

Abstract
Positron emission tomography (PET) is a relatively non-invasive imaging test that is able to detect abnormalities in different organs based on derangements in the chemical functions and/or receptor expression at the cellular level. PET imaging of the brain has been shown to be a powerful diagnostic tool for detecting neurochemical abnormalities associated with various neurologic disorders as well as to study normal brain development. Although its use in detecting neurological abnormalities has been well described in adults and pediatrics, it application in the newborn nursery has not been explored adequately. Early detection of brain injury secondary to intrauterine and perinatal insults using PET imaging can provide new insight in prognosis and in instituting early therapy. In this review, the authors describe applications of PET imaging in the newborn nursery specifically related to the detection of metabolic changes seen in hypoxic ischemic encephalopathy, neonatal seizures and neuroinflammation in the neonatal period.
Introduction
Positron emission tomography (PET) is a diagnostic imaging tool that can detect and map abnormalities in various organs related to glucose metabolism, blood flow, receptor binding, oxygen utilization, protein synthesis, neurotransmitter synthesis, release and transport. This is achieved using compounds labeled with positron emitting isotopes generated by a cyclotron; the radiotracers are injected intravenously and their distribution and fate are detected by a positron camera. Current state-of-the-art PET scanners have a spatial resolution of approximately 3 mm. PET imaging, when combined with magnetic resonance imaging (MRI) or computed tomography (CT), allow anatomical co-registration of the functional PET images to provide unique details about changes at the cellular level in different regions of the brain. Since PET imaging has been applied extensively in adult and pediatric patients, most clinical PET scanners have been designed for use in older patients and have relatively large fields-of-view.
Recently, a new generation of microPET scanners has been designed for imaging of small animals in the laboratory setting and these provide superior spatial resolution compared to clinical scanners. These microPET scanners have the advantage of being portable and can be taken to the newborn nursery for scanning the infant. One of these new devices, the Focus 220 microPET scanner (Concorde Microsystems, Knoxville, TN) when appropriately modified for use in humans, has a patient opening of 22 cm and an axial field-of-view of 8 cm that can accommodate infants weighing <15 kg [FIG 1]. This scanner has a high spatial resolution of <2mm full-width-at-half-maximum. The patient can be monitored throughout the period of the scan and can be maintained on all infusions. Most neonatal PET scans would take about 1 hour to be completed.
Figure 1. Focus 220 microPET scanner in the nursery.
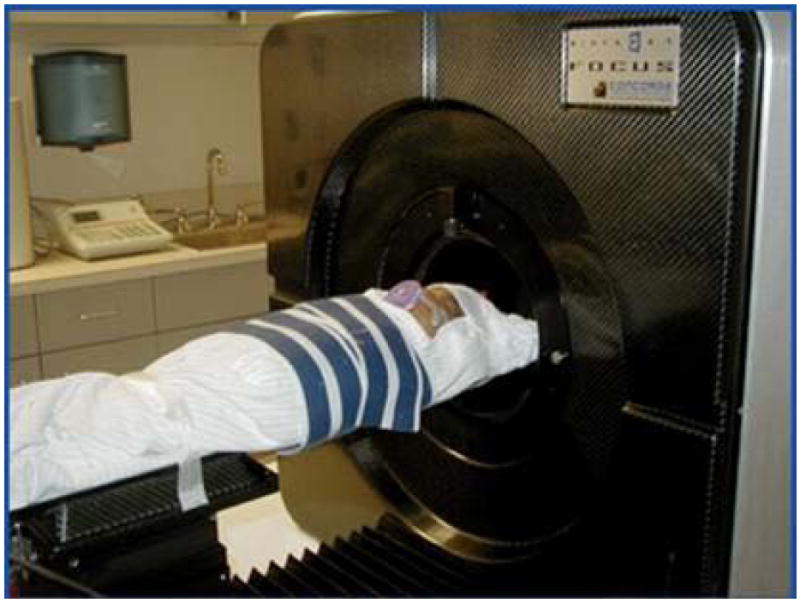
This scanner provides higher spatial resolution for imaging infants <15kg. The scanner has more than 24,000 individual LSO detectors. In order to assure safety and comfort of the infants during the PET scan, the infant is bundled with blankets and the bundled infant is then fastened to the bed with velcro straps.
