al. Checkpoint responses to replication fork barriers. Biochimie. 2005;87:591–602. doi: 10.1016/j.biochi.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 69.Kaufmann WK, et al. Cell cycle control, DNA repair and initiation of carcinogenesis. FASEB J. 1993;7:1188–1191. doi: 10.1096/fasebj.7.12.8375618. [DOI] [PubMed] [Google Scholar]
- 70.Kaufmann WK, et al. Reversible inhibition of rat hepatocyte proliferation by hydrocortisone and its effect on cell cycle-dependent hepatocarcinogenesis by N-methyl-N-nitrosourea. Cancer Res. 1981;41:4653–4660. [PubMed] [Google Scholar]
- 71.Kaufmann WK, et al. Cell cycle-dependent initiation of hepatocarcinogenesis in rats by methyl(acetoxymethyl)nitrosamine. Cancer Res. 1987;47:1263–1266. [PubMed] [Google Scholar]
- 72.Kaufmann WK, et al. Cell cycle-dependent initiation of hepatocarcinogenesis in rats by (+/-)-7r,8t-dihydroxy-9t,10t-epoxy-7,8,9,10-tetrahydrobenzo(a)pyrene. Cancer Res. 1987;47:3771–3775. [PubMed] [Google Scholar]
- 73.Kaufmann WK. Cell cycle checkpoints and DNA repair preserve the stability of the human genome. Cancer Metastasis Rev. 1995;14:31–41. doi: 10.1007/BF00690209. [DOI] [PubMed] [Google Scholar]
- 74.Watanabe M, et al. Excision repair of UV- or benzo[a]pyrene diol epoxide-induced lesions in xeroderma pigmentosum variant cells is ‘error free’. Mutat. Res. 1985;146:285–294. doi: 10.1016/0167-8817(85)90070-7. [DOI] [PubMed] [Google Scholar]
- 75.Maher VM, et al. Frequency of UV-induced neoplastic transformation of diploid human fibroblasts is higher in xeroderma pigmentosum cells than in normal cells. Proc. Natl Acad. Sci. USA. 1982;79:2613–2617. doi: 10.1073/pnas.79.8.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rude JM, et al. Semi-conservative deoxyribonucleic acid synthesis in unirradiated and ultraviolet-irradiated xeroderma pigmentosum and normal human skin fibroblasts. Mutat. Res. 1977;42:433–442. doi: 10.1016/s0027-5107(77)80047-x. [DOI] [PubMed] [Google Scholar]
- 77.Edenberg HJ. Inhibition of DNA replication by ultraviolet light. Biophys. J. 1976;16:849–860. doi: 10.1016/S0006-3495(76)85735-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaufmann WK, et al. Mechanisms of inhibition of DNA replication by ultraviolet light in normal human and xeroderma pigmentosum fibroblasts. J. Mol. Biol. 1981;149:171–187. doi: 10.1016/0022-2836(81)90297-7. [DOI] [PubMed] [Google Scholar]
- 79.Kaufmann WK, et al. Ultraviolet radiation inhibits replicon initiation in S phase human cells. Biochim. Biophys. Acta. 1980;608:191–195. doi: 10.1016/0005-2787(80)90147-1. [DOI] [PubMed] [Google Scholar]
- 80.Kaufmann WK, et al. DNA repair and replication in human fibroblasts treated with (+/-)-r-7,t-8-dihydroxy-t-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene. Biochim. Biophys. Acta. 1985;824:146–151. doi: 10.1016/0167-4781(85)90091-0. [DOI] [PubMed] [Google Scholar]
- 81.Guo N, et al. Carcinogen-induced S-phase arrest is Chk1 mediated and caffeine sensitive. Cell Growth Differ. 2002;13:77–86. [PubMed] [Google Scholar]
- 82.Painter RB, et al. X-ray-induced inhibition of DNA synthesis in Chinese hamster ovary, human HeLa, and Mouse L cells. Radiat. Res. 1975;64:648–656. [PubMed] [Google Scholar]
- 83.Park SD, et al. Recovery of DNA synthesis after ultraviolet irradiation of xeroderma pigmentosum cells depends on excision repair and is blocked by caffeine. Nucleic Acids Res. 1979;6:1151–1159. doi: 10.1093/nar/6.3.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moore PD, et al. Sites of termination of in vitro DNA synthesis on ultraviolet- and N-acetylaminofluorene-treated phi X174 templates by prokaryotic and eukaryotic DNA polymerases. Proc. Natl Acad. Sci. USA. 1981;78:110–114. doi: 10.1073/pnas.78.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lehman AR, et al. Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV-irradiation. Proc. Natl Acad. Sci. USA. 1975;72:219–223. doi: 10.1073/pnas.72.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Johnson RE, et al. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 87.Masutani C, et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 88.McCulloch SD, et al. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bienko M, et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–1824. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- 90.Lehmann AR. Replication of damaged DNA by translesion synthesis in human cells. FEBS Lett. 2005;579:873–876. doi: 10.1016/j.febslet.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 91.Bassett E, et al. The role of DNA polymerase eta in translesion synthesis past platinum-DNA adducts in human fibroblasts. Cancer Res. 2004;64:6469–6475. doi: 10.1158/0008-5472.CAN-04-1328. [DOI] [PubMed] [Google Scholar]
