

Abstract
Isoketals (IsoKs) are γ-ketoaldehydes formed via the isoprostane pathway of arachidonic acid peroxidation and are among the most reactive byproducts of lipid peroxidation. IsoKs selectively adduct to protein lysine residues and are highly cytotoxic, but the targets and molecular events involved in IsoK-induced cell death are poorly defined. Our previous work established that physiologically-relevant aldehydes induce mitochondrial dysfunction (Kristal, et. al., 1996). We therefore examined whether IsoKs induced mitochondrial dysfunction. Incubation of mitochondria with synthetic IsoKs in the presence or absence of Ca2+ was associated with alterations in mitochondrial respiration, membrane potential (ΔΨ), and pyridine nucleotide redox state. IsoKs dose-dependently (0.5–4 μM) accelerated liver mitochondria swelling induced by low concentrations of Ca2+ and Zn2+ or by the pro-oxidant tert-butylhydroperoxide, and release of cytochrome c, with similar observations in heart/brain mitochondria. The mitochondrial permeability transition (mPT) inhibitor cyclosporin A delayed IsoK-induced mitochondria dysfunction. The actions of IsoKs are consistent with interactions with cytochrome c, a protein rich in the lysine residues. Direct reaction of IsoKs with select lysines in cytochrome c was demonstrated using high resolution mass spectrometry. Overall, these results suggest that IsoKs may, in part, mediate their cytotoxic effects through induction of the mPT and subsequent activation of downstream cell death cascades.
Keywords: γ-ketoaldehydes, oxidative stress, lipid peroxidation, mitochondrial permeability transition, oxidative modification of cytochrome c, high resolution mass spectrometry
INTRODUCTION
Membrane lipid peroxidation plays a critical role in the propagation of oxidative damage and in cell death cascades, in part through the formation of reactive aldehydes [1]. These secondary products of lipid peroxidation, which include malondialdehyde, a potential cross-linking agent, and the reactive hydroxyalkenals, are known to contribute and partially mediate the effects of lipid peroxidation [1,2]. These reactive aldehydes can be derived from the most abundant class of polyunsaturated fatty acids in the membranes and can undergo a number of reactions with critical biomolecules – lipids, proteins, and DNA. Such reactions amplify and propagate the biochemical consequences of the lipid peroxidation reactions. Lipid peroxidation byproducts rapidly accumulate in tissues following a range of pathological conditions, including myocardial ischemia/reperfusion injury, arrhythmias, atherosclerosis, heart failure, chronic and acute neurodegeneration [3–11, 30,33,50,51].
More recent work has identified isoketals (IsoKs), a family of 64 regio- and stereo-isomers of levuglandin-like γ-ketoaldehydes, as some of the most reactive products of lipid peroxidation [12–16]. IsoKs are produced from the arachidonic acid via prostaglandin H2-like endoperoxide intermediates (H2-isoprostanes) that undergo concerted rearrangement to form γ-ketoaldehyde [12–16]. IsoKs react several orders of magnitude more rapidly than hydroxyalkenals with lysine residues of proteins and potently induce cross-links [16]. For this reason, IsoKs have been proposed as markers and mediators of oxidative damage [16–17]. Elevated levels of IsoKs in plasma and tissues have been observed in multiple pathological conditions, including end-stage renal disease, atherosclerosis, acute myocardial injury, hyperoxic injury, and allergic inflammation [16–19,33].
Although IsoKs are highly cytotoxic, the molecular targets and mechanisms by which these compounds induce cell death have not been fully elucidated. IsoKs react with lysine residues on proteins and induce cross-links at rates much faster than other lipid peroxidation byproducts, including hydroxyalkenals [16]. IsoK-adduction to proteins can alter protein function and IsoK- adducted proteins have also been shown to be poorly degraded by the proteasome and inhibit proteasome function [20,21].
Previous work from our group has established that other physiologically-relevant aldehydes can induce mitochondrial dysfunction [22–24]. The byproducts of norepinephrine and dopamine metabolism by monoamine oxidase - 3,4-dihydroxyphenylglycolaldehyde (DOPEGAL) and 3,4-dihydroxyphenylacetaldehyde (DOPAL), respectively, accelerate induction of the mitochondrial permeability transition (mPT), an event that sits directly upstream of some cell death cascades [25–28]. DOPAL was shown to be active in both isolated mitochondria and cultured cells. These aldehydes have been implicated in neurodegenerative disorders [29]. Furthermore, lipid peroxidation-derived aldehydes including hydroxyhexenal, hydroxynonenal, and acrolein (acrylaldehyde) also accelerate the mPT [22–24]. Hydroxyhexenal is the most potent yet discovered, with observable effects beginning at femtomolar concentrations [22]. These aldehydes primarily react with cellular thiols, including cysteine residues and gluthathione, via Michael addition, whereas IsoKs primarily react with lysine residues through pyrrole intermediates. We therefore tested whether the more reactive, lysine-targeted aldehydes, IsoKs, could also induce mitochondrial dysfunction.
We now demonstrate that IsoKs are potent mediators of mitochondrial dysfunction. Our results show that IsoKs are a previously unrecognized class of lipid peroxidation byproducts that mediate mitochondrial dysfunction and may thus contribute to disease and pathology.
MATERIAL AND METHODS
Chemicals
Tetramethylrhodamine methyl ester (TMRM) and Ca-Green-5N were purchased from Invitrogen, Inc. (Carlsbad, CA). Rat/mouse cytochrome c immunoassay kit “Quantikine®” was purchased from R&D Systems, Inc. (Minneapolis, MN). All other chemicals except IsoKs were purchased from Sigma-Aldrich Company (St. Louis, MO).
Synthesis of isoketals
There are eight regioisomers of IsoKs, and as each IsoK regioisomer has 3 stereocenters and therefore 8 stereoisomers, there are 64 total IsoK isomers. To test the effect of IsoKs, we synthesized one regioisomer of IsoK, 15-E2-IsoK, from its stable dimethoxyacetal precursor by incubation with Mont K-10 to convert it to the active aldehyde as previously described [15]. This synthesis procedure generates the eight stereoisomers of 15-E2-IsoK in approximately equal abundance. The concentration of total IsoK isomers generated was quantified using Ehrlich’s reagent and a 10 mM stock stored in DMSO at −20°C until use.
For the mitochondrial experiments, we attempted to approximate physiological levels by using concentrations of IsoKs that generated equivalent levels of IsoK protein adducts when added to cultured cell as the levels of IsoK protein adducts found in vivo [53].
Isolation of liver, heart and brain mitochondria
Liver mitochondria were isolated from ~6 month old male Fisher 344 x Brown Norway F1 rats by the standard differential centrifugation method in sucrose-based buffers as described and as used previously in our lab [34]. Liver isolation buffer contains 0.25 M sucrose, 10 mM HEPES, 1 mM EGTA, 0.5% bovine serum albumin (BSA).
Hearts were rapidly removed from sacrificed ~ 6 month old rats and placed in ice cold buffer (220 mM mannitol, 70 mM sucrose, 10 mM HEPES, pH 7.4, 1 mM EGTA, 0.05% BSA) [35,36]. Hearts were washed to remove blood, minced with a press, and homogenized with a polytron. The homogenate was centrifuged at 1000 x g for 10 min. Supernatants were removed and centrifuged at 10,000 x g for 10 min. Pellets were washed twice in the buffer and centrifuged at 10,000 x g to repellet. Following the final wash, mitochondria were resuspended in buffer without EGTA. Described technique allowed us to isolate subsarcolemmal fraction of heart mitochondria [36].
Isolation of nonsynaptosomal rat brain mitochondria from ~ 2–3 month old male Fisher 344 x Brown Norway F1 rats was achieved using a discontinuous Ficoll gradient according to the commonly used method of Lai and Clark [37], with slight modifications as previously described [38]. Mitochondrial protein concentration was determined by the Lowry method using BSA as a standard [39].