Cerebral glucose metabolism in the developing brain
PET imaging with the tracer 2-deoxy-2-[18F]fluoro-D-glucose (FDG) has provided crucial information regarding the functional maturation of the human brain. Correlations between regional cerebral glucose utilization and behavioral maturation, synaptogenesis and plasticity have provided information about normal and abnormal brain development. The pattern of brain glucose metabolism appears to be related to postconceptional age and undergoes marked changes in the first year of life 1-4. The highest rates of glucose metabolism in the newborn brain are seen in the primary sensory and motor cortex, thalamus, brain stem and vermis, hippocampus/amygdala and occasionally the basal ganglia with very low activity noted in the remaining cerebral cortex [FIG 2]. This corresponds to the development phase in the neonate with predominance of intrinsic brainstem reflexes and limited visuomotor integration. The prominence of glucose metabolic activity in the hippocampus/amygdala seen in newborns suggests that these limbic structures are functionally active. It has been hypothesized that early activity in these structures may be related to bonding and early attachment 5. During infancy, glucose metabolism patterns proceed in a phylogenetic order with functional maturation of older anatomic structures occurring first followed by the newer structures. Increased glucose metabolism is observed in the parietal, temporal and primary visual cortical regions, basal ganglia and cerebellar hemispheres by 2-3 months postnatal age. This coincides with the reorganization of primitive neonatal reflexes, improved visuospatial and visuosensorimotor integrative functions and increased cortical maturation of electroencephalographic activity 2,3. By about 6-8 months of age, the lateral and inferior portions of the frontal cortex become more functionally active followed by the dorsal and medial frontal regions, coinciding with the development of higher cortical and cognitive maturation. This period corresponds with an expansion of dendritic fields and an increase in capillary density in the frontal cortex. Adult patterns of glucose utilization are achieved by about 1 year of age. These regional variations in glucose metabolism help explain the pattern of injury (e.g., from hypoxia-ischemia) at different postnatal ages, where areas of higher metabolic demands have a greater propensity for injury or greater selective vulnerability 2,3.
Figure 2. Cerebral glucose metabolism during infancy.
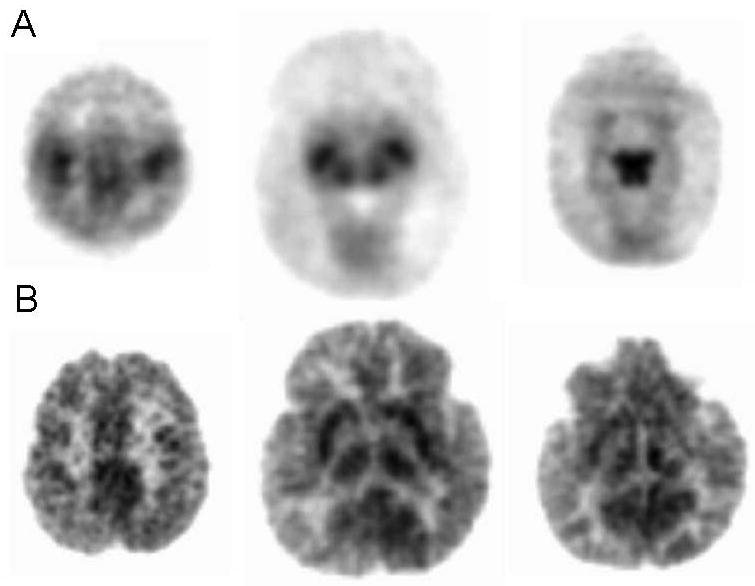
Panel A represents PET images of glucose metabolism using 2-deoxy-2[18F] fluoro-D-glucose (FDG) in a term newborn infant at postnatal day 4 of age. Panel B represents images of glucose uptake in a 12 month old infant. Increased glucose uptake is noted in the primary sensory- and motor cortex, thalamus, basal ganglia and brainstem with decreased activity in the cerebral cortex in the newborn.
