

Abstract
The aim of this study was to investigate the influence of drying methods and low range of degrees of substitution (DS) on the structural, physicochemical, and drug-release properties of carboxymethyl high-amylose starch (CMS). CMS with three DS of 0.03, 0.14, and 0.25 was synthesized and dried by either solvent precipitation (SP), spray drying (SD), or lyophilization (Ly). DS had an influence on the crystalline structure of CMS. It was found that a DS of 0.14 or higher induced a modification of polymorphism. The drying method and the DS had both an impact on the physical properties of the CMS powder which can further influence the formulation characteristics and drug-release properties from monolithic tablets. The CMS with DS of 0.14 and 0.25 dried by SP or SD presented good excipient properties in terms of compressibility. With acetaminophen (20%) as tracer, the monolithic CMS tablets showed controlled drug release over 17 h for DS of 0.14 and 10 h for DS of 0.25, almost independent of pH, suggesting interesting properties for sustained release applications.
Key words: carboxymethyl starch, controlled delivery, lyophilisation, solvent precipitation, spray drying
INTRODUCTION
Hydrophilic polymers, swellable, or gel forming matrices, are largely used as excipient for oral formulations affording controlled drug delivery (1). The dissolution kinetics and mechanisms of drug release from a hydrophilic matrix can be adjusted by tailoring factors that govern gel-layer formation, gel network structure, and resulting characteristics such as water penetration, polymer swelling, drug dissolution and diffusion, and matrix erosion. All these aspects can be related to the physical and structural properties of the polymer itself or to polymer chains–chains and polymer chains–dissolution medium interactions (2). Modified starch hydrophilic polymers are advantageous for controlled drug delivery systems due to relatively low cost, accessibility, biocompatibility, and good in vivo performance (3). A major factor influencing the starch properties is its crystalline structure (4). Depending on source, starch is composed of different ratio of two distinct polysaccharides: amylose, unramified chains of 200 to 2,000 glucopyranose units (GU) linked through α(1–4) glucosidic bonds and amylopectin, ramified polymer with sequences of 20–30 GU linked through α(1–4) glucosidic bonds and branching points with α(1–6) glucosidic bonds, for a total of about 3 × 106 GU (5). In native starch, these two polysaccharides can form crystallites in two polymorphic forms, A- and B-type structures, which are based on left-handed double-stranded helices parallel-packed into, respectively, monoclinic and hexagonal cells units, interspersed by amorphous region (4,6). Crystalline regions inside modified starch can adopt a further polymorphic form, the V-type structure, based on left-handed single-stranded helices close-packed orthorhombicly (4). V-type structure is generally showing increased gelatinization temperatures, a better dispersibility, a lesser swelling, and a higher solubility in water versus A and B types (4).
Carboxymethyl high amylose starch (CMS) was recently introduced as a hydrophilic ionic matrix for oral tablets to control the release of small (7) and large (8) active molecules or bioactive agents (9,10). CMS can be synthesized by etherification of the hydroxyl groups (–OH) with monochloroacetic acid (MCA) or its sodium salt (SMCA) under basic conditions (NaOH) to obtain carboxymethyl groups (CM, –CH2COONa) (11), as follows:
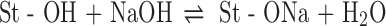 |
1 |
 |
2 |
The degree of substitution (DS, the average number of CM groups per GU) can vary between 0 and 3 and depends on GU/NaOH/MCA (or SMCA) ratio and reaction yield. After the carboxymethylation in aqueous medium, CMS needs to be precipitated, washed (removal of residual salts), and finally dried. It is known that drying procedures can have an effect on the crystalline structure and physicochemical properties of solid materials and thus have an impact on subsequent formulations and drug-release characteristics (12–14). The concept of carboxymethylation of starch was first intended to add pH sensitivity to the matrix and thus modulate the drug release throughout the gastrointestinal tract. In dissolution media below the pKa of CMS (approximately 4.2), an additional stabilization by hydrogen bonding through dimerized carboxylic protonated groups (–COOH) will enhance the strength of the outer gel layer, thus reducing the drug release rate. Differently, in dissolution media above the pKa, repulsive forces between dissociated carboxylate groups (–COO−) will reduce the gel-layer strength and increase the drug-release rate. It was previously shown that CMS with a DS of 0.3 was more suitable for controlled release than CMS with lower DS (e.g., 0.06), which often presented a capping during dissolution (9). On the other hand, dissolution profiles at higher DS (e.g., 0.66) were faster due to higher water penetration in the matrix, particularly in simulated intestinal fluid (SIF). The DS may also have an effect on the physical and crystalline structure of the CMS since hydrogen bonding between the hydroxyl groups of the GU plays an important role in the organization of the starch crystallites. Structural modifications according to the degree of cross-linking have been reflected in the drug-release kinetics for the cross-linked high amylose starch excipient used for controlled release (15).
The aim of the study was to investigate the role of drying process and of low range of DS (up to 0.25) on the structural and physical characteristics of CMS powders and on drug-release properties of CMS monolithic tablets. The drying methods evaluated were solvent precipitation (SP), spray drying (SD), and lyophilization (Ly; freeze-drying). These procedures were selected since they are common drying and particle generation methods used in the pharmaceutical industry.
MATERIALS AND METHODS
Materials
High amylose corn starch (HAS; Hylon VII) was supplied by National Starch and Chemical Co. (Bridgewater, NJ). Acetaminophen and MCA (Sigma–Aldrich, St. Louis, MO) as well as the other chemicals were reagent grade and used without further purification.
Synthesis of CMS
The CMS were synthesized according to Mulhbacher et al. (16) with slight modifications. Briefly, 100 g of HAS were suspended in 245 mL of H2O at 50°C in a 2-L jacketed reaction glass vessel (Chemglass, Vineland, NJ) equipped with a servo-controlled speed mixer head (Cole-Palmer Instrument, model 5000-40, Niles, IL) fitted with an anchor agitator to ensure constant and vigorous stirring during the synthesis. Subsequently, 335 mL of preheated 1.5 M NaOH solution was added in the medium and stirred for 20 min to induce starch gelatinization and solubilization. CMS with different DS were synthesized by adding different amounts of MCA, dissolved in minimal volumes of H2O, and added to the medium for a 1-h reaction. Three products were synthesized: CMS1, CMS2, and CMS3 by adding 10, 45, and 70 g of MCA, respectively. As control, nonreacted HAS (hereto called HAS-0) was prepared under the same conditions but without adding MCA to the medium. During the reaction, pH was continuously monitored (Fisher Scientific, model AR20 pH/conductivity meter, Waltham, MA) and maintained between 9.5 ± 0.5 by adding small volumes of 10 M NaOH when required. After carboxymethylation, the medium was neutralized at pH 7.0 ± 0.2 with acetic acid (5 M) and cooled to room temperature.