- 92.McGregor WG, et al. Abnormal, error-prone bypass of photoproducts by xeroderma pigmentosum variant cell extracts results in extreme strand bias for the kinds of mutations induced by UV light. Mol. Cell. Biol. 1999;19:147–154. doi: 10.1128/mcb.19.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maher VM, et al. Frequency of ultraviolet light-induced mutations is higher in xeroderma pigmentosum variant cells than in normal human cells. Nature. 1976;261:593–595. doi: 10.1038/261593a0. [DOI] [PubMed] [Google Scholar]
- 94.Wang Y, et al. Evidence that in xeroderma pigmentosum variant cells, which lack DNA polymerase eta, DNA polymerase iota causes the very high frequency and unique spectrum of UV-induced mutations. Cancer Res. 2007;67:3018–3026. doi: 10.1158/0008-5472.CAN-06-3073. [DOI] [PubMed] [Google Scholar]
- 95.Painter RB. Inhibition and recovery of DNA synthesis in human cells after exposure to ultraviolet light. Mutat. Res. 1985;145:63–69. doi: 10.1016/0167-8817(85)90041-0. [DOI] [PubMed] [Google Scholar]
- 96.Canman CE, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 97.Bullock SK, et al. Enhanced S phase delay and inhibition of replication of an undamaged shuttle vector in UVC-irradiated xeroderma pigmentosum variant. Carcinogenesis. 2001;22:233–241. doi: 10.1093/carcin/22.2.233. [DOI] [PubMed] [Google Scholar]
- 98.Meyerson M, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 99.Nakamura TM, et al. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 100.Counter CM, et al. Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene. 1998;16:1217–1222. doi: 10.1038/sj.onc.1201882. [DOI] [PubMed] [Google Scholar]
- 101.Heffernan TP, et al. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol. Cell. Biol. 2002;22:8552–8561. doi: 10.1128/MCB.22.24.8552-8561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miao H, et al. Regulation of cellular and SV40 virus origins of replication by Chk1-dependent intrinsic and UVC radiation-induced checkpoints. J. Biol. Chem. 2003;278:4295–4304. doi: 10.1074/jbc.M204264200. [DOI] [PubMed] [Google Scholar]
- 103.Chastain PD, 2nd, et al. Checkpoint regulation of replication dynamics in UV-irradiated human cells. Cell Cycle. 2006;5:2160–2167. doi: 10.4161/cc.5.18.3236. [DOI] [PubMed] [Google Scholar]
- 104.Falck J, et al. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat. Genet. 2002;30:290–294. doi: 10.1038/ng845. [DOI] [PubMed] [Google Scholar]
- 105.Sorensen CS, et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3:247–258. doi: 10.1016/s1535-6108(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 106.Kim ST, et al. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xie G, et al. Requirements for p53 and the ATM gene product in the regulation of G1/S and S phase checkpoints. Oncogene. 1998;16:721–736. doi: 10.1038/sj.onc.1201793. [DOI] [PubMed] [Google Scholar]
- 108.Krude T. Initiation of human DNA replication in vitro using nuclei from cells arrested at an initiation-competent state. J. Biol. Chem. 2000;275:13699–13707. doi: 10.1074/jbc.275.18.13699. [DOI] [PubMed] [Google Scholar]
- 109.Dulic V, et al. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 110.Chen S, et al. Suppression of WEE1 and stimulation of CDC25A correlates with endothelin-dependent proliferation of rat aortic smooth muscle cells. J. Biol. Chem. 2004;279:13755–13763. doi: 10.1074/jbc.M310064200. [DOI] [PubMed] [Google Scholar]
- 111.Onn I, et al. Sister chromatid cohesion: a simple concept with a complex reality. Annu. Rev. Cell Dev. Biol. 2008;24:105–129. doi: 10.1146/annurev.cellbio.24.110707.175350. [DOI] [PubMed] [Google Scholar]
- 112.Unal E, et al. DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7) Science. 2007;317:245–248. doi: 10.1126/science.1140637. [DOI] [PubMed] [Google Scholar]