PET Imaging for Neonatal Seizures
Seizures in the newborn are the most common clinical manifestation of neurologic insults sustained during the perinatal or neonatal period. They may be triggered by perinatal hypoxia-ischemia, metabolic disturbances, intracerebral bleeds, infections, stroke, trauma and maternal drug abuse. Neonatal seizures are a risk factor for refractory epilepsy during childhood 6-8. Moreover, seizures may impair normal brain development contributing to an adverse neurologic outcome 9. Hence, early diagnosis and aggressive treatment to obtain seizure control is necessary for normal cognitive development. Although MRI remains the imaging modality of choice in determining the etiology of seizures, PET imaging may assist in identifying the epileptic focus and areas of cerebral dysgenesis in newborns with refractory epilepsy where subtle structural abnormalities may not be optimally visualized on MRI because of the relatively high water content in the newborn brain. The most common tracers used in the imaging of epilepsy for clinical purposes are FDG and [11C]-flumazenil followed by alpha-[11C]methyl-L-tryptophan (AMT) 10, but a large number of other PET ligands have been applied in the research setting to better understand the basic mechanisms of epilepsy.
FDG PET scans can detect areas of hypometabolism consistent with epileptogenic foci even in patients with normal MRI scans 11,12. The tracer [11C]-flumazenil binds to GABAA receptors and has been shown to improve localization of epileptic foci in select groups of patients with intractable epilepsy especially when FDG PET shows large areas of glucose hypometabolism 13-15. In addition, [11C]-flumazenil PET is very sensitive in detecting mesial temporal sclerosis. Recent studies have shown that AMT PET can pinpoint epileptic foci in children with cortical developmental malformations and cortical dysplasia 16. Abnormalities in metabolic function and/or neuroreceptor expression determined by PET imaging, when correlated with electrophysiological data, help in accurate localization of epileptogenic foci for presurgical evaluation of these patients 17. Another condition where FDG PET studies have been helpful is in West Syndrome which is an age-specific debilitating seizure disorder beginning in early infancy and is characterized by uncontrolled seizures (infantile spasms), hypsarrhythmia on EEG and developmental delay. FDG PET studies have shown that many of these children have focal cortical dysplasias indicated by severe hypometabolism. These areas of cortical dysplasias can trigger the brainstem resulting in activation of the striatum, subsequently leading to multifocal discharges on EEG and symmetric spasms. Surgical resection of the focal cortical dysplasia results in alleviation of the seizures 18.
In the neonate, when seizures are poorly controlled and no obvious etiology for the seizures is apparent, the most likely causes are malformation of cortical development or an inborn error of metabolism. In this setting, if the MRI scan is normal, PET scanning with FDG may be very helpful in depicting an epileptic focus [FIG 3]. However, if a diffuse pattern of abnormality is shown on the PET scan, a metabolic disorder would be more likely.
Figure 3. FDG PET scan in 8 day old term infant with seizures during the scan.
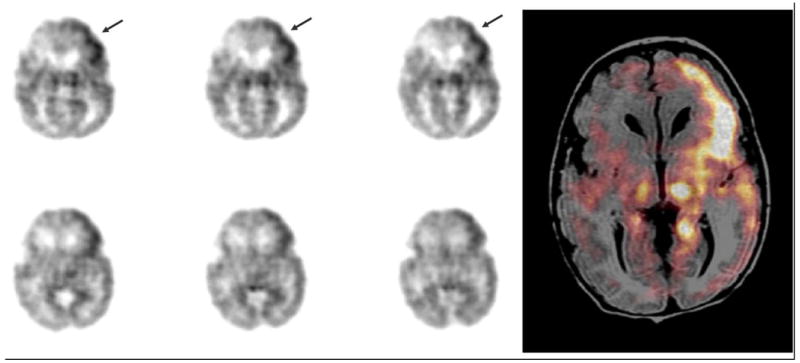
Ictal PET images in a term neonate with intractable seizures show activation of the left frontal cortex (arrows). The image on the right is a representative PET image that has been co-registered with the patient's MRI. This shows that the left thalamus and parahippocampal regions are involved in the propagation without any obvious structural abnormalities seen on MRI.