CMS Purification and Drying Method
Solvent Precipitation
Solubilized CMS derivatives (including HAS-0) were extracted from the neutralized reaction medium by solvent precipitation. A volume of 700 mL of an aqueous acetone solution (90% v/v) was slowly added to the CMS solution under continuous and vigorous stirring. After 1 h, the CMS suspensions were vacuum-filtered (Büchner with grade 54 cellulose filter paper, Whatman, Kent, UK), and the wet masses were retained and purified by at least four successive resuspension/vacuum filtration cycles with acetone/water (70% v/v) solution. Purification was continued until the filtrate conductivity was less than 25 μS/m (Fisher Scientific, model AR20 pH/conductivity meter, Waltham, MA) in order to ensure the removal of residual salt ions. Purified wet CMS were then resuspended twice in acetone for 2 h for dehydration and recovered by vacuum filtration. CMS powder was finally left under vacuum drying (−70 kPa) for 12 h at room temperature in order to remove residual solvents. The resulting powder was screened over a 300-μm sieve for deagglomeration prior to use.
Spray Drying
SD was performed using a mini-spray dryer model B-290 (Buchi, Zurich, Switzerland). The CMS and HAS-0 previously obtained and purified by SP were suspended in Nanopure® water, at a concentration of 25 g/L and homogenized (Heidolph, model Diax 600 homogenizer, Kelheim, Germany) at 8,000 rpm for 2 min at 25°C. The CMS mixtures were then spray-dried using the following operating parameters: 0.5-mm nozzle; 10.0 ± 0.5 mL/min spray rate; 190 ± 5°C inlet temperature; 85 ± 2°C outlet temperature; 470 ± 50 NL/h atomization flow; and 100% air flow (≈37 m3/h). The resulting powder was screened over a 300-μm sieve for deagglomeration and comparison with powders obtained by other drying methods.
Lyophilization
Ly was performed using a Freeze Mobile 24 (Virtis Company Inc., Gardiner, NY). The CMS and HAS-0 previously obtained and purified by SP were suspended and homogenized as described for SD but at a higher concentration (150 g/L). The CMS preparations were separated into 35 mL aliquots, frozen at −80°C, and then dried at −55°C and 0.2 mbar for 48 h. Finally, lyophilized cakes were gently ground with a mortar/pestle and screened over a 300-μm sieve for deagglomeration and comparison with powders obtained by other drying methods.
Determination of the Degree of Substitution
DS was determined by back titration (17) of the protonated form (–COOH) obtained by acid treatment of the polymer and calculated from:
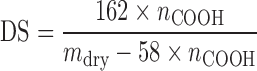 |
3 |
Where 162 g/mol is the molar mass of GU, nCOOH (mol) is the amount of COOH determined by titration (n = 3), 58 g/mol is the net increase in the mass of one GU for each CM group added, and mdry is the mass of the dry sample.
CMS Particles and Powder Characterization
X-ray Crystallography
X-ray diffraction patterns were obtained with a Siemens D-5000 diffractometer (Munich, Germany) operating in reflectance mode at a Co–Kα wavelength (λ) of 1.78897 Å, over an angular range 2θ from 5° to 35° and a scan rate of 2°/min. The recorded diffractograms were smoothed by a local second-order polynomial regression (Savitzky-Golay 11 points) using the software GRAMS/AI v7.0 (Thermo Galactic, Waltham, MA). The peak positions were computed according to Bragg’s law (12).
Relative Solubility
The relative solubility of CMS and HAS-0 dried with the different methods was determined following Chen and Jane (18) and Volkert et al. (19). The method consisted of precise weighing a dry CMS sample (0.2 g) and dissolving it in 10 mL of neutral Nanopure® water at 25°C. The solution was vortexed for 1 min and stored at room temperature for 2 h. The solution was then centrifuged for 15 min at 4,000 rpm (IEC, model HN-II centrifuge, Needham, MA), and 5 mL of the supernatant liquid was evaporated in an oven (Blue M, model OV-12A, Blue Island, IL) at 105°C until a constant mass was reached. The weight of the dry residue was used to calculate the relative solubility of the sample.
Particle Size and Size Distribution
The granulometry of the particles was determined by optical microscopy using a Leica microscope (model DM2500M, Frankfurt, Germany) interfaced with a color camera (Clemex, model L 2.0C CL-13-211, Longueuil, Canada) and an image analysis software (Clemex Vision PE, Longueuil, Canada). Powder samples were randomly spread on a glass slide to obtain a representative sample. At least 105 particles were observed under ×200 or ×500 magnification and analyzed by digital image analysis. This image processing incorporated gray thresholding and geometric constraints to first differentiate the particles from the background and then to eliminate overlapped or partial particles. Particles were then characterized by their mean size 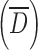 and cumulative size distribution (D10, D50, and D90), where the size of one particle represents the average value of 64 feret diameters (i.e., distance between two parallel tangents on each side of the particle).
and cumulative size distribution (D10, D50, and D90), where the size of one particle represents the average value of 64 feret diameters (i.e., distance between two parallel tangents on each side of the particle).
Particle Morphology and Surface Characteristics
The morphology and surface characteristics of particles were examined with a Hitachi S-4300SE/N variable pressure scanning electron microscope (SEM; Hitachi High Technologies America, Pleasanton, CA), at 10.0 or 25.0 kV and magnification of ×300, ×10,000, or ×25,000. Samples were prepared on metallic stubs using double-sided conductive tape and then made electrically conductive by gold coating.
Bulk and Tapped Densities and Powder Flow Properties
The bulk (ρB) and the tapped (ρT) densities of the powder were determined with a Vankel tap density tester (Varian Inc., Cary, NC) according to USP method <616> (20) (n = 3). For each sample, the Carr index (CI, also called compressibility index) (21) and the Hausner ratio (HR) (22) were, respectively, calculated from Eqs. 4 and 5 and interpreted as per USP method <1174> (20)
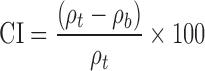 |
4 |
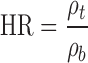 |
5 |
Thermogravimetric Analysis
Thermogravimetric analysis (TGA) measurements were carried out with a Seiko TG/DTA 200 system (Seiko Instrument Inc., Chiba-shi Chiba, Japan). CMS samples (8–12 mg) were heated from 30°C to 800°C at 10°C/min in platinum open crucible under nitrogen flow (200 mL/min; n = 2).
CMS Tablet Characterization
Tablet Preparation
Tablets (500 mg, 12 mm diameter) of CMS and HAS-0 were formulated with acetaminophen (20% drug loading) as drug model, by dry powder blending, and direct compression at 24.5 kN (2.5 T force) using flat faced tooling and a hydraulic press (Carver, model Mini C, Wabash, IN). Tablets were dedusted over a 600-μm screen.