Mitochondrial respiratory assays
Mitochondrial respiration rates were measured using a Clark electrode and a Strathkelvin Oxygraph or Oroboros Systems, as described [40]. Briefly, isolated liver mitochondria (0.25 mg/ml) were added to buffer A (250 mM sucrose, 10 mM HEPES, pH 7.4, 2 mM KH2PO4) containing the complex I substrate glutamate/malate (5 mM) or the complex II substrate succinate (5 mM) and rotenone (1 μM). Respiration in the presence of these substrates only corresponds to state 2 respiration (V2). Addition of ADP (200 μM) initiated ATP synthesis coupled to proton reentry across the membrane, which corresponds to state 3 (V3). ADP exhaustion led to a reduction of the respiratory rate and corresponds to state 4 (V4). Dinitrophenol (DNP) (20 μM) was added after state 4 respiration had proceeded for at least 2 min. The respiratory control ratio (RCR) was calculated as the ratio between rates of respiration in state 3 and 4.
Measurement of mitochondrial Ca2+ uptake capacity, membrane potential, NAD(P)H oxidation, and swelling
Liver and heart mitochondria were incubated in buffer containing 5 mM succinate or glutamate/malate. Liver mitochondria were used at a concentration of 0.25 mg/ml; heart mitochondria were used at 0.15 mg/ml. Brain mitochondria (0.15–0.2 mg/ml) were incubated in buffer containing 100 mM sucrose, 65 mM KCl, 10 mM HEPES, pH 7.4, 2 mM KH2PO4, 150 μM ATP, 150 μM MgCl2, 3 μM EDTA, and 5 mM glutamate/malate.). Mitochondria were challenged to single or sequential Ca2+ additions. For liver mitochondria, each addition was ~30 nmol Ca2+/mg mitochondrial protein; for heart ~34 nmol Ca2+/mg protein; and for brain ~50 nmol Ca2+/mg mitochondrial protein.
All fluorescence measurements were performed simultaneously on a multichannel fluorimeter (C&L Instruments, Inc., www.fluorescence.com) as described in [41]. Mitochondrial membrane potential (ΔΨ) was estimated as the change in TMRM (60 nM) fluorescence intensity (λex= 543 nm and λem = 590 nm). TMRM was used at a concentration sufficient to cause self-quenching of the fluorescence in the mitochondrial matrix. TMRM as a rhodamine 123 derivative can accumulate in mitochondria to levels that exceed those predicted by the Nernst equation (42). Excess accumulation may be due to fluorophore stacking and formation of aggregates. Formation of aggregates causes fluorescence quenching and a red shift of absorbance. In the experimental design we used (cuvette and multiwell assays), decreased fluorescence due to quenching signifies an increase of Δϕm, and an increase in TMRM fluorescence corresponds to depolarization of mitochondria (43). Mitochondrial Ca2+ uptake and release capacities were measured as changes in CaGreen-5N (125 nM) fluorescence intensity. (λex= 482 and λem = 535 nm). The redox state of pyridine nucleotides were measured as the NAD(P)H autofluorescence (λex= 350 nm and λem = 450 nm). Mitochondrial swelling was measured as a function of light scattering at excitation and emission wavelengths of 587 nm or by a standard spectroscopic assay at 540 nm.
Cytochrome c release was measured using immunoassay kit Quantikine® (R&D Systems, Inc). Aliquots for detection of release of cytochrome c were taken every 3 min after Ca2+ addition in parallel with the measurement of other parameters. Mitochondrial samples (50 μl) were placed into 150 μl buffer containing 125 mM KCL, 10 mM HEPES, pH 7.4 and 15 μl Protease Inhibitor Cocktail (Sigma-Aldrich). Samples were then centrifuged for 2 min at 14,000 x g. The pellet and supernatant were separated and stored at −80 °C as described [44] for further analysis.
Kinetic model of mPT in populations of isolated mitochondria
Kinetic analysis was performed using a recently introduced kinetic model [41]. Intensity of Ca-Green-5N fluorescence is modulated by Ca2+. Since the dye is nonpermeable, changes in fluorescence intensity reflect changes in concentration of extramitochondrial Ca2+. Thus, the concentration of Ca2+ in the assay medium was determined according to a calibration curve. The calibration plot of intensity of CaGreen-5N fluorescence vs. Ca2+ concentration was done as described previously in [41]. The experimental data on Ca2+ -induced mPT were fitted into this model using numerical fitting routines implemented into Matlab (Mathworks, www.mathworks.com). Such fitting parameters as k1, k2′ and n were found using Levenberg–Marquardt algorithm and quality of fitting determined as adjusted R2. In all the cases R2 was >0.97. The model presented below functionally resolves possible sites of action for different mPT modulators:
Here [MH-(Ca2+)i]A represents the population of active, unswollen mitochondria ([MH]A) and i represents the number of Ca2+ ions already adsorbed by a given mitochondrion. Ca2+ absorption occurs during the k1 step shown in the scheme as formation of [MH-(Ca2+)i+1]A, and it initiates the mPT process that we describe as a sequence of two steps. The kinetic parameter k2 represents the rate of formation of the intermediate state [MH]I. k2 is a complex function of the number of Ca2+ ions absorbed by the mitochondria, , where [Ca2+ is the concentration of Ca2+ that has been absorbed by mitochondria. Thus, the ratio [Ca2+]M/[MH]A is essentially the average number of Ca2+ ions absorbed per active mitochondrion. The parameter n is an apparent order of the k2 step with respect to Ca2+ and k2′ is a reaction constant of this step. The kp step is fast and leads to the formation of a population of inactive (swollen) mitochondria, represented as [MH]N, with all Ca2+ released back to the medium. [Ca2+]out is the concentration of free Ca2+ in the medium, available for absorption by mitochondria.
Mass spectrometry
The procedure for modification of cytochrome c was adapted from Isom et al. [48]. Briefly, bovine heart cytochrome c (2 mg/ml) was incubated with IsoKs (4 μM) in a buffer containing 250 mM sucrose, 10mM HEPES, pH 7.4 at 37°C for 2 h. The control sample was treated with vehicle (DMSO). The reaction was terminated by the addition of 25 μl of 1% formic acid. Samples were then desalted by ultrafiltration using Amicon Ultra-4 devices with a 10k molecular weight cutoff (Millipore Corp.). The desalted samples were reconstituted into a 1:1 acetonitrile:water solution containing 0.2% formic acid. Measurements were performed on an LTQ-Orbitrap mass spectrometer (MS) (Thermo Electron) in the positive ion mode. Samples were introduced into the MS by direct infusion with a flow rate of 5 μl/min. MS2 fragments of the unmodified and modified cytochrome c were produced by collision-induced dissociation (CID) in the LTQ with an isolation width of 6 m/z and normalized collision energy of 35%. Both the full scan and MS2 were acquired in the orbitrap at a resolving power of 60,000 and 30,000, respectively. The orbitrap was externally mass calibrated immediately before the analyses. Scans were collected with automated gain control settings as 2×106 for full scan and 2×105 for MS2, and microscan settings as 5 and 2, respectively. To obtain the exact mass of the intact protein, Xtract software from Thermo was used to convert the m/z values of the multiply charged ions to the molecular masses of the uncharged molecular ions.
Statistical analysis
The statistical significance between population means was estimated using two-sample t-test in OriginPro v.8 (OriginLab Corp., Northhampton, MA). Differences were considered statistically significant when p < 0.05.