- 113.Strom L, et al. Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science. 2007;317:242–245. doi: 10.1126/science.1140649. [DOI] [PubMed] [Google Scholar]
- 114.Heffernan TP, et al. Cdc7-Dbf4 and the human S checkpoint response to UVC. J. Biol. Chem. 2007;282:9458–9468. doi: 10.1074/jbc.M611292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hengstschlager M, et al. Cyclin-dependent kinases at the G1-S transition of the mammalian cell cycle. Mutat. Res. 1999;436:1–9. doi: 10.1016/s1383-5742(98)00022-2. [DOI] [PubMed] [Google Scholar]
- 116.Sheu YJ, et al. Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell. 2006;24:101–113. doi: 10.1016/j.molcel.2006.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tsuji T, et al. Essential role of phosphorylation of MCM2 by Cdc7/Dbf4 in the initiation of DNA replication in mammalian cells. Mol. Biol. Cell. 2006;17:4459–4472. doi: 10.1091/mbc.E06-03-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Costanzo V, et al. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol. Cell. 2003;11:203–213. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 119.Dierov J, et al. BCR/ABL translocates to the nucleus and disrupts an ATR-dependent intra-S phase checkpoint. Cancer Cell. 2004;5:275–285. doi: 10.1016/s1535-6108(04)00056-x. [DOI] [PubMed] [Google Scholar]
- 120.Tenca P, et al. Cdc7 is an active kinase in human cancer cells undergoing replication stress. J. Biol. Chem. 2007;282:208–215. doi: 10.1074/jbc.M604457200. [DOI] [PubMed] [Google Scholar]
- 121.Liu P, et al. The Chk1-mediated S-phase checkpoint targets initiation factor Cdc45 via a Cdc25A/Cdk2-independent mechanism. J. Biol. Chem. 2006;281:30631–30644. doi: 10.1074/jbc.M602982200. [DOI] [PubMed] [Google Scholar]
- 122.Moyer SE, et al. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl Acad. Sci. USA. 2006;103:10236–10241. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Merrick CJ, et al. Visualization of altered replication dynamics after DNA damage in human cells. J. Biol. Chem. 2004;279:20067–20075. doi: 10.1074/jbc.M400022200. [DOI] [PubMed] [Google Scholar]
- 124.Painter RB. Inhibition of initiation of HeLa cell replicons by methyl methanesulfonate. Mutat. Res. 1977;42:299–303. doi: 10.1016/s0027-5107(77)80031-6. [DOI] [PubMed] [Google Scholar]
- 125.Noguchi E, et al. Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol. Cell. Biol. 2004;24:8342–8355. doi: 10.1128/MCB.24.19.8342-8355.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tercero JA, et al. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–557. doi: 10.1038/35087607. [DOI] [PubMed] [Google Scholar]
- 127.Gotter AL. Tipin, a novel timeless-interacting protein, is developmentally co-expressed with timeless and disrupts its self-association. J. Mol. Biol. 2003;331:167–176. doi: 10.1016/s0022-2836(03)00633-8. [DOI] [PubMed] [Google Scholar]
- 128.Gotter AL, et al. Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork-associated factors. J. Mol. Biol. 2007;366:36–52. doi: 10.1016/j.jmb.2006.10.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yoshizawa-Sugata N, et al. Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J. Biol. Chem. 2007;282:2729–2740. doi: 10.1074/jbc.M605596200. [DOI] [PubMed] [Google Scholar]
- 130.Chou DM, et al. Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc. Natl Acad. Sci. USA. 2006;103:18143–18147. doi: 10.1073/pnas.0609251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Errico A, et al. Tipin is required for stalled replication forks to resume DNA replication after removal of aphidicolin in Xenopus egg extracts. Proc. Natl Acad. Sci. USA. 2007;104:14929–14934. doi: 10.1073/pnas.0706347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kumagai A, et al. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell. 2000;6:839–849. doi: 10.1016/s1097-2765(05)00092-4. [DOI] [PubMed] [Google Scholar]
- 133.Lin SY, et al. Human Claspin works with BRCA1 to both positively and negatively regulate cell proliferation. Proc. Natl Acad. Sci. USA. 2004;101:6484–6489. doi: 10.1073/pnas.0401847101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Myers MP, et al. Positional cloning and sequence analysis of the Drosophila clock gene, timeless. Science. 1995;270:805–808. doi: 10.1126/science.270.5237.805. [DOI] [PubMed] [Google Scholar]