Physical Characterization of Tablets
Crushing strength (hardness) of tablets was determined (n = 6) with a Vankel hardness tester (model VK200, Varian Inc., Cary, NC) according to USP method <1217> (20). Tablet friability was determined (n = 2) with a Vankel friabilator (Varian Inc., Cary, NC) according to USP method <1216> (20). Finally, the thickness of the tablets was measured (n = 6) with a digital caliper.
In Vitro Drug-Release Properties
The dissolution kinetics of CMS tablets (n = 3) were recorded in USP dissolution apparatus II (Distek 5100, North Brunswick, NJ) at 50 rpm for 2 h in 900 mL pepsin-free simulated gastric fluid (SGF; pH 1.2) at 37°C and then in pancreatin-free SIF (pH 6.8) until the complete release of the drug. This change of dissolution medium simulated the pH change in the gastrointestinal tract. Acetaminophen (a weak organic acid with pKa 9.7) was selected as drug model since it is not ionizable at physiological pH values. It also shows minor differences in solubility as a function of pH (e.g., 21.3 mg/mL in pH 1.2 and 17.8 mg/mL in pH 6.8 at 37°C) (23). This behavior limits the influence of drug solubility on the drug-release rate. Acetaminophen-release profiles were measured by UV spectroscopy at 280 nm (Hewlett–Packard spectrophotometer, model 8452A, Palo Alto, CA).
The drug-release mechanism from the CMS tablets was evaluated by the empirical model of Ritger and Peppas (24):
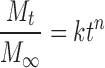 |
6 |
where 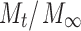 is the fraction of drug released in time t, k is a kinetic constant incorporating characteristics of the macromolecular network system, and n is the release exponent which is indicative of the transport mechanism. The fraction of released drug was fitted, up to
is the fraction of drug released in time t, k is a kinetic constant incorporating characteristics of the macromolecular network system, and n is the release exponent which is indicative of the transport mechanism. The fraction of released drug was fitted, up to 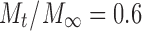 (60% release), by nonlinear regression of Eq. 6 using SigmaPlot v. 11 (Systat Software Inc., Chicago, IL; R2 > 0.96, p < 0.01) to determine the values of k and n.
(60% release), by nonlinear regression of Eq. 6 using SigmaPlot v. 11 (Systat Software Inc., Chicago, IL; R2 > 0.96, p < 0.01) to determine the values of k and n.
Statistical Analysis
Two-way analysis of variance according to drying method and DS for CMS particles (except X-ray diffraction and morphology) and CMS tablets characterization were followed by Bonferroni post-test for individual P values using GraphPad Prism v. 5 (GraphPad Software Inc., San Diego, California). Statistical significance was assessed for P < 0.05.
RESULTS AND DISCUSSION
CMS Synthesis
The DS was verified for each CMS derivative after each drying method. The DS of HAS-0 (control), CMS1, CMS2, and of CMS3 were, respectively, determined as 0.00, 0.03 ± 0.01, 0.14 ± 0.02, and 0.25 ± 0.02 regardless of the drying method. After solvent precipitation but prior to SD and Ly steps, different types of CMS aqueous systems were observed: suspension-like for HAS-0 and CMS1, opalescent solution for CMS2, and clear solution for CMS3.
CMS Particles and Powder Characterization
X-ray Diffraction
Native starch is a semi-crystalline polymer with X-ray pattern characterized by minor diffraction peaks superposed on a large amorphous background (25) (Fig. 1). Since the aim of this study was to investigate the effect of the drying method and the influence of DS on the crystallinity of CMS, only the position of peaks and their relative intensities were considered for discussion.
Fig. 1.

X-ray diffractograms of native starch and CMS obtained by SP, SD, or Ly at various degrees of substitution DS
DS was found to exert a marked effect on the crystalline structure of CMS. Peaks at 15.9, 6.1, 5.2 4.5, 4.0, and 3.7 Å of the native Hylon VII are typical of corn starch containing more than 65% amylose and correspond to the B-type pattern (double-stranded helix) (26). Crystallites inside the HAS-0(SP) and CMS1(SP) also presented elements of the B-type pattern with a diffractogram similar but not identical to that of Hylon VII. At higher DS, CMS2 and CMS3 showed a different structure with increased amorphous portion and minor peaks at 7.1 and 4.5 Å characteristic of the V-type pattern (single-stranded helix) (4).
CMS polymorphism can be correlated with physical and chemical transformations that occurred during gelatinization, carboxymethylation, and precipitation steps. During the gelatinization under heat and alkaline conditions, the initial crystalline structure of starch was changed from an ordered state to a disordered state. However, under certain conditions used in this study, not chemically modified (HAS-0) or slightly modified i.e. CMS1 (low DS) can be gelified in its initial B-type pattern (Fig. 1). The carboxymethylation with increased DS was found to induce a different organization (V-type). Substitution probably induces a disentanglement of the initial B-type pattern of amylose by altering the hydrogen bonding between the hydroxyl groups. Furthermore, disentangled CMS helix chains can, to a certain extent, complex with acetone during the precipitation, favoring the V-type pattern at high DS (0.25). At low DS (0.03), the initial hydrogen bonding was still possible. Thus, the B-type was still predominant in CMS1(SP). At moderate DS (0.14), the hydrogen bonding is partially altered, favoring the V-type pattern as for CMS2(SP) where both B- and V-patterns coexist but with a weak B-type peak at 5.2 Å and V-type 7.1 and 4.5 Å peaks of lower relative intensities compared to the CMS3(SP) (Fig. 1). Therefore, a DS of at least 0.14 was sufficient to induce a modification of the polymorphism of CMS synthesized from Hylon VII. Furthermore, increasing DS appears to reduce network self-assembling by hydrogen association between hydroxyl groups and to promote a reorganization of the network via dimerization of carboxylic groups located at a proper distance (as for CMS3).
Figure 1 also shows the crystalline structure of the different CMS as a function of the drying procedure. It was found that drying process did not induce modifications of the peaks positions but influenced their relative intensities. Differences in water content of the CMS as result of different drying methods may explain the variations of the peak intensities where higher water content resulted in higher peak intensities. This aspect is also discussed below in thermogravimetric analysis section. It was previously reported that conversion from the V- to B-type pattern can be readily accomplished by hydration (4,27). In our case, CMS (SD) and CMS (Ly) powders, differing in moisture content, showed no difference in peak positions. It was, therefore, assumed that the presence of sufficient CM groups on the GU induced a stabilization of the CMS crystalline structure.