RESULTS
IsoKs impair mitochondrial calcium metabolism and accelerate large amplitude swelling
IsoKs (0.5–4 μM) dose-dependently accelerated swelling of rat liver mitochondria as much as 75% as compared with 5 μM Ca2+ alone (Fig. 1A, section d and Fig. 1B). Rat liver mitochondria were chosen for the initial studies as they are the best characterized system with which to study the effect of different agents on mitochondrial functions, especially with respect to the mPT. IsoK-induced mitochondrial swelling was also associated with (i) a dose-dependent decrease in ΔΨ (Fig. 1A, section a); (ii) an accelerated oxidation of NAD(P)H to NAD(P)+ after Ca2+ addition (Fig. 1A, section c); and (iii) a reduction of Ca2+ retention time measured as the time between Ca2+ addition and Ca2+-induced Ca2+ release (Fig. 1A, section b and 1C). Extended dose-response analysis (from 0.1 nM to 40 μM IsoKs) revealed a sigmoidal effect curve between 0.5 to 20 μM IsoKs (Fig. 1B). The maximal effect on mitochondrial swelling was observed in the presence of 10–20 μM IsoKs. The half maximal effect was observed at 1.56±0.16 μM. IsoKs induced visible mitochondrial precipitation at 40 μM and above.
Fig. 1.

IsoKs induce concentration-dependent dysfunction in liver mitochondria. A. Basic mitochondrial parameters were measured in the presence of Ca2+ either alone or in the presence of IsoK (0.5–4 μM). Section a. Mitochondrial ΔΨ was measured as changes in TMRM fluorescence signal Addition of mitochondria into incubation buffer, indicated by arrow, induced the drop of TMRM signal (i.e., TMRM concentration in the buffer) due to the accumulation of the dye by mitochondria. The addition of Ca2+ induced an increase of TMRM signal (TMRM release from mitochondria), which corresponds to lower ΔΨ, and a subsequent decrease of the TMRM signal, which indicates a restoration of ΔΨ. The final gradual increase of TMRM signal corresponds to gradual loss of ΔΨ due to Ca2+ induced damage to mitochondria. Section b. Ca2+ concentration in the mitochondrial suspension was measured as Ca-Green 5N fluorescence signal. A higher signal corresponds to an increase of Ca2+ concentration in the buffer. Addition of Ca2+ into incubation buffer induced an increase of Ca-Green 5N fluorescent signal following by its decrease due to the uptake of Ca2+ by mitochondria, and gradual increase of Ca-Green 5N signal due to Ca2+ release from mitochondria. Section c. NAD(P)H autofluorescence signal. A lower signal corresponds to a more oxidized state of NAD(P)H. The addition of Ca2+ to mitochondria was associated with gradual decrease in NAD(P)H autofluorescence. Section d. Mitochondrial swelling was measured as changes in light scattering of the mitochondrial suspension. A decrease in the fluorescence signal indicates an increase in swelling. Mitochondria were incubated with succinate (5 mM) under conditions as noted in the Experimental Procedures. Spikes resulting from additions of Ca2+ and IsoKs to the assay chamber and from removing an aliquot for cytochrome c detection were manually reduced; arrows are used to indicate where these manipulations occurred. Applied spike reduction did not affect data analysis. Representative traces from 6 experiments are shown. B. An extended concentration-response curve for the swelling response, as determined by following the absorbance signal. Mitochondria (1mg/ml) were treated with IsoKs (0.1–100 μM) in the presence of Ca2+ (5 μM). IsoKs concentrations above 40 μM are not plotted as they appear to precipitate mitochondria. Data were calculated as the integrated area under the swelling curve normalized to the control. A sample without IsoKs was used as the control. Data were expressed in arbitrary units and plotted as the mean ± sem, N=7, *, p<0.05. C. Calcium retention time is plotted as a function of IsoKs concentration. Retention time is defined as the time between Ca2+ addition and complete Ca2+ release. Data were plotted as the mean ± sem, N=6, *, p<0.05.
IsoKs were found to induce dysfunction in brain (Fig. 2A and B) and heart (see supplemental) mitochondria similar to that observed in liver mitochondria. Brain mitochondria were challenged with sequential Ca2+ additions until spontaneous Ca2+ release from mitochondria occurred (Fig. 2A, section b), which in turn was associated with swelling (Fig. 2A, section d), decreased ΔΨ (Fig. 2A, section a), and oxidation of pyridine nucleotides (Fig. 2A, section c). Concentration dependence was qualitatively and quantitatively similar on a μM/mg of mitochondria basis for brain and heart mitochondria.
Fig. 2.

IsoKs induce concentration-dependent dysfunction in nonsynaptosomal brain mitochondria. Mitochondria were challenged with sequential Ca2+ additions (5 μM each) in the presence of IsoKs (0.5–2 μM, lines 2–5) until Ca2+ was spontaneously released. Mitochondria were incubated with glutamate/malate (5 mM). To induce complete swelling a non specific pore forming agent, alamethicin (Ala) was added at the end of each incubation. Sections are the same as in Fig. 1. Representative traces from 6 experiments are shown. B. Dependence of brain mitochondria Ca2+ retention capacity on IsoKs concentration. Data were plotted as the mean ± sem, N=3, *, p<0.05.
IsoKs induce mitochondrial dysfunction in the absence of endogenous Ca2+
Mitochondria were incubated with IsoKs (4 μM) in the presence of EDTA (50 μM) to eliminate potential effects of low concentrations of Ca2+ or other divalent cations in the buffer. Under these conditions, IsoKs behaved as weak mitochondrial toxins, initiating swelling and other swelling-associated dysfunction only after 40 min incubation (Fig. 3A, data can be compared with effects at 10–15 minutes when Ca2+ is present). Decreases in ΔΨ and oxidation of pyridine nucleotides also occurred slowly. Similar observations were made in isolated brain and heart mitochondria (data not shown). To determine whether IsoKs action might involve endogenous calcium stores, liver mitochondria were incubated with IsoK in the presence of the Ca2+ ionophore A23187 (0.5 μg/ml) and EDTA (50 μM). IsoKs were observed to induce slowly evolving dysfunction similar to that described above (Fig. 3B), suggesting that in the absence of another inducer of mPT, such as Ca2+, IsoKs are weak inducers.
Fig. 3.

IsoKs compromise liver mitochondria function in the absence of Ca2+. A. Basic mitochondrial parameters were measured in the presence of IsoKs (4 μM). Section a. Mitochondrial ΔΨ was measured as changes in the TMRM fluorescence signal, as described in Fig. 1. Section b. NAD(P)H autofluorescence signal. Section c. Mitochondrial swelling was measured as changes in the fluorescence of the mitochondrial suspension. B. The mitochondrial parameters in the presence of IsoKs (4 μM), ionophore A 23187 (0.5 μg/ml), and EDTA (50 μM). Sections are the same as in panel A. Representative traces from 3 experiments are shown.
Classical inhibitors and co-inducers provide evidence that IsoK-induced mitochondrial dysfunction results from induction of the mPT
The mPT has been largely defined and investigated in isolated liver mitochondria. Therefore, we used this model to test the hypothesis that IsoKs mediate mitochondrial dysfunction, at least in part, by facilitating induction of the mPT. Addition of the mPT inhibitor cyclosporin A (CsA) delayed IsoK-mediated mitochondrial swelling and prevented loss of ΔΨ, Ca2+ release from mitochondria, and the oxidation of pyridine nucleotides (Fig. 4). Two other classical mPT inhibitors, ADP in the presence of oligomycin and Mg2+, exerted similar protection (data not shown). Two FDA-approved tricyclic drugs, promethazine and nortriptyline, recently shown in our lab to be mPT inhibitors [25,34] also provided protection in liver (data not shown) and brain mitochondria (see supplemental). Surprisingly, these drugs were not effective against the mPT in heart mitochondria (data not shown).
Fig. 4.

Cyclosporin A delays IsoK-mediated mitochondrial dysfunction in liver tissue. Mitochondrial parameters were measured in the presence of Ca2+ (5 μM) and IsoKs (2 μM) in the presence or absence of cyclosporin A (0.5 μM, line 2). Sections are the same as on Fig. 1. Representative traces from 3 experiments are shown.