PET imaging in cerebral palsy
Cerebral palsy is a broad term encompassing a group of non-progressive disorders of posture and motor impairment including spasticity, movement disorders, muscle weakness, ataxia, and rigidity that occurs with damage to the developing brain 19. Clinical evidence indicates that the presence of periventricular leukomalacia (PVL) is one of the most identifiable risk factors for developing cerebral palsy 20. MRI has been widely used as the neuroimaging tool of choice in detecting brain lesions in the neonatal period and children with CP. Although MRI has been used to reveal macrostructural brain abnormalities such as periventricular leukomalacia, ventricular enlargement, porencephalic cysts and cerebral atrophy 21-23, conventional MRI is unable to detect micropathology in patients with CP 24,25. Indeed, conventional MRI was unable to detect any structural abnormalities in 17% of patients with CP 26. Functional imaging techniques such as PET imaging may be helpful in detecting metabolic abnormalities, changes in regional blood flow and in receptor expression at much earlier stage, before the development of structural and morphological abnormalities 12,27-29. Abnormalities in glucose metabolism that extend beyond the site of the structural lesion may help determine the actual extent of injury [FIG 4].
Figure 4. Glucose metabolism in a 6 week old premature neonate born at 28 weeks with grade III left -intraventricular hemorrhage (IVH).
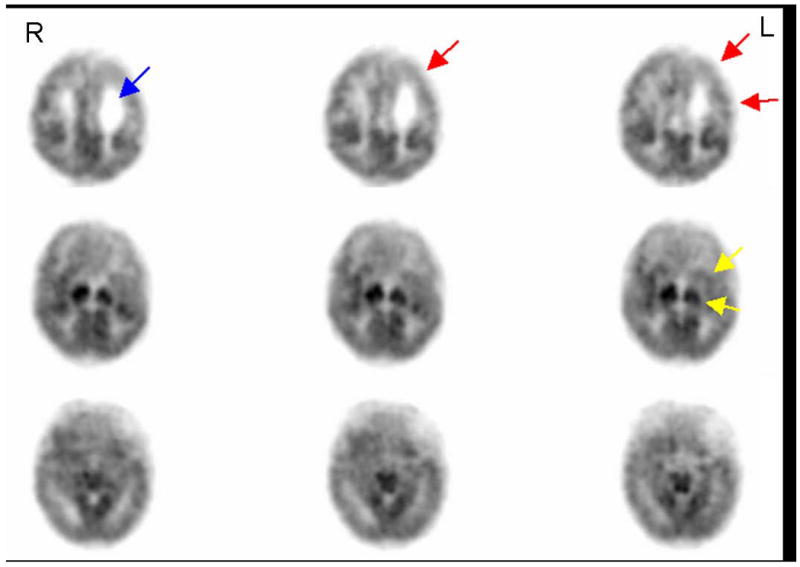
FDG PET images at 34 weeks post-conceptional age in a neonate born at 28 weeks gestation with grade III left IVH demonstrates not only a dilated left lateral ventricle (arrow in top panel) indicative of structural changes, but also widespread metabolic changes in the left hemisphere indicated by hypometabolism of the left frontal, parietal and superior temporal cortex (arrows in top panel), as well as left basal ganglia and thalamus (arrows in middle panel). R and L indicate right and left side of the brain respectively.