Relative Solubility
Substitution of the starch hydroxyl groups with the CM groups induced higher solubility in cold water in function of DS (Fig. 2). The relative solubility of CMS1 (13%) was slightly higher compared to the almost insoluble HAS-0 (4%) Differently, the CMS2 and CMS3 were freely soluble showing the impact of CM groups. The relative solubilities of the CMS were independent of the drying methods. The larger amorphous ratio at increasing DS could also play a role in increased relative solubility due to weaker stabilization by hydrogen association and higher hydrophilicity of carboxylic groups.
Fig. 2.

Relative solubility (%) of CMS obtained by SP, SD, or Ly at various DS in Nanopure® water (pH 7.0; n = 3)
Particle Size and Size Distribution
Table I shows a significant decrease of mean size particle 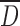 with the increase in DS from CMS1 to CMS3 for the SP process. The decrease of particle
with the increase in DS from CMS1 to CMS3 for the SP process. The decrease of particle 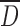 from 96 to 25 μm was probably caused by a modification of interfacial energy between surface boundaries water-solvent polymer due to the larger amount of acetone required to induce dehydration and precipitation of polymer chains. Indeed, with increased DS and hydrophilicity of CMS, there is reduction of agglomeration between gelified particles and production of CMS powder with the largest size distributions of particles. The decrease of particle
from 96 to 25 μm was probably caused by a modification of interfacial energy between surface boundaries water-solvent polymer due to the larger amount of acetone required to induce dehydration and precipitation of polymer chains. Indeed, with increased DS and hydrophilicity of CMS, there is reduction of agglomeration between gelified particles and production of CMS powder with the largest size distributions of particles. The decrease of particle 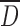 of CMS (SP) particles at higher DS could be also the result of more compaction of the particles due to carboxyl–carboxyl or carboxyl–hydroxyl stabilizations.
of CMS (SP) particles at higher DS could be also the result of more compaction of the particles due to carboxyl–carboxyl or carboxyl–hydroxyl stabilizations.
Table I.
Particle Size and Size Distribution (n = 2, m > 10,000 Particles), ρ B and ρ T, CI and HR (n = 3) of CMS Obtained by SP, SD, or Ly at Various DS (n = 3)
| Drying method | Material | Granulometry (μm) | Densities (g/cm3) and powder flow | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
D 10 | D 50 | D 90 | Span | ρ B a | ρ T a | CI (%) | HR | ||
| SP | HAS-0 | 109.9 ± 84.4 | 21.6 | 80.6 | 238.6 | 2.69 | 0.52 | 0.72 | 28 | 1.38 |
| CMS1 | 95.8 ± 75.8 | 19.2 | 69.2 | 216.1 | 2.85 | 0.64 | 0.87 | 26 | 1.36 | |
| CMS2 | 46.1 ± 55.3 | 11.3 | 25.8 | 112.6 | 3.93 | 0.43 | 0.62 | 31 | 1.44 | |
| CMS3 | 24.8 ± 20.8 | 9.6 | 18.9 | 45.1 | 1.88 | 0.24 | 0.54 | 56 | 2.25 | |
| SD | HAS-0 | 13.3 ± 7.6 | 4.7 | 11.5 | 23.5 | 1.63 | 0.42 | 0.73 | 42 | 1.74 |
| CMS1 | 4.9 ± 3.1 | 2.7 | 5.5 | 8.7 | 1.10 | 0.34 | 0.64 | 47 | 1.88 | |
| CMS2 | 5.5 ± 5.8 | 2.5 | 5.0 | 8.7 | 1.24 | 0.14 | 0.33 | 58 | 2.36 | |
| CMS3 | 12.2 ± 8.9 | 2.8 | 7.7 | 19.0 | 2.10 | 0.15 | 0.31 | 52 | 2.07 | |
| Ly | HAS-0 | 96.7 ± 52.4 | 32.1 | 86.9 | 171.5 | 1.60 | 0.45 | 0.58 | 22 | 1.29 |
| CMS1 | 103.4 ± 71.1 | 33.7 | 86.5 | 203.8 | 1.97 | 0.43 | 0.60 | 28 | 1.40 | |
| CMS2 | 63.1 ± 58.4 | 10.9 | 58.9 | 147.2 | 2.31 | 0.30 | 0.57 | 47 | 1.90 | |
| CMS3 | 57.9 ± 56.4 | 11.3 | 47.7 | 142.2 | 2.74 | 0.30 | 0.63 | 52 | 2.10 | |
ρ B bulk density, ρ T tapped density, CI Carr’s Index, HR Hausner Ratio, CMS carboxymethyl high-amylose starch, SP solvent precipitation, SD spray drying, Ly lyophilization, DS degrees of substitution
aStandard deviation equal or under 0.02
For CMS (SD) and CMS (Ly) results were typical of the drying process used. It was also found that the DS had no significant influence on the mean particle size 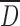 and on particle size distribution (Table I). Spray drying produced micronized particles with the narrowest particle size distribution ranging from 2 to 20 μm. The
and on particle size distribution (Table I). Spray drying produced micronized particles with the narrowest particle size distribution ranging from 2 to 20 μm. The 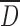 of CMS (Ly) probably depended on the cake grinding which produced particles with similar values compared to CMS (SP) but with a narrower particle size distribution.
of CMS (Ly) probably depended on the cake grinding which produced particles with similar values compared to CMS (SP) but with a narrower particle size distribution.
Particle Morphology and Surface Characteristics
Morphology and surface characteristics of CMS (Fig. 3) revealed interesting features in terms of both powder properties and particle formation mechanism. Micrographs of CMS (SP) particles showed particles with irregular shapes and large particle size distribution at a magnification of ×300. Surface roughness was revealed at magnification of ×10,000 where agglomeration of individual particles on the surface of larger particles was also observed.
Fig. 3.

Scanning electron micrographs of CMS obtained by SP, SD, or Ly, at various DS (SEM at a voltage of 10.0 or 25.0 kV, magnification and scale bar value indicated on the pictures)
For HAS-0(SD) and CMS(SD), the micrographs showed spherical micronized particles with indented rough surfaces. At higher DS, CMS2(SD) and CMS3(SD) micrographs showed particles with very a different morphology. Spherical and elliptic cross-section of the CMS2(SD) and CMS3(SD) particles, respectively, appeared as a result of the collapsed atomized droplets. The HAS-0(Ly) and CMS1(Ly) particles had morphologies similar to those obtained by SP. Lyophilization of CMS solution yielded to a particle morphology that differed to that of particles obtained from the lyophilization of suspensions. Indeed, CMS2(Ly) and CMS3(Ly) particles presented prismatic morphology with some apparent microporosity (micrographs at ×25,000) probably resulting from the removal of ice crystals during the lyophilization process.