Conversely, IsoKs accelerated tert-butylhydroperoxide (tBH)-induced swelling of mitochondria (Fig. 5A and B). Similar effects were observed with Zn2+, which has been more recently described as an mPT inducer [49,50]. The dose dependence for co-induction by IsoKs with tBH and Zn2+ had the same pattern as observed for Ca2+. The half maximal effect of IsoKs was observed at 2.97 ±0.25 and 1.85±0.12 μM for Zn2+ and tBH, respectively.
Fig. 5.

Dependence of Zn2+- and tBH-induced swelling of liver mitochondria on the presence of IsoKs. A. An extended concentration-response curve for the swelling response, as determined by following the absorbance signal. Mitochondria were treated with IsoKs (0.1–100 μM) in the presence of Zn2+ (2 μM). IsoKs concentrations above 40 μM are not plotted as they appear to precipitate mitochondria. Data were calculated as the integrated area under the swelling curve normalized to the control. A sample without IsoKs was used as the control. Data were expressed in arbitrary units and plotted as the mean ± sem, N=5, *, p < 0.05. B. An extended concentration-response curve for the swelling response, as determined by following the absorbance signal. Mitochondria were treated with IsoKs (0.1–100 μM) in the presence of tBH (100 μM) and Ca2+ (2 μM). IsoKs concentrations above 40 μM are not plotted as they appear to precipitate mitochondria. Data were calculated as described above and plotted as the mean ± sem, N=3, *, p < 0.05 compare with IsoKs untreated mitochondria.
IsoKs effects on mitochondrial calcium dysregulation might be attributed to impairment of the mitochondrial Ca2+ transport system (which mediates the uptake, release, and intra-mitochondrial sequestration of Ca2+). A kinetic analysis approach that functionally distinguishes possible sites of action for mPT modulators was used to further localize the functional effects of IsoKs on the calcium system. Analysis revealed that IsoK (0.5–4 μM) did not affect the kinetic parameter k1, which directly relates to the rate of the net Ca2+ uptake (Fig. 6A). This result suggests that IsoKs do not affect the Ca2+-uniporter system. In contrast, IsoK-mediated acceleration of Ca2+-induced mPT was associated with an increase of parameter k2. Kinetic parameter k2 is related to the ability of the Ca2+ sequestered by mitochondria to induce the mPT, and is proportionally dependent on the reaction constant k2′ (see equation in “Experimental Procedures”). IsoK-mediated increase in parameters k2 (Fig. 6C) through the parameter k2′, thus suggests an increase in [Ca2+] available for mPT induction. IsoKs did not affect kinetic parameter n (Fig. 6B), which appears to relate to the number of Ca2+ ions needed to initiate the mPT [41].
Fig. 6.

IsoKs accelerate the rate of the mPT induction but not the order of the reaction. A. Dependence of k1 (the Ca2+ uptake rate) on the presence of IsoKs. B. Dependence of n (the reaction order) on the presence of IsoKs. C. Dependence of k2′ (rate of the mPT induction) on the presence of IsoKs. The kinetic analysis was performed using a recently introduced kinetic model (54). The model functionally resolves possible sites of action for different mPT modulators.
where [MH-(Ca2+)i]A represents the population of active, unswollen mitochondria ([MH]A), i being the number of Ca2+ already adsorbed by a given mitochondria. Ca2+ uptake occurs during the k1 step shown in scheme as formation of [MH-(Ca2+)i+1]A, and it initiates the mPT process that we describe as a sequence of two steps. The k2 step is rate-limiting step of the mPT induction that results in the formation of the intermediate state [MH]I and depends on the reaction constant k2′. The next step (kp) leads to the formation of a population of inactive (swollen) mitochondria, represented as [MH]N, with all Ca2+ released back to the medium. [Ca2+]out is the concentration of free Ca2+ in the medium, available for absorption by mitochondria. Data were plotted as the mean ± sem, N=3, *, p < 0.05 compare with IsoKs untreated mitochondria.
IsoKs also facilitate release of cytochrome c from mitochondria through an mPT dependent pathway
Induction of an mPT is also generally associated with release of cytochrome c, which can then activate downstream stages of cell death. To determine if IsoKs could also accelerate cytochrome c release, mitochondria were incubated with 0.5–4 μM IsoKs added 1 min prior to 5 μM Ca2+, and the extent of cytochrome c release determined. Mitochondria treated with IsoKs released cytochrome c faster than mitochondria incubated with Ca2+ alone. Samples treated with 2–4 μM IsoKs (Fig. 7, samples 4,5) underwent observable release of cytochrome c within 2 min after Ca2+ addition (Fig. 7C). Release of cytochrome c preceded complete swelling of mitochondria (Fig. 7D), and was inhibited by inhibitors of the mPT.
Fig. 7.

IsoKs accelerate cytochrome c release from liver mitochondria. A. Changes in the absorbance of a mitochondrial suspension treated with Ca2+, either in the absence (line 1) or presence of IsoKs (0.5–4 μM), (lines 2–5). Samples (50 μL) were taken every 3 min before or after Ca2+ addition. Cytochrome c concentration was measured by immunoassay kit. B. Dependence of the time of maximum cytochrome c release on the presence of IsoKs. C. Cytochrome c was released into the medium after the addition of Ca2+ and IsoKs. D. Relationship between the concentration of cytochrome c released into the medium and mitochondrial swelling. Data were plotted as the mean ± sem, N=3, *, p < 0.05 compare with IsoKs untreated mitochondria.
Modification of mitochondrial respiration is a potential target for IsoKs
We further examined the effect of IsoKs on mitochondrial respiration to provide insight into potential intra-mitochondrial targets of IsoKs and estimate whether IsoKs affect mitochondrial respiration with the same efficacy as they act on mitochondrial parameters tested above. The effects of IsoKs on mitochondrial respiratory parameters were measured in different metabolic states [state 2 (V2), state 3 (V3), state 4 (V4), and DNP-stimulated respiration (VDNP)]. Glutamate/malate was used to stimulate respiration initiated at complex I and succinate and rotenone were used to stimulate respiration initiated at complex II. Results observed were qualitatively similar for both substrates (Fig. 8A, data for succinate/rotenone not shown). IsoKs dose-dependently impaired respiration in active states, including state 3 (coupled to ADP phosphorylation, V3) and DNP-stimulated respiration (VDNP), suggesting a direct effect of IsoKs on the portion of the electron transport chain between the Q-cycle and complex IV. In contrast, IsoKs dose-dependently accelerated respiration in resting states, including state 2 (before ADP addition, V2) and state 4 (after ADP phosphorylation, (V4), suggesting an increase in futile cycling of calcium or a generalized proton leak. The RCR, measured as the ratio between rates of respiration in state 3 and 4 (V3/V4), was thus dose-dependently reduced by IsoKs with a maximum effect at 4 μM for both substrates (Fig. 8B).
Fig. 8.

IsoKs affect mitochondrial respiratory parameters. Mitochondrial respiration was measured in the presence of IsoKs (0.5–4 μM). A. Isolated liver mitochondria (0.25 mg/ml) were supplemented with the complex I substrate glutamate/malate (5 mM). The incubation buffer also contained 2 mM KH2PO4, 3 μM EDTA. The rate of mitochondrial respiration in the presence of substrates corresponds only to state 2 (V2). Addition of ADP (200 μM) initiated ATP synthesis coupled to proton reentry across the membrane, which corresponds to state 3 (VADP or V3). ADP exhaustion led to the reduction of respiratory rate and corresponds to state 4 (V4). Addition of DNP (20 μM) is followed by state 4. Data are expressed as a percentage change of respiration parameters in the presence of IsoKs compare with IsoKs untreated mitochondria and plotted as a mean ± sem, N=3, *, p < 0.05. B. RCR was calculated as the ratio between the rates of respiration in state 3 and 4. Data plotted as a mean ± sem, N=3, *, p < 0.05 compare with IsoKs untreated mitochondria.