The clinical phenotype of CP which later emerges may be predicted in the newborn by the patterns of hypometabolism noted on FDG PET scan. Bilateral thalamic hypometabolism (particularly in the lateral thalamic nuclei) predicts the spastic diplegic type of CP. In such patients, the cerebral cortex may appear normal or may show focal areas of hypometabolism without any apparent structural abnormalities on the MRI. The topography of these focal cortical areas of hypometabolism correlate with specific cognitive deficits and are likely to be due to an interruption of thalamocortical projections resulting from the injury 30. This interpretation was later supported by volumetric MRI studies demonstrating regional cortical volume loss in such patients 31.
Newborns that show a predominantly unilateral pattern of hypometabolism on FDG PET are likely to develop the infantile hemiplegic type of CP 30. Most of these patients have had their injury in utero in the third trimester, often the result of various coagulopathies. Unlike adult patients who have suffered a unilateral cerebral cortical injury, these newborns do not typically show crossed cerebellarhypometabolism (diaschisis), presumably because of reorganization of corticopontocerebellar tracts.
Transient hypermetabolism followed by severe hypometabolism in the basal ganglia and thalamus appears to be related to the development of dystonic/choreoathetoid CP 32. It has been hypothesized that this transient hypermetabolism in the basal ganglia may be related to a transient increase in vascularity in that region or increased energy demand of cells that are attempting to reinstate their ionic balance following perinatal asphyxial injury, which is usually an acute total injury from insults such as placental abruption or uterine rupture. Late secondary processes such as delayed excitotoxicity may lead to neuronal loss resulting in the severe hypometabolism seen at an older age.
With partial prolonged hypoxic-ischemic brain injury in the perinatal period, the clinical type of CP is usually a spastic quadriplegia which, on MRI, is seen as multifocal cystic encephalomalacia and on FDG PET as multifocal hypometabolism. In the most severe cases, glucose metabolic activity remains only in the basal ganglia, brainstem and cerebellum30. The relative preservation of glucose metabolism in the cerebellum in the absence of activity in supratentorial structures may be due to the fact that cell division in cerebellum occurs until the second postnatal year.
One of the advantages of PET technology is that it may be used to image and measure a wide variety of chemical processes other than glucose metabolism. In our laboratory, PET imaging of GABAA receptor binding using 11C-flumazenil is frequently applied in the localization of epileptic foci during evaluation for epilepsy surgery. Since GABAergic signaling appears to play an important role in the integration of sensory and motor cortical impulses and dissociation between the sensory and motor inputs, a disruption of this process may at least partially account for the motor dysfunction in CP patients 33,34. Differences in regional GABAA receptor binding on PET imaging with [11C]-flumazenil characterized by increased binding in the motor and visual cortices, and decreased binding in the brainstem were noted in patients with spastic cerebral palsy when compared to normal controls34. Under normal circumstances, a significant change in GABAA receptor binding and subunit expression occurs with increase in postnatal age. Indeed, we have shown that there is a postnatal decline of GABAA receptor binding in most brain regions and the adult levels are reached at about age 18 to 24 years 35. A better understanding of the normal distribution of GABAA receptor binding and expression in the neonate will be helpful in elucidating the pathophysiology of neurodevelopmental disorders where GABAergic mechanisms may play a key role. This would also help in modifying pharmacological therapy to specifically target these mechanisms at various stages of development.