Bulk and Tapped Densities and Powder Flow Properties
The ρB and ρT as well as resulting flow characteristics are not intrinsic powder physical properties, but they depend on interparticulate interactions (28). These physical properties were greatly influenced by both the drying method and the DS (Table I). The SP drying produced the particles with the highest overall particle 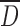 and thus the powder with the highest ρB and ρT. The decrease of particle
and thus the powder with the highest ρB and ρT. The decrease of particle 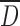 associated with increasing DS led to a significant decrease of ρB from 0.6 to 0.2 g/mL and of ρT from 0.9 to 0.5 g/mL for CMS(SP) powder. CMS(SD) micronized particles resulted in powders with the lowest ρB and ρT. For instance, CMS2(SD)-collapsed particles induced a two-fold diminution of density values. The larger CMS(Ly) particles at low DS induced lower ρB and ρT values than for CMS(SP) particles, even though they had comparable
associated with increasing DS led to a significant decrease of ρB from 0.6 to 0.2 g/mL and of ρT from 0.9 to 0.5 g/mL for CMS(SP) powder. CMS(SD) micronized particles resulted in powders with the lowest ρB and ρT. For instance, CMS2(SD)-collapsed particles induced a two-fold diminution of density values. The larger CMS(Ly) particles at low DS induced lower ρB and ρT values than for CMS(SP) particles, even though they had comparable 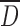 . The apparent microporosity observed for the CMS2(Ly) and CMS3(Ly) led to lower ρB and ρT values compared to those of corresponding SP powders.
. The apparent microporosity observed for the CMS2(Ly) and CMS3(Ly) led to lower ρB and ρT values compared to those of corresponding SP powders.
According to CI and HR values (Table I), the flowability of CMS(SP) particles ranged in the USP scale (20) between Poor Flow for CMS1(SP) and CMS2(SP) and Very Very Poor Flow for the CMS3(SP). The SD produced powder with Very Very Poor Flow since micronized particles are known to be more influenced by triboelectric charge phenomena that increase cohesive forces between the particles and thus decrease powder flow properties. Finally, freeze-drying produced powders with Poor Flow properties for CMS1(Ly) and Very Very Poor Flow properties for CMS2(Ly) and CMS3(Ly). Flow properties of CMS dried with different methods were poorer or similar to those of the respective HAS-0. Powders with improved flow properties (lower CI and HR values) such as CMS1(SP), CMS1(Ly), and especially CMS2(SP) showed a wide particle size distribution which can favor particle segregation during powder handling or manufacturing process of oral solid dosage forms. These results suggest that the drying step may limit the use of CMS powder as such in dry blend and direct compression applications. Powder flow properties can be improved by optimization of drying process or by standard densification procedures such as dry or wet granulation.
Thermogravimetric Analysis
Thermal stability of CMS was reported to be low compared to that of amylose (29). TGA and differential thermogravimetric (DTG) measurements of selected representative CMS samples are presented in Fig. 4, and complete thermogravimetric data are presented in Table II. The reproducibility of the results was considered acceptable since the differences in normalized weight (%) measured versus temperature were in the range of 0.02% to 1.7%. Each CMS powder sample showed similar TG and DTG curves (Fig. 4) with two stage events occurring at increasing temperature. An initial stage between 25°C and 170°C, corresponding to the removal of residual solvents and water, that was followed by CMS decomposition stage (180°C to 405°C). Thermal stability of CMS was independent of drying methods but decreased with increasing DS. Temperature range and temperature at maximal degradation rate (Tmax; Table II and insert Fig. 4b) decreased at increasing DS. These results suggest that thermal stability of CMS was mainly controlled by the starch chemical backbone (links between the GU) rather than by the drying methods. The lowest temperature of degradation was observed for CMS3(SP) (180°C) which was still higher than typical temperatures involved in standard drying process and pharmaceutical operations.
Fig. 4.

TG (a) and DTG (b) thermograms of representative CMS samples obtained by SP, SD, or Ly at various DS: HAS-0(SP), CMS1(SP), CMS2(SD), and CMS3(Ly) (n = 2)
Table II.
Thermogravimetric Data of Carboxymethyl High-Amylose Starch (CMS) Obtained by Solvent Precipitation (SP), Spray Drying (SD) or Lyophilisation (Ly) at Various Degrees of Substitution (DS) (n = 2)
| Drying method | Material | Stage 1 | Stage 2 | % Residue at 600°Cc | ||||
|---|---|---|---|---|---|---|---|---|
| Temperature range (°C)a | Tmax (°C)a | % Water contentb | Temperature range (°C)a | Tmax (°C)a | % Weight lossc | |||
| SP | HAS-0 | 37–145 | 72 | 10.5 | 208–398 | 300 | 69.1 | 21.6 |
| CMS1 | 35–162 | 91 | 6.0 | 194–395 | 298 | 71.9 | 21.0 | |
| CMS2 | 34–155 | 73 | 11.4 | 182–388 | 293 | 68.5 | 22.8 | |
| CMS3 | 32–132 | 65 | 9.5 | 180–367 | 285 | 68.1 | 23.8 | |
| SD | HAS-0 | 31–164 | 74 | 11.4 | 197–408 | 301 | 71.9 | 20.1 |
| CMS1 | 31–156 | 75 | 14.7 | 195–398 | 299 | 72.9 | 21.0 | |
| CMS2 | 30–141 | 65 | 12.1 | 188–384 | 292 | 69.5 | 22.3 | |
| CMS3 | 31–131 | 77 | 7.5 | 186–365 | 286 | 69.2 | 24.4 | |
| Ly | HAS-0 | 32–169 | 93 | 4.3 | 203–403 | 300 | 72.1 | 21.5 |
| CMS1 | 31–158 | 92 | 4.5 | 195–398 | 299 | 73.2 | 20.3 | |
| CMS2 | 32–147 | 86 | 4.8 | 192–386 | 293 | 66.2 | 25.7 | |
| CMS3 | 33–141 | 87 | 5.4 | 187–385 | 285 | 64.3 | 27.1 | |
CMS carboxymethyl high-amylose starch, SP solvent precipitation, SD spray drying, Ly lyophilization, DS degrees of substitution
aStandard deviation equal or under 4°C.
bStandard deviation equal or under 1.5%.
cNormalized with dry mass, standard deviation equal, or under 1.5%.