Therefore, our data indicated that IsoKs affect mitochondrial respiration with the same efficacy as they act on mitochondrial, Δψ, redox state of pyridine nucleotides, swelling and cytochrome c release, suggesting a possible target within respiratory chain as well. It is widely accepted that mPT induction is associated with an increase of the respiration rate while we observed quite opposite effect - the inhibition of ADP or DNP-stimulated respiration. This might suggest the uniqueness of IsoKs as mPT inducers, and represent an additional evidence of Isok-induced impairment of the respiratory chain.
Exogenously added cytochrome c restores affected by IsoKs and Ca2+ mitochondrial respiration
The respiration data described above suggest that there is an effect of IsoKs on the portion of the electron transport chain between the Q-cycle and complex IV Cytochrome c sits in this portion of the respiratory chain, has 19 lysine residues and one of these lysines has been previously shown to be modified by hydroxynonenal [59]. We hypothesized that cytochrome c might be one of the possible targets of IsoKs within mitochondria. Specifically, we hypothesized that IsoKs modified cytochrome c, reducing its affinity for the electron transport chain complex, leading to its release from the complex and thereby disrupting of respiration. To test our hypothesis, we measured mitochondrial respiration under conditions when mitochondria undergo mPT, e.g., in the presence of IsoKs (4 μM) and Ca2+ (20μM) (Fig. 9). As in the above experiments, this treatment was associated with an impairment of maximal respiration. The addition of unmodified cytochrome c essentially restored maximal respiration, consistent with our working hypothesis that it is the modification of cytochrome c that is inhibiting maximal respiration.
Fig. 9.

Addition of exogenous cytochrome c restores mitochondrial respiration affected by IsoKs and Ca2+. Line 1 represents mitochondrial respiration measured in the presence of Ca2+ (20 μM), line 2 – in the presence of IsoKs (4 μM) and Ca2+, line 3 - in the presence of IsoKs (4 μM) and Ca2+ and exogenously added cytochrome c (10 μM). Experimental conditions were the same as on Fig. 1. Representative traces from 3 experiments are shown.
Modification of cytochrome c by IsoKs
To further explore our working hypothesis, we used high resolution mass spectrometry to examine whether IsoKs could adduct with cytochrome c under our assay conditions (i.e., incubation of purified cytochrome c with IsoKs), and whether this in vitro modification would be site-specific. If IsoK-induced modification of cytochrome c is responsible for its release, then IsoKs would be expected to modify lysine residues that are important for the interaction of cytochrome c with cardiolipin. Full scans of intact cytochrome c after incubation with and without IsoKs were acquired in the orbitrap (Fig. 10A and B), and the results show that IsoK-modified cytochrome c is formed (as indicated by the “*” marks in Fig. 10B). The molecular masses of unmodified and modified cytochrome c were determined by Xtract software as 12222.3359 and 12520.5110, respectively. This mass shift of +298.1751 (Fig. 10C) corresponds to the anhydro pyrrole form of IsoKs (+298.1933) with a measured mass error of 1.45 ppm. The structures and exact masses of the known forms of IsoK-lysyl adducts are shown in Fig. 10D as follows from Davies et al. [16].
Fig. 10.
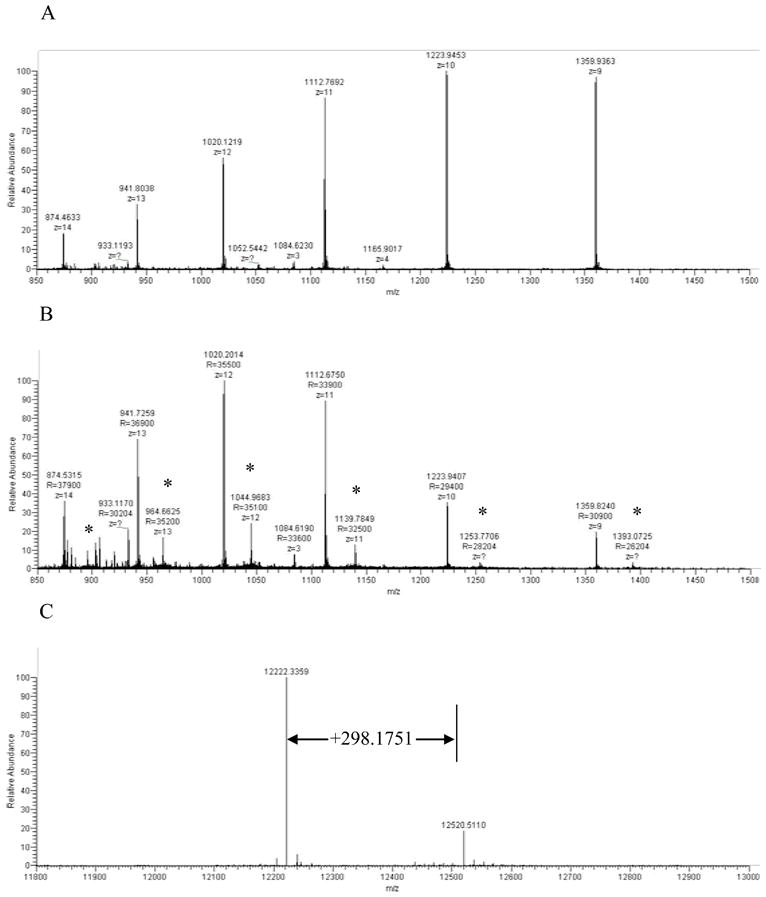

High resolution mass spectrometry reveals IsoKs adducts on intact cytochrome c. Full scans of intact cytochrome c after incubating with and without pre-incubations with IsoKs. A. The full scan of the cytochrome c control sample (containing no IsoKs). B. The full scan of the cytochrome c sample preincubated with IsoKs. Peaks labeled with ‘*’ represent the IsoK-modified cytochrome c. Representative traces from 3 experiments are shown. C. The result of B analyzed by Xtract software. The masses shown in C are uncharged molecular masses. D. Structures and masses of known IsoK-lysyl adducts.
MS2 spectra generated by selecting the most abundant charge states of unmodified and IsoK-modified cytochrome c to CID fragmentation provide clues to the possible locations of the IsoK-lysyl-adduct. Theoretical fragmentation (b and y) ions of cytochrome c were generated by using the Protein Prospector MS-Product tool (http://prospector.ucsf.edu/cgi-bin/msform.cgi?form=msproduct), and the measured b and y ions were compared to the theoretical b and y ions to reveal the location of modification. Bovine cytochrome c has the sequence GDVEKGKKIFVQKCAQCHTVEKGGKHKTGPNLHGLFGRKTGQAPGFSYTDANKNKGITWGEETLMEYLENPKKYIPGTKMIFAGIKKKGEREDLIAYLKKATNE as obtained from the NCBI protein database (Locus# ABA06541), which has a theoretical molecular mass of 11565.0280. When this theoretical mass and the measured mass (12222.3359) are compared, there is a mass discrepancy of 657.3079, which corresponds to the N-acetyl group (42.0106) and the heme group (615.2973) of cytochrome c. The N-terminal acetylation of cytochrome c has been well documented in the literature, and the mass of the heme group agrees with the mass (615±0.4) reported elsewhere [59,60]. The MS2 fragmentation of the unmodified cytochrome c generated a series of y ions such that their masses are in agreement with that of the theoretical y ions. However, all of the b ions measured (from b21 to b62) have a mass shift of 657.1779±0.0044 (see supplemental, Table 1), which is likely due to the fact that the heme group is bound to cytochrome c by three covalent linkages to Cys14, Cys17, and His18 [61,62]. The MS2 spectra of the IsoK-modified cytochrome c contain a series of y ions, the masses of which again agree with those of the theoretical y ions, but the b ions (b21to b50) have an additional mass shift of +298.1933 (see supplemental, Table 2), indicating that IsoKs bind to the region spanning amino acid residues 1–20. There are four lysine residues in this region, namely Lys5, 7, 8, and 13. This region, particularly the section spanning residues 1–12, appears to be on the surface of the molecule based on the reported three-dimensional structure of cytochrome c determined by X-ray crystallography [61,63].