Imaging of neuroinflammation in Cerebral Palsy
In recent years a number of in vitro and in vivo studies have implicated a neuroinflammatory response characterized by activated microglial cells in the development of PVL and CP 36-40.Microglia constitute about 10% to 12% of the total cells of the brain and are located predominantly in the grey matter, including the hippocampus, olfactory telencephalon, basal ganglia, and substantianiagra in the normal adult brain 41,42. In the developing brain, microglia are present in the white matter tracts from late second trimester until term, decreasing in density in the postnatal period and eventually migrating to the cortex to assume their typical ramified form 43. In the healthy brain, microglia interact with other cortical elements such as astrocytes, neurons, and blood vessels, thereby monitoring neuronal well-being. The resident microglia in adults are characterized by ramified morphology and are highly dynamic structures sensing subtle changes in the microenvironment with their motile processes and protrusions 41,42,44. On activation after an injury, microglia undergo a pronounced change in morphology from ramified to an amoeboid structure. Activated microglia secrete proinflammatory mediators such as prostaglandins, tumor necrosis factor-α, interleukin-1β, chemokines, reactive oxygen species, nitric oxide, and anti-inflammatory substances such as interleukin10 and neurotrophic factors41.
Haynes et al. (2003) have shown the increased presence of activated microglia diffusely throughout the white matter in autopsy specimens of patients with PVL indicating that activated microglia are involved in causing white matter damage by oxidative and nitrosative stress45. Activated microglial cells may induce oligodendrocyte injury by releasing oxidative and nitrosative products39, excitotoxic metabolites such as glutamate and quinolinic acid, that may cause glutamate receptor, or NMDA receptor mediated injury to oligodendrocytes 46,and by producing a variety of pro-inflammatory cytokines many of which are cytotoxic47. The low resistance of oligodendrocytes to oxidative stress, presence of calcium permeable glutamate receptors and presence of NMDA receptors in their myelinating processes make them highly susceptible to injury 48,49.
An accumulating body of evidence suggests that in addition to hypoxic/ischemic injury, intrauterine infection or inflammation plays a central role in the etiology of PVL and CP 50-52. Pro-inflammatory cytokines such as IL-1β have been shown to induce microglial activation in vitro53. Lipopolysaccharide treatment results in oligodendrocyte damage only in the presence of microglial cells supporting the role of microglial cell activation in white matter injury 54. The fetal inflammatory response after intra-uterine infection/inflammation may lead to activation and recruitment of microglial cells present in the white matter regions of the fetal brain, subsequently resulting in widespread production of pro-inflammatory mediators and leading to the death of surrounding oligodendrocytes and, hence, white matter injury38.
Activated microglial cells express peripheral benzodiazepine binding sites or receptors (PBR) which are multimeric protein complexes comprised of three subunits 55. These receptors are most commonly located in the outer mitochondrial membrane of activated microglia in the brain and have been used as a sensitive marker to visualize and measure glial cell activation associated with various neuroinflammatory disorders 56-57. Though the exact function of the peripheral benzodiazepine receptor has not been clearly elucidated, it appears to be involved with physiologic processes such as cell proliferation, apoptosis, steroidogenesis and immunomodulation59. The normal healthy brain does not express peripheral benzodiazepine binding sites except in areas such as the choroid plexus, ependymal layer and perivascular cells 56. The activation of microglial cells is typically localized to the site of the injured neuron with extension along the anterograde or retrograde axonal pathway. This characteristic response helps localize the site and distribution of injury accurately when imaging of activated microglia is performed in order to provide information about the temporal and spatial progression of various neuroinflammatory disorders. Isoquinolineligands such as PK11195 (1-[2-chlorophenyl]-N-methyl-N-[1-methyl-propyl]-3-isoquinoline carboxamide) bind the 18kDA subunit of the peripheral benzodiazepine binding site that are expressed on activated microglial cells 59. When labeled with carbon-11, PK 11195 can be effectively used as a ligand for PET studies, indicating the presence of activated microglia in acute inflammatory and neurodegenerative disorders 56-58.
Animal studies in our laboratory have demonstrated an increase in the retention of [11C]PK11195 indicating specific binding of the tracer to peripheral benzodiazepine receptors in activated microglial cells in the newborn rabbit brain exposed to endotoxin in utero. [FIG 5] Our results show that this is not only confirmed by histology but is also associated with neurobehavioral changes, neuronal cell death and loss of myelin in these animals 60,38. These results indicate that detection of activated microglial cells may be used as an effective tool to determine the presence of a neuroinflammatory response in the neonatal brain following a perinatal insult, resulting in PVL. Thus, PET imaging with [11C] PK11195 may be an effective tool in the early detection of PVL in the neonate before structural changes occur. Moreover, the longitudinal assessment of [11C] PK11195 binding kinetics can be used as an effective tool in following the evolution of microglial activation in PVL and to determine the optimal time period for institution of therapy and follow response to treatment.