Table II presents the moisture content (%) determined by TGA during the initial thermal stage. The drying methods used during this study efficiently removed water to values similar to those of native starch and other modified commercial starches (5% to 15%) (30,31). The SP and SD produced CMS powder with comparable moisture content of approximately 10%. Ly produced CMS powder with the lowest water content at approximately 5%. The influence of DS on CMS powder moisture was negligible. CMS1(SP) had a lower water content than CMS2(SP) and CMS3(SP) since for SP samples, hydrophilicity and V-type structure of the CMS increased with DS as well as the powder susceptibility to hydration. The difference in water content of CMS2(SP) and CMS3(SP) compared to HAS-0(SP) typical value of 10% was less important than the difference between HAS-0(SP) and CMS1(SP). For SD, the water content was consistent with particle characteristics. At higher DS, the water content decreased with the reduction of the particle diameter thus reducing the mass transfer path for CMS(SD) particles. For Ly, moisture content slightly increased with DS and hydrophilicity of CMS due to the presence of more regions of V-type structure within the crystallites of the CMS2(Ly) and CMS3(Ly). These results showed that water content of CMS particles was mainly related to the drying procedure and process parameters and only slightly influenced by the DS which modified the hydrophilicity and crystalline structure of the CMS. The relatively high water content of CMS(SP) and CMS(SD) particles as well as their hydrophilicity show that special attention should be paid to long-term stability of dosage forms using CMS particularly in presence of moisture sensitive active pharmaceutical ingredients. Addition of a desiccant will probably be required for storage.
Characterization of CMS Tablet
As presented above, both DS and drying method had a significant impact on the particle size 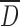 , size distribution, particle morphology, and surface properties of CMS powders. It is known that these properties, as well as powder structural organization, water content, and solubility are among the factors that can influence the compression process and the drug-release kinetics from oral solid dosage forms (13).
, size distribution, particle morphology, and surface properties of CMS powders. It is known that these properties, as well as powder structural organization, water content, and solubility are among the factors that can influence the compression process and the drug-release kinetics from oral solid dosage forms (13).
Physical Characterization of Tablets
For starch derivatives, compression properties are mainly function of the plastic deformation of the particles under the action of the forces that lead to formation of bonds between the surface of the deformed particles by Van der Waal’s forces, solid bridges, and mechanical interlocking (32). In general, a decrease in 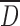 as well as increasing of particle roughness and water content, until an optimal value, will result in higher bonding between the deformed particles and, subsequently, higher tablet strength (14). Particle compressibility was assessed by tablet thickness, crushing strength, and friability (Fig. 5). For CMS(SP), the decrease of particles
as well as increasing of particle roughness and water content, until an optimal value, will result in higher bonding between the deformed particles and, subsequently, higher tablet strength (14). Particle compressibility was assessed by tablet thickness, crushing strength, and friability (Fig. 5). For CMS(SP), the decrease of particles 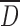 with increasing DS resulted in a significant increase of crushing strength up to 345 N and a decrease of thickness and friability. Compared to HAS-0(SP), CMS1(SP) had a lower crushing strength probably related to a lower water content which can reduce the plastic deformation of the particles and thus lead to increased particle fragmentation similarly to that of acetaminophen particles as shown by Garekani et al. (33).
with increasing DS resulted in a significant increase of crushing strength up to 345 N and a decrease of thickness and friability. Compared to HAS-0(SP), CMS1(SP) had a lower crushing strength probably related to a lower water content which can reduce the plastic deformation of the particles and thus lead to increased particle fragmentation similarly to that of acetaminophen particles as shown by Garekani et al. (33).
Fig. 5.

Physical and mechanical characteristic of direct compression tablets from CMS obtained by SP, SD, or Ly at various DS loaded with acetaminophen (20% w/w): a thickness (n = 6), b crushing strength (n = 6), and c friability (n = 2, m = 10)
CMS1(SD) tablets had higher crushing strength probably due to smaller particles 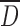 and higher water content compared to CMS2(SD) and CMS3(SD) tablets (Fig. 5b). The rough surface of CMS1(SD) particles compared to the smooth surface of CMS2(SD) particles resulted in a significantly higher crushing strength for particles with similar
and higher water content compared to CMS2(SD) and CMS3(SD) tablets (Fig. 5b). The rough surface of CMS1(SD) particles compared to the smooth surface of CMS2(SD) particles resulted in a significantly higher crushing strength for particles with similar 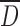 , water content and tablet thickness. Further increase of the DS to CMS3(SD) induced higher crushing strength.
, water content and tablet thickness. Further increase of the DS to CMS3(SD) induced higher crushing strength.
For the same DS, CMS(Ly) tablets had the lowest crushing strength and higher thickness compared to other drying methods, except for CMS1(Ly). The poor compression properties of the CMS(Ly) appeared related to the lower water content. However, at increased DS, the crushing strength was considerably higher while thickness and friability remained comparable. These results, added to the increase in crushing strength from CMS2(SD) to CMS3(SD) tablets, suggest that the stabilization via carboxylic groups and its influence in the amorphous region of the CMS in function of DS may play a role on the compaction properties of the material.
No significant differences in compression properties were found (Fig. 5) for B-type (CMS1) versus V-type structures (CMS2 and CMS3). All CMS tablets showed low friability (less than 0.5%), except for CMS1(SP) where insufficient hardness resulted in high friability. Higher DS generated particles with improved compression when compared to the corresponding HAS-0 dried with the same methods. These results suggest that these CMS derivatives may be convenient excipient for tablet dosage forms as per their adequate compressibility.
In Vitro Drug-Release Properties
Figure 6 presents dissolution profiles obtained for CMS and control HAS-0 tablets in (a) SGF (pH 1.2) only and (b) SGF for the first 2 h and then in SIF (pH 6.8) until complete release of the model drug acetaminophen. Table inserts present drug release times (T100%), kinetic constants (k), and release exponents (n) of selected representative tablet samples. A fast dissolution in SGF occurred for all HAS-0 and CMS1 tablets regardless of the drying method and despite certain differences in their physical properties. The immediate drug release from CMS1 tablets with almost instantaneous disintegration of the tablet in acidic dissolution medium seems due to rapid water penetration between the slightly soluble CMS1 particles, annihilating the cohesive forces between the particles that have been created during the compression of the tablets. Therefore, CMS1 acted as a disintegrant such as several other starch forms in oral solid dosages. This observation is consistent with similar crystalline patterns observed for CMS1 and HAS-0 (Fig. 1).
Fig. 6.