DISCUSSION
Enhanced production of isoprostanes has been shown to be associated with a broad range of pathological conditions, including ischemia/reperfusion injury, diabetes, inflammatory vascular diseases, atherosclerosis, and chronic neurodegenerative conditions [14,16–19,30,51,52]. Since IsoKs are formed as products of the isoprostane pathway, some of the injurious effects in these conditions may in part be due to the formation of IsoKs owing to their ability to irreversibly adduct to and crosslink proteins. While the pathobiology of IsoKs is likely quite complex, in these studies we focused on the effects of IsoKs on mitochondrial function as mitochondrial dysfunction is a common feature of many disorders involving oxidative damage.
IsoKs – evidence for action at the level of the mPT
Mitochondria are well established as both a major source and a major target for free radicals and reactive species. Studies noted in the introduction describe the effects of other aldehydes on mitochondrial dysfunction, including induction of the mPT. The sensitivity of IsoK-mediated dysfunction to the classical mPT inhibitor cyclosporin A suggests the involvement of an mPT-dependent mechanism [54]. This conclusion was strengthened by the data obtained with other well established mPT inhibitors that act via different mechanisms, ADP and Mg2+, as well as nortriptyline and promethazine [34]. Although it is difficult to compare concentrations across studies from different labs and different systems, we note that the concentrations that accelerate mPT induction are comparable to those that inhibit the Na+ channel [53]. Whether induction of mitochondrial dysfunction by IsoKs could potentially contribute to IsoK-induced Na+ channel dysfunction is unknown.
IsoKs as probes of the mPT
Mitochondrial calcium metabolism ranges from physiological responses to stimuli to induction of the mPT by pathophysiological levels of Ca2+. The ability of mitochondria to take up and retain calcium is in close functional relationship with all-important mitochondrial functions such as maintenance of ΔΨ and the redox state of pyridine nucleotides [54,55]. IsoKs dose-dependently reduced mitochondrial ability to maintain ΔΨ, thus compromising their Ca2+ transporting function and redox state. Compromising the redox state of pyridine nucleotides has also long been known to increase mitochondrial sensitivity to other inducers, like Ca2+, and to promote mPT induction [54–58].
Maintaining ΔΨ, in turn, directly depends on respiratory chain function, and the results of the respiration studies are consistent with the notion that IsoKs affect the mitochondrial respiratory chain within or downstream of the Q-cycle portion of complex III (Fig. 8). In our respiration assays, effects at the level of cytochrome c are indistinguishable from those on complex III itself, indicating feasibility that our respiration effects could be explained by IsoK-modification of cytochrome c. This working hypothesis is consistent with the nineteen lysines of cytochrome c, which make it a chemically attractive target. Our data showed that at least in vitro, IsoKs primarily adducts to a lysine in the N-terminus of the protein, the portion of the cytochrome c previously established to participate in electrostatic interaction with cardiolipin, and thereby bind cytochrome c to the outer surface of the inner membrane. This interaction is known to be sensitive to peroxidation of cardiolipin, ionic strength, surface-charge density, and pH [64,65]. It appears a reasonable speculation that modification of positively charged lysine residues by the negatively charged IsoKs might potentially disrupt this interaction and facilitate a relatively early cytochrome c release (See Figure 7D, mitochondria exposed to calcium plus IsoKs release cytochrome c earlier relative to swelling than those mitochondria exposed to calcium alone). Future studies will, however, clearly be needed to determine whether cytochrome c or other interacting proteins are targets of IsoKs modification in mitochondria and how such modification alters their function. Nevertheless, IsoKs appear to offer insight into involvement of a previously unrecognized critical lysine residues and the mPT, in much the way as previous studies have implicated critical thiol, arginine and histidine residues [55–57]. We are continuing to examine these interactions.
Current Limitations and Future Studies
Further studies are needed in two areas: (i) to extend these findings from the isolated systems described here to progressively more (patho)physiologically relevant systems, e.g., intact isolated mitochondria, cell culture, and eventually animal models of cell death, and; (ii) to determine whether the apparent effect on cytochrome c is sufficient to underlie the observed effects on mPT or whether there are other direct targets. For example, it remains possible that cytochrome c release is mediated by IsoK-induced modification of other structural (protein) components involved in the mPT that contain lysine residues. For instance, Bernadi et al [54–57] have proposed that modification of lysine residues of mitochondrial pore structures by any agent may lead to structural remodeling of mitochondrial domains and subsequent induction of the mPT.
Summary
In summary, the data presented suggest that IsoKs facilitate mitochondrial calcium dysregulation and promote entry into cell death cascades under pathophysiological conditions. These effects are broadly conserved across at least three tissues and respond to known physiological inhibitors and inducers. Although their mechanisms of reaction vary distinctly from other previously characterized aldehydes that are formed by lipid peroxidation and disrupt mitochondrial function, their net effect is quite similar. These data are consistent with a role for IsoK-induced mitochondrial dysfunction during pathophysiological conditions. The results obtained herein may help shed light on the mechanisms underlying lipid peroxidation-induced changes in mitochondrial function.
Supplementary Material
Acknowledgments
This work was supported by funds from the Department of Neurosurgery, Brigham and Women’s Hospital (BSK), from Burke Medical Research Institute (BSK), where the studies were initiated, and from NIH MERIT Award GM42056 (LJR).