- 135.Sehgal A, et al. Loss of circadian behavioral rhythms and per RNA oscillations in the Drosophila mutant timeless. Science. 1994;263:1603–1606. doi: 10.1126/science.8128246. [DOI] [PubMed] [Google Scholar]
- 136.Gotter AL, et al. A time-less function for mouse timeless. Nat. Neurosci. 2000;3:755–756. doi: 10.1038/77653. [DOI] [PubMed] [Google Scholar]
- 137.Urtishak KA, et al. Timeless maintains genomic stability and suppresses sister chromatid exchange during unperturbed DNA replication. J. Biol. Chem. 2009;284:8777–8785. doi: 10.1074/jbc.M806103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chan RC, et al. Chromosome cohesion is regulated by a clock gene paralogue TIM-1. Nature. 2003;423:1002–1009. doi: 10.1038/nature01697. [DOI] [PubMed] [Google Scholar]
- 139.Bekker-Jensen S, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Petermann E, et al. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol. Cell. Biol. 2006;26:3319–3326. doi: 10.1128/MCB.26.8.3319-3326.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Petermann E, et al. Claspin promotes normal replication fork rates in human cells. Mol. Biol. Cell. 2008;19:2373–2378. doi: 10.1091/mbc.E07-10-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang X, et al. Involvement of Hus1 in the chain elongation step of DNA replication after exposure to camptothecin or ionizing radiation. Nucleic Acids Res. 2004;32:767–775. doi: 10.1093/nar/gkh243. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 143.Seiler JA, et al. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol. Cell. Biol. 2007;27:5806–5818. doi: 10.1128/MCB.02278-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Boyer JC, et al. Defective postreplication repair in xeroderma pigmentosum variant fibroblasts. Cancer Res. 1990;50:2593–2598. [PubMed] [Google Scholar]
- 145.Unsal-Kacmaz K, et al. Preferential binding of ATR protein to UV-damaged DNA. Proc. Natl Acad. Sci. USA. 2002;99:6673–6678. doi: 10.1073/pnas.102167799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Byun TS, et al. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lupardus PJ, et al. A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev. 2002;16:2327–2332. doi: 10.1101/gad.1013502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cortez D, et al. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 149.Cimprich KA, et al. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kumagai A, et al. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 151.Choi JH, et al. The human ATR-mediated DNA damage checkpoint in a reconstituted system. Methods. 2009;48:3–7. doi: 10.1016/j.ymeth.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 152.Cordeiro-Stone M, et al. Structure of the replication fork in ultraviolet light-irradiated human cells. Biophys. J. 1979;27:287–300. doi: 10.1016/S0006-3495(79)85218-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cordeiro-Stone M, et al. Analysis of DNA replication forks encountering a pyrimidine dimer in the template to the leading strand. J. Mol. Biol. 1999;289:1207–1218. doi: 10.1006/jmbi.1999.2847. [DOI] [PubMed] [Google Scholar]
- 154.Kaufmann WK. Pathways of human cell post-replication repair. Carcinogenesis. 1989;10:1–11. doi: 10.1093/carcin/10.1.1. [DOI] [PubMed] [Google Scholar]
- 155.Chen B, et al. Human papilloma virus type16 E6 deregulates CHK1 and sensitizes human fibroblasts to environmental carcinogens independently of its effect on p53. Cell Cycle. 2009;8:1775–1787. doi: 10.4161/cc.8.11.8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Li Z, et al. hREV3 is essential for error-prone translesion synthesis past UV or benzo[a]pyrene diol epoxide-induced DNA lesions in human fibroblasts. Mutat. Res. 2002;510:71–80. doi: 10.1016/s0027-5107(02)00253-1. [DOI] [PubMed] [Google Scholar]
- 157.Gibbs PE, et al. The relative roles in vivo of Saccharomyces cerevisiae Pol eta, Pol zeta, Rev1 protein and Pol32 in the bypass and mutation induction of an abasic site, T-T (6-4) photoadduct and T-T cis-syn cyclobutane dimer. Genetics. 2005;169:575–582. doi: 10.1534/genetics.104.034611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gibbs PE, et al. The function of the human homolog of Saccharomyces cerevisiae REV1 is required for mutagenesis induced by UV light. Proc. Natl Acad. Sci. USA. 2000;97:4186–4191. doi: 10.1073/pnas.97.8.4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yoo HY, et al. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell. 2004;117:575–588. doi: 10.1016/s0092-8674(04)00417-9. [DOI] [PubMed] [Google Scholar]
