
Figure 6. The midazolam paradox.
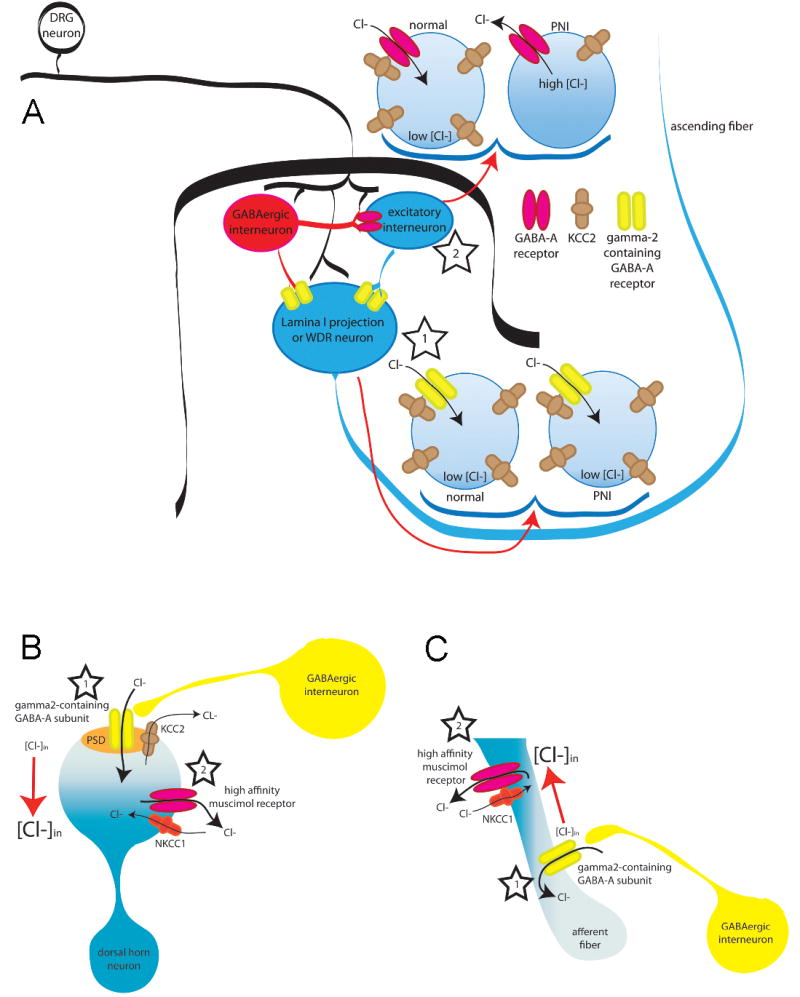
There are at least 3 models that could explain the apparent paradox associated with the continued analgesic efficacy of benzodiazepine receptor agonist in the face of a shift in the valence of GABAA receptor antagonist effects in the presence of persistent inflammation. All 3 require a functional separation between depolarizing shifts in Cl− equilibrium potential and γ2 containing GABAA receptors (i.e., those receptors responsive to benzodiazepines). Panel A: Separation in two subpopulations of dorsal horn neurons: In this model, γ2 containing GABAA receptors are enriched in a subpopulation of dorsal horn neurons critical for the establishment of nociceptive threshold and/or the perception of pain (i.e., dorsal horn projection neuron, star 1). There is no shift in Eanion in these neurons, so that GABAA receptor activation is inhibitory, and facilitation of this receptor activation by benzodiazepines is analgesic (star 1). The γ2-containing neuron is post-synaptic to both GABAergic input and excitatory interneurons. A depolarizing shift in Eanion in these excitatory interneurons results in a shift in the valence of GABAA signaling and a GABA-mediated facilitation of nociceptive behavior (star 2). The density of γ2-containing GABAA receptors in these interneurons would have to be relatively low such that benzodiazepines do not facilitate excitatory GABAergic transmission. The majority of lamina I–II neurons are interneurons (~90%) and these neurons may account for reduced KCC2 expression and excitatory synaptic GABAA responses (star 2) observed following peripheral nerve injury (PNI). Such a model could also account for changes observed in the presence of inflammation if the GABAA receptors on these excitatory interneurons have a higher affinity for muscimol. Panel B: Separation within a dorsal horn neuron: A differential distribution of γ2 containing GABAA receptors and cation-Cl− co-transporters within a dorsal horn neuron critical for nociceptive behavior and the perception of pain could underlie relatively localized changes in Eanion. In this model, there would be little or no change in Eanion around γ2 containing receptors following tissue injury (star 1), whereas a depolarizing shift in Eanion near receptors with a high affinity for muscimol would confer the facilitation of nociceptive transmission observed in the presence of persistent inflammation (star 2). Importantly, such a model is unlikely to account for changes observed following PNI given that a depolarizing shift in Eanion appears to impact the response to both exogenous and endogenous GABAA receptor activation, suggesting that the shift in Eanion is relatively uniform in these neurons. Panel C: Separation with the central terminals of primary afferent neurons. This model is analogous to that described for Panel B, except that the separation of γ2-containing receptors and depolarizing shifts in Eanion occur in the primary afferent. A subset of dorsal horn interneurons make axo-axonic synapses onto incoming afferent fibers (star 1). While the subcellular localization of GABAA subunits and/or NKCC1 is not known in these afferent axons, mechanisms involving the localization of midazolam-sensitive (γ2-containing) GABAA receptors coupled with differences in the localization of inflammation-induced plasticity in Cl− gradients (via changes in NKCC1 expression, localization or activity) could yield pharmacological profiles that account for the preservation of antinociceptive effects of midazolam observed in the presence of both inflammation and nerve injury (star 2).