ABBREVIATIONS
- IsoK
isoketals, γ-keroaldehydes
- mPT
mitochondrial permeability transition
- ΔΨ
membrane potential
- tBH
tert-butylhydroperoxide
- TMRM
Tetramethyl Rhodamine Methyl Ester
- DNP
dinitrophenol
- BSA
bovine serum albumin
- Cs A
cyclosporine A
- RCR
respiratory control ratio
- MS
mass spectrometry
- CID
collision induced dissociation
- FDA
Food and Drug Administration
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fai Poon H, Calabrese V, Scapagnini G, Butterfield DA. Free radical and brain aging. Clin Geriatr Med. 2004;20:329–359. doi: 10.1016/j.cger.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Comporti M. Lipid peroxidation and biogenic aldehydes: from the identification of 4-hydroxynonenal to further achievements in biopathology. Free Radic Res. 1998;28:623–35. doi: 10.3109/10715769809065818. [DOI] [PubMed] [Google Scholar]
- 3.Blasig IE, Tilman G, Schonheit K, Rohde E, Jakstadt M, Haseloff RF, Siems WG. 4-Hydroxyninenal, a novel indicator of lipid peroxidation for reperfusion injury of the myocardium. Am J Physiol. 1995;269:H14–H22. doi: 10.1152/ajpheart.1995.269.1.H14. [DOI] [PubMed] [Google Scholar]
- 4.Korantzopoulos P, Kolettis TM, Galaris D, Goudevenos JA. The role of oxidative stress in the pathogenesis and perpetuation of atrial fibrillation. Int J Cardiol. 2007;115:135–143. doi: 10.1016/j.ijcard.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 5.Boyden PA, Davies SS, Vismanathan PC, Amarnath V, Balser JR, Roberts LJ., 2nd Potential role of isoketals formed via the isoprostane pathway of lipid peroxidation in ischemic arrhythmias. J Cardiovasc Pharmacol. 2007;50:480–486. doi: 10.1097/FJC.0b013e31815a0564. [DOI] [PubMed] [Google Scholar]
- 6.Srivastava S, Chandrasekar B, Bhatnagar A, Prabhu S. Lipid-peroxidation-derived aldehydes and oxidative stress in the failing heart: role of aldose reductase. Am J Physiol. 2002;283:H2612–2619. doi: 10.1152/ajpheart.00592.2002. [DOI] [PubMed] [Google Scholar]
- 7.Beal MF. Aging, energy and oxidative stress in neurodegenerative diseases. Ann Of Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 8.Montine TJ, Beal MF, Robertson D, Cudkowicz ME, Biaggioni I, O’Donnell H, et al. Cerebrospinal fluid F2-isoprostanes are elevated in Huntington’s disease. Neurobiology. 1999;52:1104–1105. doi: 10.1212/wnl.52.5.1104. [DOI] [PubMed] [Google Scholar]
- 9.Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiology Aging. 2001;22:187–194. doi: 10.1016/s0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- 10.Springer JE, Azbill RD, Mark RJ, Begley JG, Waeg G, Mattson MP. 4-Hydroxynonenal, a lipid peroxidation product, rapidly accumulates following traumatic spinal cord injury and inhibits glutamate uptake. J Neurochem. 1997;68:2469–2476. doi: 10.1046/j.1471-4159.1997.68062469.x. [DOI] [PubMed] [Google Scholar]
- 11.Luo J, Uchida K, Shi R. Accumulation of acrolein-protein adducts after trauatic spinal cord injury. Neurochem Res. 2005;30:291–295. doi: 10.1007/s11064-005-2602-7. [DOI] [PubMed] [Google Scholar]
- 12.Morrow JD, Hill KE, Burk RF, Nammour TM, Bard KF, Roberts LJ. A series of prostaglandin-like compounds produced in vivo in humans by a non-cyclooxygenase free radical catalyzed mechanism. Proc Natl Acad Sci U S A. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brame CJ, Salomon RG, Morrow JD, Roberts LJ., 2nd Identification of extremely reactive gamma-ketoaldehydes (isolevuglandines) as products of the isoprostane pathway and characterization of their lysyl protein adducts. J Biol Chem. 1999;274:13139–13146. doi: 10.1074/jbc.274.19.13139. [DOI] [PubMed] [Google Scholar]
- 14.Davies S, Amarnath V, Roberts LJ., II Isoketals highly reactive gamma-ketoaldehydes formed from the H2-isoprostane pathway. Chem Phys Lipids. 2004;128:85–99. doi: 10.1016/j.chemphyslip.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 15.Amarnath V, Amarnath K, Matherson T, Davies S, Roberts LJ., 2nd A simplified synthesis of diasteriomers of levuglandin E2. Synthetic Communications. 2005;35:397–408. [Google Scholar]
- 16.Davies SS, Amarnath V, Brame CJ, Boutaud O, Roberts LJ., 2nd Measurement of chronic oxidative and inflammatory stress by quantification of isoketals/levuglandine γ–ketoaldehyde protein adducts using liquid chromatography tandem mass spectrometry. Nature Protocols. 2007;2:2079–2091. doi: 10.1038/nprot.2007.298. [DOI] [PubMed] [Google Scholar]
- 17.Montuschi P, Barnes P, Roberts LJ., 2nd Isoprostanes: markers and mediators of oxidative stress. FASEB J. 2004;18:1791–1800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- 18.Morrow J. Quantification of isoprostanes as indices of oxidant stress and the risk of atherosclerosis in humans. Arterioscler Thromb Vasc Biol. 2005;25:279–286. doi: 10.1161/01.ATV.0000152605.64964.c0. [DOI] [PubMed] [Google Scholar]
- 19.Montine TJ, Morrow JD. Fatty acid oxidation in the pathogenesis of Alzheimer’s Disease. Am J Patology. 2005;166:1283–1289. doi: 10.1016/S0002-9440(10)62347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brame CJ, Boutand O, Davies SS, Yang T, Oates JA, et al. Modification of proteins by isoketal-containing oxidized phospholipids. J Biol Chem. 2004;279:13447–13451. doi: 10.1074/jbc.M313349200. [DOI] [PubMed] [Google Scholar]
- 21.Davies SS, Amarnath V, Montine KS, Bernoud-Huback N, Boutaud O, Montine TJ, Roberts LJ., 2nd Effect of reactive gamma-ketoaldehydes formed by isoprostane pathway (isoketals) and cyclooxygenase pathway (levugandines) on proteosome function. FASEB J. 2002;16:715–717. doi: 10.1096/fj.01-0696fje. [DOI] [PubMed] [Google Scholar]
- 22.Kristal BS, Park BK, Yu BP. 4-Hydroxynonenal is a potent inducer of the mitochondrial permeability transition. J Biol Chem. 1996;271:6033–8. doi: 10.1074/jbc.271.11.6033. [DOI] [PubMed] [Google Scholar]
- 23.Burke WJ, Kristal BS, Yu BP, Li SW, Lin TS. Norepinephrine transmitter metabolite generates free radicals and activates mitochondrial permeability transition: a mechanism for DOPEGAL-induced apoptosis. Brain Res. 1998;787:328–32. doi: 10.1016/s0006-8993(97)01488-1. [DOI] [PubMed] [Google Scholar]
- 24.Kristal BS, Conway AD, Brown AM, Jain JC, Ulluci PA, Li SW, Burke WJ. Selective dopaminergic vulnerability:3,4-dihydroxyphenylacetaldehyde targets mitochondria. Free Rad Biol Med. 2001;30:924–31. doi: 10.1016/s0891-5849(01)00484-1. [DOI] [PubMed] [Google Scholar]
- 25.Stavrovskaya IG, Kristal BS. The powerhouse takes control of the cell: is the mitochondrial permeability transition a viable therapeutical target against neuronal dysfunction and death. Free Rad Biol Med. 2005;38:687–97. doi: 10.1016/j.freeradbiomed.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 26.Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12:815–33. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- 27.Halestrap A. Biochemistry: a pore way to die. Nature. 2006;434:578–9. doi: 10.1038/434578a. [DOI] [PubMed] [Google Scholar]
- 28.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilisation in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 29.Burke WJ, Li SW, Chung HD, Ruggiero DA, Kristal BS, Johnson EM, Lampe P, Kumar VB, Franko M, Williams EA, Zahm DS. Neurotoxicity of MAO metabolites of catecholamine neurotransmitters: role of neurodegenerative diseases. Neurotoxicol. 2004;25:101–115. doi: 10.1016/S0161-813X(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 30.Calingasan NY, Uchida K, Gibson GE. Protein-bound acrolein: a novel marker of oxidative stress in Alzheimer’s disease. J Neurochem. 1999;72:751–6. doi: 10.1046/j.1471-4159.1999.0720751.x. [DOI] [PubMed] [Google Scholar]