Dissolution profiles of acetaminophen (20% w/w) from CMS tablets obtained by SP, SD, or lyophilisation (Ly) at various degrees of substitution (DS): a in SGF for 1.5 h for HAS-0 (SP, SD, Ly), CMS1 (SP, SD, Ly), CMS2(Ly), and CMS3(Ly) and b in SGF for 2 h followed by SIF for CMS2(SP, SD) and CMS3(SP, SD) (n = 3)
Figure 6 also showed that a DS of 0.14 (CMS2) or higher can control drug release over 1 h for Ly, 17 h for SD, and 18 h for SP. This minimal DS (0.14) of CMS also corresponds to the crystalline structure modification from predominant B-type to V-type (Fig. 1). Hydration of CMS2 and CMS3 powders induced the formation of a gel layer by the entanglement of the swollen single-helix chains (V-type structure) through attractive intermolecular interactions and water bridging. For CMS, these intermolecular interactions are pH dependent and also related to hydrogen bonding between hydroxyl groups, hydroxyl groups, and carboxylic or carboxylate groups and between dimerized carboxylic groups. In CMS2(SP) gel layer, polymer chains were strongly entangled, limiting water penetration within the tablet and thus leading to the lowest values of k and n. In this case, drug release was more influenced by a slow diffusion of the drug through the gel layer (n ≈ 0.60). This value is closer to the 0.45 limit for diffusion-controlled, than to the 0.89 limit ascribed for swelling-controlled drug release (24). For CMS2 tablets, neither cracking nor capping were observed during dissolution irrespective of drying methods, suggesting that a more uniform tablet hydration at increased DS ensures a better preservation of the gel-layer integrity. At further increasing DS, higher hydrophilicity and a greater amorphous ratio were most probably responsible for a slight T100% decrease from 18 to 14 h for CMS2(SP) and CMS3 (SP) tablets, respectively. Higher water content in the gel layer reduced the compactness of the gel network leading to an increase of k value to 0.21 h−0.7 and n value to 0.70. These results indicate an increase of both diffusion and swelling contributions to sustained drug release mechanism at higher DS (0.25).
The drug release from CMS tablets with a DS between 0.14 and 0.25 presented almost pH independent release rates since no statistically significant discontinuity of the drug-release rate was observed after changing the dissolution medium after 2 h from SGF to SIF (Fig. 6b). Thus, the CMS hydrogel formed and the release profile support the hypothesis of hydrogen bonding between hydroxyl groups and between hydroxyl groups and carboxylic groups (at low pH) or carboxylate groups (at neutral pH). Thus, CMS at moderate–high DS (0.14–0.25) appears more appropriate as excipient for sustained release than for delayed, pH-dependent release formulation.
Similar drug-release profiles between CMS2(SP) and (SD) as well as CMS3(SP) and (SD) tablets (Fig. 6) confirmed that for these two CMS, the drying method had no significant influence on the crystalline structure, even if they generated differences in physical properties. Compared to corresponding CMS(SP), these different powder properties markedly influenced the drug release kinetics for CMS(Ly) and CMS(SD) tablets. For CMS(SD) tablets, the slightly lower T100% by 1 h for CMS2 and 3 h for CMS3 was probably due to the lower crushing strength compared to CMS(SP) which allowed faster water penetration in the tablets.
The microporosity of CMS2(Ly) and CMS3(Ly) particles significantly increased water penetration rate into the tablet matrix resulting in a dramatic decrease of T100% to 0.8 and 1.25 h for CMS2(Ly) and CMS3(Ly) tablets, respectively. Faster water penetration inside the tablet matrix did not allow formation of hydrogel by the polymer chain entanglement. This phenomenon induced a fast erosion of the tablet matrix as showed by the high k (0.77 h−1.08) and n (1.08) values for CMS2(Ly) (Fig. 6 a). Lower T100% of CMS2(Ly) tablets can be ascribed to smaller number and size of micropores compared to CMS3(Ly) particles (Fig. 3). However, dissolution profiles of CMS3(Ly) tablets confirmed that the amount of entanglement between the polymer chains increased with DS (regardless to the drying method) since the drug release from CMS3(Ly) tablets was still driven by a contribution from diffusion mechanism (n ≈ 0.64).
CONCLUSION
The influence of drying methods (solvent precipitation, spray-drying, and lyophilization) and DS up to 0.25 was investigated on the solid-state and drug release properties of CMS. A DS of at least 0.14 was necessary to induce a modification of the polymorphism from B-type to V-type patterns whereas the drying method had no influence on the crystalline structure of the CMS particles. Relative solubility and hydrophilicity of CMS were also function of the DS. A minimal DS of 0.14 allowed CMS to become freely soluble in cold water. Particle size, size distribution, particle morphology, and surface characteristics as well as the bulk/tapped densities and powder flow characteristics were function of both drying process and DS. Thermogravimetric analysis showed that thermal stability of CMS was predominantly driven by the starch chemical backbone, partially dependent of the DS, and independent of the drying method. The water content of CMS powder was mainly dependant on the drying processing and only slightly influenced by DS. These structural and physicochemical differences influenced the properties of tablets made from CMS of different DS and dried by various procedures. Most of the CMS evaluated presented adequate compression properties. Finally, CMS with a DS of 0.14 or 0.25 obtained either by solvent precipitation or spray-drying process appeared suitable for sustained release applications over 10 to 17 h almost irrespective to pH.
Acknowledgements
Natural Sciences and Engineering Research Council of Canada (NSERC) and Canada Foundation for Innovation (CFI) support to M. A. Mateescu is gratefully acknowledged. Ph. D. graduate studentship from NSERC and Corealis Pharma Inc. (Montreal, Canada) awarded to M. Lemieux is also gratefully acknowledged. Special thanks are due to Dr. Roch Thibert for insightful comments on the manuscript and to F. Byette, R. Mineau and M. Preda for providing technical support for this work.