- 160.Lloyd DR, et al. p53-dependent global genomic repair of benzo[a]pyrene-7,8-diol-9,10-epoxide adducts in human cells. Cancer Res. 2000;60:517–521. [PubMed] [Google Scholar]
- 161.Garinis GA, et al. Transcriptome analysis reveals cyclobutane pyrimidine dimers as a major source of UV-induced DNA breaks. EMBO J. 2005;24:3952–3962. doi: 10.1038/sj.emboj.7600849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rogakou EP, et al. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 163.Marti TM, et al. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl Acad. Sci. USA. 2006;103:9891–9896. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ward IM, et al. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 165.Ward IM, et al. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J. Biol. Chem. 2004;279:9677–9680. doi: 10.1074/jbc.C300554200. [DOI] [PubMed] [Google Scholar]
- 166.Rathmell WK, et al. DNA-dependent protein kinase is not required for accumulation of p53 or cell cycle arrest after DNA damage. Cancer Res. 1997;57:68–74. [PubMed] [Google Scholar]
- 167.Watanabe K, et al. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004;23:3886–3896. doi: 10.1038/sj.emboj.7600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kannouche PL, et al. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- 169.Bi X, et al. DNA polymerase kappa is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J. Biol. Chem. 2005;280:22343–22355. doi: 10.1074/jbc.M501562200. [DOI] [PubMed] [Google Scholar]
- 170.Yang XH, et al. Chk1 and Claspin potentiate PCNA ubiquitination. Genes Dev. 2008;22:1147–1152. doi: 10.1101/gad.1632808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Niimi A, et al. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc. Natl Acad. Sci. USA. 2008;105:16125–16130. doi: 10.1073/pnas.0802727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chini CC, et al. Chk1 is required to maintain claspin stability. Oncogene. 2006;25:4165–4171. doi: 10.1038/sj.onc.1209447. [DOI] [PubMed] [Google Scholar]
- 173.Kaufmann WK, et al. Inhibition of replicon initiation by 12-O-tetradecanoylphorbol-13-acetate. Biochem. Biophys. Res. Commun. 1981;103:82–89. doi: 10.1016/0006-291x(81)91663-6. [DOI] [PubMed] [Google Scholar]
- 174.Stiff T, et al. Replication independent ATR signalling leads to G2/M arrest requiring Nbs1, 53BP1 and MDC1. Hum. Mol. Genet. 2008;17:3247–3253. doi: 10.1093/hmg/ddn220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jiang G, et al. Recruitment of DNA damage checkpoint proteins to damage in transcribed and nontranscribed sequences. Mol. Cell. Biol. 2006;26:39–49. doi: 10.1128/MCB.26.1.39-49.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Reardon JT, et al. Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 2003;17:2539–2551. doi: 10.1101/gad.1131003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Reardon JT, et al. Thermodynamic cooperativity and kinetic proofreading in DNA damage recognition and repair. Cell Cycle. 2004;3:141–144. [PubMed] [Google Scholar]
- 178.Graser S, et al. Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 2007;179:321–330. doi: 10.1083/jcb.200707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pan YR, et al. UV-dependent interaction between Cep164 and XPA mediates localization of Cep164 at sites of DNA damage and UV sensitivity. Cell Cycle. 2009;8:655–664. doi: 10.4161/cc.8.4.7844. [DOI] [PubMed] [Google Scholar]
- 180.Seguin LR, et al. Ultraviolet light-induced chromosomal aberrations in cultured cells from Cockayne syndrome and complementation group C xeroderma pigmentosum patients: lack of correlation with cancer susceptibility. Am. J. Hum. Genet. 1988;42:468–475. [PMC free article] [PubMed] [Google Scholar]
- 181.Chipchase MD, et al. The formation of UV-induced chromosome aberrations involves ERCC1 and XPF but not other nucleotide excision repair genes. DNA Repair (Amst) 2002;1:335–340. doi: 10.1016/s1568-7864(02)00010-1. [DOI] [PubMed] [Google Scholar]