- 31.Sun L, Luo C, Long J, Wei D, Liu J. Acrolein is a mitochondrial toxin: effect on respiratory function and and enzyme activities in isolated rat liver itochondria. Mitochondrion. 2006;6:136–142. doi: 10.1016/j.mito.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 32.Stevens JF, Maier CS. Acrolein: sources, metabolism, biomolecular interactions relevant to human health and disease. Mol Nutr Food Res. 2008;52:7–25. doi: 10.1002/mnfr.200700412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeiger SLH, Musiek ES, Zanoni G, Vidari G, Morrow JD, Milne GJ, McLaughlin B. Neurotoxic lipid peroxidation species formed by ischemic stroke increase injury. Free Rad Biol Med. 47:1422–1431. doi: 10.1016/j.freeradbiomed.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stavrovskaya IG, Narayanan MV, Zhang W, Krasnikov BF, Heemskerk J, Young SS, Blass JP, Brown AM, Beal MF, Friedlander RM, Kristal BS. Clinically approved heterocyclics act on mitochondrial target and reduce stroke-induced pathology. J Exp Med. 2004;200:211–222. doi: 10.1084/jem.20032053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crompton M, Kessar P, Al-Nasser I. The alpha-adrenergic –mediated activation of the cardiac mitochondrial Ca2+ uniporter and its role in the control of intramitochondrial Ca2+ in vivo. Biochem J. 1983;216:333–342. doi: 10.1042/bj2160333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmer JW, Tandler B, Hoppel CL. Biochemical properies of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J B C. 1977;252:8731–8739. [PubMed] [Google Scholar]
- 37.Lai JC, Clark JB. Preparation of synaptic and nonsynaptic mitochondria from mammalian brain. Methods Enzymol. 1979;55:51–60. doi: 10.1016/0076-6879(79)55008-3. [DOI] [PubMed] [Google Scholar]
- 38.Krasnikov BF, Kim SY, McConoughey SJ, Ryu H, Xu H, Stavrovskaya IG, et al. Transglutaminase activity is present in highly purified nonsynaptosomal mouse brain and liver mitochondria. Biochem J. 2005;44:7830–43. doi: 10.1021/bi0500877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 40.Kristal BS, Jackson CT, Chung H-Y, Matsuda M, Nguyen H, Yu BP. Defects at center P underlie diabetes-associated itochondrial dysfunction. Free Rad Biol Med. 1997;22:823–833. doi: 10.1016/s0891-5849(96)00428-5. [DOI] [PubMed] [Google Scholar]
- 41.Baranov SV, Stavrovskaya IG, Brown AM, Tyryshkin AM, Kristal BS. Kinetic model for Ca2+-induced permeability transition in energized liver mitochondria discriminates between inhibitor mechanisms. J Biol Chem. 2008;283:665–76. doi: 10.1074/jbc.M703484200. [DOI] [PubMed] [Google Scholar]
- 42.Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as aprobe of transmembrane potential in isolated rat- liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta. 1986;850:436–448. doi: 10.1016/0005-2728(86)90112-x. [DOI] [PubMed] [Google Scholar]
- 43.Ward MW, Rego AC, Frenguelli BG, Nicholls DG. Mitochondrial membrane potential and glutamate excititoxicity in cultured cerebellar granule cells. J Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andreyev A, Fiskum G. Calcium induced release of mitochondrial cytochrome c by different mechanisms selective for brain versus liver. Cell Death Differ. 1999;6:825–832. doi: 10.1038/sj.cdd.4400565. [DOI] [PubMed] [Google Scholar]
- 45.Kristal BS, Dubinsky JM. Mitochondrial permeability transition in the central nervous system: induction by calcium cycling-dependent and independent pathways. J Neurochem. 1997;69:524–38. doi: 10.1046/j.1471-4159.1997.69020524.x. [DOI] [PubMed] [Google Scholar]
- 46.Brustovetsky N, Dubinsky JM. Limitations of cyclosporin A inhibition of the permeability transition in CNS mitochondria. J Neurosci. 2000;20:103–13. doi: 10.1523/JNEUROSCI.20-22-08229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kristal BS, Staats PN, Shestopalov AI. Biochemical characterization of the mitochondrial permeability transition in isolated forebrain mitochondria. Dev Neurosci. 2000;22:376–83. doi: 10.1159/000017463. [DOI] [PubMed] [Google Scholar]
- 48.Isom AL, Barnes S, Wilson L, Kirk M, Coward L, Darley-Usmar V. Modification of cytochrome c by 4-hydroxy-2-nonenal: evidence for histidine, lysine, and arginine-aldehyde adducts. J Am Soc Mass Spectrom. 2004;15:1136–1147. doi: 10.1016/j.jasms.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 49.Brown AM, Kristal BS, Effron MS, Shestopalov AI, Ullucci PA, Sheu KF, Blass JP, Cooper AJ. Zn2+ inhibits alpha-ketoglutarate stimulated mitochondrial respiration and isolated alpha-ketoglutarate dehydrogenase complex. J Biol Chem. 2000;275:13441–7. doi: 10.1074/jbc.275.18.13441. [DOI] [PubMed] [Google Scholar]
- 50.Gazaryan IG, Krasinskaya IP, Kristal BS, Brown AM. Zinc irreversibly damages major enzymes of energy production and antioxidant defense prior to mitochondrial permeability transition. J Biol Chem. 2007;282:24373–80. doi: 10.1074/jbc.M611376200. [DOI] [PubMed] [Google Scholar]
- 51.Zagol-Ikapitte I, Masterson T, Amarnath V, Montine T, Andreasson K, Boutaud O, Oates J. Prostaglandine H(2)-derived adducts of proteins correlate with Alzheimer’s disease severity. J Neurochem. 2005;94:1140–1145. doi: 10.1111/j.1471-4159.2005.03264.x. [DOI] [PubMed] [Google Scholar]
- 52.Cracowsky J-L. Isoprostanes: an emerging role in vascular physiology and disease. Chemistry and physics of lipids. 2004;128:75–83. doi: 10.1016/j.chemphyslip.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 53.Fukuda K, Davies SS, Nakajima T, Ong BH, Kupershmidt S, et al. Oxidative mediated lipid peroxidation recapitulates proarrhytmic effects on cardiac sodium channels. Circ Res. 2005;97:1262–9. doi: 10.1161/01.RES.0000195844.31466.e9. [DOI] [PubMed] [Google Scholar]
- 54.Petronilli V, Cola C, Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. J Biol Chem. 1993;268:1011–1016. [PubMed] [Google Scholar]
- 55.Bernardi P, Vassanelli S, Veronese P, Colona R, Szabo I, Zoratti M. Modulation of the mitochondrial permeability transition pore. Effect of divalent cations and protons. J Biol Chem. 1992;267:2934–2939. [PubMed] [Google Scholar]
- 56.Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem. 1994;269:16638–43. [PubMed] [Google Scholar]
- 57.Constantini P, Chernyak BV, Petronilli V, Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine mucleotides and dithiol oxidation at two separate sites. J Biol Chem. 1996;271:6746–51. doi: 10.1074/jbc.271.12.6746. [DOI] [PubMed] [Google Scholar]
- 58.Vercessi AE, Kowaltowski AJ, Grijalba MT, Meinicke AR, Castilho RF. The role of reactive oxygen species in mitochondrial permeability transition. Biosci Rep. 1997;17:43–52. doi: 10.1023/a:1027335217774. [DOI] [PubMed] [Google Scholar]
- 59.Nakashima T, Higa H, Matsubara H, Benson AM, Yasunobu KT. The amino acid sequence of bovine heart cytochrome c. J Biol Chem. 1966;241:1166–1177. [PubMed] [Google Scholar]
- 60.Mortz E, O’connor PB, Roepstorff P, Kelleher NL, Wood TD, McLaffery FW, Mann M. Sequence tag identification of intact proteins by matching tandem mass spectral data against sequence data bases. Proc Natl Acad Sci. 1996;93:8264–8267. doi: 10.1073/pnas.93.16.8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bushnell GW, Louie GV, Brayer GD. High-resolution three-demensional structure of horse heart cytochrome c. J Mol Biol. 1990;214:585–595. doi: 10.1016/0022-2836(90)90200-6. [DOI] [PubMed] [Google Scholar]
- 62.Yamamoto Y. A 1H NMR study of structurally relevant inter-segmental hydrogen bond in cytochrome c. Biochim Biophys Acta. 1997;1343:193–202. doi: 10.1016/s0167-4838(97)00109-x. [DOI] [PubMed] [Google Scholar]
- 63.Gao Y, Lee DJ, Willians RJP, Willians G. The effect of multiple amino acid substitutions of the polypeptide backbone of tuna and horse cytochrome c. Eur J Biochem. 1989;182:57–65. doi: 10.1111/j.1432-1033.1989.tb14800.x. [DOI] [PubMed] [Google Scholar]
- 64.Ott M, Robertson J, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sc. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sinibaldi F, Fiorucci L, Patriarca A, Lauceri R, Ferri T, Coletta M, Santucci R. Insights into cytochrome c-cardiolipin interaction. Role played by anionic strength. Biochemistry. 2008;47:6928–35. doi: 10.1021/bi800048v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.