Figure 5. [11C]PK11195 uptake in postnatal day 1 neonatal rabbit brain.
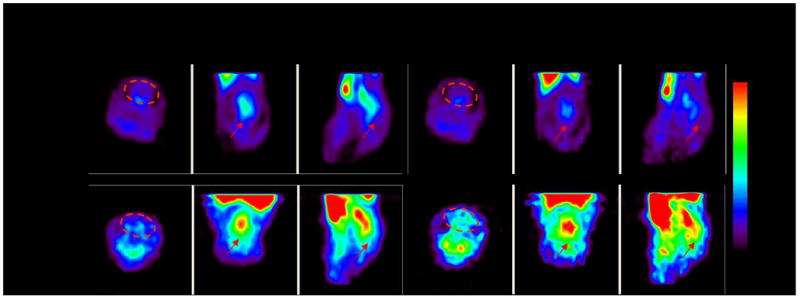
PET imaging of [11C]PK11195 uptake in a neonatal rabbit brain is compared between the 1st 10 minutes (0-10 mins) to the last 10 minutes (50-60 mins) of the scan. Endotoxin kits that were born to dams exposed to 20ug/kg of endotoxin in utero showed an increase in tracer uptake over time while control kits born to dams that were injected saline in utero showed a decrease in tracer uptake over time. This indicates specific binding of the tracer to activated microglial cells in the brain of endotoxin kits.
Ethical considerations in PET imaging in neonates
The greatest impediment in the widespread use of PET imaging in the neonatal period has been due to concerns regarding radiation exposure. Although large radiation exposures in the neonatal period is a matter for concern, most PETscans in neonates could be accomplished using effective doses approximately equal to the yearly background radiation exposure, and doses that are typically less than the exposure of a clinical CT scan of the head.CT scans currently result in an effective dose of around 30-90 mSv (3.0-9.0 rem) per scan to the organ scanned61. Exposure to these doses in the newborn period is associated with a 0.04-0.06% lifetime attributable risk of radiation associated cancer61. A substantial decrease in the risk occurs with a decrease in the dose used. With PET scans, the typical neonatal doses would be around 5mSv which is 6-18 times less than that reported with CT scans. Given the very serious debilitating consequences from perinatal brain injury, PET scanning for early detection of neurochemical changes in the neonate may potentially have a high benefit to risk ratio.