References
- 1.Siepmann J, Peppas NA. Hydrophilic matrices for controlled drug delivery: an improved mathematical model to predict the resulting drug release kinetics (the “sequential layer” model) Pharm Res. 2000;17:1290–1298. doi: 10.1023/A:1026455822595. [DOI] [PubMed] [Google Scholar]
- 2.Colombo P, Bettini R, Santi P, Peppas NA. Swellable matrices for controlled drug delivery: gel-layer behaviour, mechanisms and optimal performance. Pharm Sci Technol Today. 2000;3:198–204. doi: 10.1016/S1461-5347(00)00269-8. [DOI] [PubMed] [Google Scholar]
- 3.Brouillet F, Bataille B, Cartilier L. High-amylose sodium carboxymethyl starch matrices for oral, sustained drug-release: formulation aspects and in vitro drug-release evaluation. Int J Pharm. 2008;356:52–60. doi: 10.1016/j.ijpharm.2007.12.039. [DOI] [PubMed] [Google Scholar]
- 4.Zobel HF. Starch crystal transformations and their industrial importance. Starch - Stärke. 1988;40:1–7. doi: 10.1002/star.19880400102. [DOI] [Google Scholar]
- 5.Wurzburg OB. Introduction. In: Wurzburg OB, editor. Modified starches: properties and uses. Boca Raton: CRC; 1986. pp. 4–15. [Google Scholar]
- 6.Crochet P, Beauxis-Lagrave T, Noel TR, Parker R, Ring SG. Starch crystal solubility and starch granule gelatinisation. Carbohydr Res. 2005;340:107–113. doi: 10.1016/j.carres.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Ispas-Szabo P, De Koninck P, Calinescu C, Mateescu MA (2007) Novel carboxymethyl starch excipients for oral dosage forms. Transaction of the 34th Annual meeting and exposition of the Controlled Release Society; Long Beach, California
- 8.Calinescu C, Nadeau E, Mulhbacher J, Fairbrother JM, Mateescu MA. Carboxymethyl high amylose starch for F4 fimbriae gastro-resistant oral formulation. Int J Pharm. 2007;343:18–25. doi: 10.1016/j.ijpharm.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 9.Calinescu C, Mulhbacher J, Nadeau E, Fairbrother JM, Mateescu MA. Carboxymethyl high amylose starch (CM-HAS) as excipient for Escherichia coli oral formulations. Eur J Pharm Biopharm. 2005;60:53–60. doi: 10.1016/j.ejpb.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Calinescu C, Mateescu MA. Carboxymethyl high amylose starch: Chitosan self-stabilized matrix for probiotic colon delivery. Eur J Pharm Biopharm. 2008;70:582–589. doi: 10.1016/j.ejpb.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Heinze T, Koschella A. Carboxymethyl ethers of cellulose and starch - A review. Macromol Symp. 2005;223:13–40. doi: 10.1002/masy.200550502. [DOI] [Google Scholar]
- 12.Brittain HG, Bogdanowich SJ, Bugay DE, DeVincentis J, Lewen G, Newman AW. Physical characterization of pharmaceutical solids. Pharm Res. 1991;8:963–973. doi: 10.1023/A:1015888520352. [DOI] [PubMed] [Google Scholar]
- 13.Jamzad S, Tutunji L, Fassihi R. Analysis of macromolecular changes and drug release from hydrophilic matrix systems. Int J Pharm. 2005;292:75–85. doi: 10.1016/j.ijpharm.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 14.Korhonen O, Pohja S, Peltonen S, Suihko E, Vidgren M, Paronen P, et al. Effects of physical properties for starch acetate powders on tableting. AAPS PharmSciTech. 2002;3:1–9. doi: 10.1208/pt030434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ispas-Szabo P, Ravenelle F, Hassan I, Preda M, Mateescu MA. Structure-properties relationship in cross-linked high-amylose starch for use in controlled drug release. Carbohydr Res. 1999;323:163–175. doi: 10.1016/S0008-6215(99)00250-5. [DOI] [PubMed] [Google Scholar]
- 16.Mulhbacher J, Ispas-Szabo P, Lenaerts V, Mateescu MA. Cross-linked high amylose starch derivatives as matrices for controlled release of high drug loadings. J Control Release. 2001;76:51–58. doi: 10.1016/S0168-3659(01)00425-4. [DOI] [PubMed] [Google Scholar]
- 17.Stojanovic Z, Jeremic K, Jovanovic S, Lechner MD. A comparison of some methods for the determination of the degree of substitution of carboxymethyl starch. Starch - Stärke. 2005;57:79–83. doi: 10.1002/star.200400342. [DOI] [Google Scholar]
- 18.Chen J, Jane J. Preparation of granular cold-water-soluble starches by alcoholic-alkaline treatment. Cereal Chem. 1994;71:618–622. [Google Scholar]
- 19.Volkert B, Loth F, Lazik W, Engelhardt J. Highly substituted carboxymethyl starch. Starch - Stärke. 2004;56:307–314. doi: 10.1002/star.200300266. [DOI] [Google Scholar]
- 20.US Pharmacopeia XXXI. US Pharmacopeial Convention. Rockville, MD2008.
- 21.Carr RL. Classifying flow properties of solids. Chem Eng. 1965;72:69–72. [Google Scholar]
- 22.Hausner HH. Friction conditions in a mass of metal powders. Int J Powder Metall. 1967;3:7–13. [Google Scholar]
- 23.Takahashi H, Chen R, Okamoto H, Danjo K. Acetaminophen particle design usingchitosan and a spray-drying technique. Chem Pharm Bull. 2005;53:37–41. doi: 10.1248/cpb.53.37. [DOI] [PubMed] [Google Scholar]
- 24.Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Control Release. 1987;5:37–42. doi: 10.1016/0168-3659(87)90035-6. [DOI] [PubMed] [Google Scholar]
- 25.Cairns P, Bogracheva TY, Ring SG, Hedley CL, Morris VJ. Determination of the polymorphic composition of smooth pea starch. Carbohydr Polym. 1997;32:275–282. doi: 10.1016/S0144-8617(96)00115-4. [DOI] [Google Scholar]
- 26.Cheetham NWH, Tao L. Variation in crystalline type with amylose content in maize starch granules: an X-ray powder diffraction study. Carbohydr Polym. 1998;36:277–284. doi: 10.1016/S0144-8617(98)00007-1. [DOI] [Google Scholar]
- 27.Shiftan D, Ravenelle F, Mateescu MA, Marchessault RH. Change in the V/B polymorph ratio and T1 relaxation of epichlorohydrin crosslinked high amylose starch excipient. Starch - Stärke. 2000;52:186–195. doi: 10.1002/1521-379X(200007)52:6/7<186::AID-STAR186>3.0.CO;2-8. [DOI] [Google Scholar]
- 28.Fuhrer C. Interparticulate attraction mechanisms. In: Aldernborn G, Nystrom C, editors. Pharmaceutical powder compaction technology. New York: Marcel Dekker; 1996. pp. 1–15. [Google Scholar]
- 29.Desai DH, Patel KC, Patel RD. Thermal properties of amylose and its derivatives part I. Starch - Stärke. 1976;28:377–381. doi: 10.1002/star.19760281104. [DOI] [Google Scholar]
- 30.Daniel JR, Whistler RL, Röper H, Elvers B. Starch. In: Bohnet M, Bellussi G, Bus J, Cornils B, Drauz K, Greim H, et al., editors. Ullmann's Encyclopedia of Industrial Chemistry, Electronic release. 7th ed. Wiley-VCH Verlag GmbH & Co. 2007. Accessed 25 mar 2009.
- 31.Rowe RC, Sheskey PJ, Owen SC. Handbook of Pharmaceutical Excipients. 5. London: Pharmaceutical; 2006. [Google Scholar]
- 32.Adolfsson Å, Olsson H, Nyström C. Effect of particle size and compaction load on interparticulate bonding structure for some pharmaceutical materials studied by compaction and strength characterisation in butanol. Eur J Pharm Biopharm. 1997;44:243–251. doi: 10.1016/S0939-6411(97)00136-7. [DOI] [Google Scholar]
- 33.Garekani HA, Ford JL, Rubinstein MH, Rajabi-Siahboomi AR. Effect of compression force, compression speed, and particle size on the compression properties of paracetamol. Drug Dev Ind Pharm. 2001;27:935–942. doi: 10.1081/DDC-100107674. [DOI] [PubMed] [Google Scholar]