- 182.Ahmad A, et al. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell. Biol. 2008;28:5082–5092. doi: 10.1128/MCB.00293-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Al-Minawi AZ, et al. The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells. Nucleic Acids Res. 2008;36:1–9. doi: 10.1093/nar/gkm888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Brown EJ, et al. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 185.Mirkin EV, et al. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007;71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hu F, et al. Asf1 links Rad53 to control of chromatin assembly. Genes Dev. 2001;15:1061–1066. doi: 10.1101/gad.873201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wang TC, et al. Postreplication repair in ultraviolet-irradiated human fibroblasts: formation and repair of DNA double-strand breaks. Carcinogenesis. 1986;7:389–392. doi: 10.1093/carcin/7.3.389. [DOI] [PubMed] [Google Scholar]
- 188.Higgins NP, et al. A model for replication repair in mammalian cells. J. Mol. Biol. 1976;101:417–425. doi: 10.1016/0022-2836(76)90156-x. [DOI] [PubMed] [Google Scholar]
- 189.West SC. The search for a human Holliday junction resolvase. Biochem. Soc. Trans. 2009;37:519–526. doi: 10.1042/BST0370519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sorensen CS, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 191.Wilsker D, et al. Essential function of Chk1 can be uncoupled from DNA damage checkpoint and replication control. Proc. Natl Acad. Sci. USA. 2008;105:20752–20757. doi: 10.1073/pnas.0806917106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lehmann AR, et al. Repair of ultraviolet light damage in a variety of human fibroblast cell strains. Cancer Res. 1977;37:904–910. [PubMed] [Google Scholar]
- 193.McCormick JJ, et al. Abnormal sensitivity of human fibroblasts from xeroderma pigmentosum variants to transformation to anchorage independence by ultraviolet radiation. Cancer Res. 1986;46:489–492. [PubMed] [Google Scholar]
- 194.Myhr BC, et al. Ultraviolet mutagenesis of normal and xeroderma pigmentosum variant human fibroblasts. Mutat. Res. 1979;62:341–353. doi: 10.1016/0027-5107(79)90089-7. [DOI] [PubMed] [Google Scholar]
- 195.Cistulli CA, et al. p53-dependent signaling sustains DNA replication and enhances clonogenic survival in 254 nm ultraviolet-irradiated human fibroblasts. Cancer Res. 1998;58:1993–2002. [PubMed] [Google Scholar]
- 196.Kaufmann WK, et al. DNA repair endonuclease activity during synchronous growth of diploid human fibroblasts. Mutat. Res. 1990;236:107–117. doi: 10.1016/0921-8777(90)90038-7. [DOI] [PubMed] [Google Scholar]
- 197.Herrlich P, et al. Supreme enlightenment: damage recognition and signaling in the mammalian UV response. Mol. Cell. 2008;29:279–290. doi: 10.1016/j.molcel.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kaufmann WK, et al. DNA signals for G2 checkpoint response in diploid human fibroblasts. Mutat. Res. 1998;400:153–167. doi: 10.1016/s0027-5107(98)00041-4. [DOI] [PubMed] [Google Scholar]
- 199.Bulavin DV, et al. p38 and Chk1 kinases: different conductors for the G(2)/M checkpoint symphony. Curr. Opin. Genet. Dev. 2002;12:92–97. doi: 10.1016/s0959-437x(01)00270-2. [DOI] [PubMed] [Google Scholar]
- 200.Manke IA, et al. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell. 2005;17:37–48. doi: 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 201.Mouret S, et al. Differential repair of UVB-induced cyclobutane pyrimidine dimers in cultured human skin cells and whole human skin. DNA Repair (Amst) 2008;7:704–712. doi: 10.1016/j.dnarep.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 202.Kraemer KH, et al. DNA repair protects against cutaneous and internal neoplasia: evidence from xeroderma pigmentosum. Carcinogenesis. 1984;5:511–514. doi: 10.1093/carcin/5.4.511. [DOI] [PubMed] [Google Scholar]
- 203.Mouret S, et al. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl Acad. Sci. USA. 2006;103:13765–13770. doi: 10.1073/pnas.0604213103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Girard PM, et al. Inhibition of S-phase progression triggered by UVA-induced ROS does not require a functional DNA damage checkpoint response in mammalian cells. DNA Repair (Amst) 2008;7:1500–1516. doi: 10.1016/j.dnarep.2008.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.