Conclusions and Future Directions
Early detection of the presence of cellular abnormalities in the brain using a noninvasive technique such as PET imaging could potentially provide great benefit in directing relevant supportive therapies and follow up for these patients. For example, based on changes in metabolism or receptor expression, specific pharmacological interventions may be attempted. The presence of neuroinflammation as determined by [11C] PK11195 uptake in the early neonatal period may help identify patients who may benefit from therapy with anti-inflammatory agents. Similarly, abnormalities in tryptophan metabolism detected by PET imaging may be treated by inhibiting kynurenine pathway enzymes. Response to treatment with hypothermia in hypoxic ischemic injury may be determined by evaluating patterns of glucose metabolism or GABAA receptor binding and expression following therapy. Hence, judicious use of PET scanning in the neonatal period may help in the development of early therapeutic interventions and their response can be monitored non-invasively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chugani HT, Phelps ME. Maturational changes in cerebral function in infants determined by 18FDG positron emission tomography. Science. 1986;231(4740):840–3. doi: 10.1126/science.3945811. [DOI] [PubMed] [Google Scholar]
- 2.Chugani HT, Phelps ME, Mazziotta JC. Positron emission tomography study of human brain functional development. Ann Neurol. 1987;22(4):487–97. doi: 10.1002/ana.410220408. [DOI] [PubMed] [Google Scholar]
- 3.Chugani HT. A critical period of brain development: studies of cerebral glucose utilization with PET. Prev Med. 1998;27(2):184–8. doi: 10.1006/pmed.1998.0274. [DOI] [PubMed] [Google Scholar]
- 4.Kinnala A, Suhonen-Polvi H, Aärimaa T, et al. Cerebral metabolic rate for glucose during the first six months of life: an FDG positron emission tomography study. Arch Dis Child Fetal Neonatal Ed. 1996;74(3):F153–7. doi: 10.1136/fn.74.3.f153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chugani HT, Behen ME, Muzik O, et al. Local brain functional activity following early deprivation: a study of postinstitutionalized Romanian orphans. Neuroimage. 2001;14(6):1290–301. doi: 10.1006/nimg.2001.0917. [DOI] [PubMed] [Google Scholar]
- 6.Berg AT, Levy SR, Novotny EJ, Shinnar S. Predictors of intractable epilepsy in childhood: a case-control study. Epilepsia. 1996;37(1):24–30. doi: 10.1111/j.1528-1157.1996.tb00507.x. [DOI] [PubMed] [Google Scholar]
- 7.Sun Y, Vestergaard M, Christensen J, et al. Prenatal exposure to maternal infections and epilepsy in childhood: a population-based cohort study. Pediatrics. 2008;121(5):e1100–7. doi: 10.1542/peds.2007-2316. [DOI] [PubMed] [Google Scholar]
- 8.Whitehead E, Dodds L, Joseph KS, et al. Relation of pregnancy and neonatal factors to subsequent development of childhood epilepsy: a population-based cohort study. Pediatrics. 2006;117(4):1298–306. doi: 10.1542/peds.2005-1660. [DOI] [PubMed] [Google Scholar]
- 9.Thibeault-Eybalin MP, Lortie A, Carmant L. Neonatal seizures: do they damage the brain. Pediatr Neurol. 2009;40(3):175–80. doi: 10.1016/j.pediatrneurol.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 10.Juhász C, Chugani HT. Imaging the epileptic brain with positron emission tomography. NeuroimagingClin N Am. 2003;13(4):705–16. viii. doi: 10.1016/s1052-5149(03)00090-x. Review. [DOI] [PubMed] [Google Scholar]
- 11.Chugani HT. The role of PET in childhood epilepsy. J Child Neurol. 1994 Oct;9 1 doi: 10.1177/0883073894009001131. [DOI] [PubMed] [Google Scholar]
- 12.Da Silva EA, Chugani DC, Muzik O, et al. Identification of frontal lobe epileptic foci in children using positron emission tomography. Epilepsia. 1997;38:1198–208. doi: 10.1111/j.1528-1157.1997.tb01217.x. [DOI] [PubMed] [Google Scholar]
- 13.Juhász C, Chugani DC, Muzik O, et al. Relationship of flumazenil and glucose PET abnormalities to neocortical epilepsy surgery outcome. Neurology. 2001;56(12):1650–8. doi: 10.1212/wnl.56.12.1650. 26. [DOI] [PubMed] [Google Scholar]
- 14.Savic I, Thorell JO, Roland P. [11C]flumazenil positron emission tomography visualizes frontal epileptogenic regions. Epilepsia. 1996;36(12):1225–32. doi: 10.1111/j.1528-1157.1995.tb01066.x. [DOI] [PubMed] [Google Scholar]
- 15.Muzik O, da Silva EA, Juhasz C, et al. Intracranial EEG versus flumazenil and glucose PET in children with extratemporal lobe epilepsy. Neurology. 2000;54(1):171–9. doi: 10.1212/wnl.54.1.171. 11. [DOI] [PubMed] [Google Scholar]
- 16.Juhász C, Chugani DC, Muzik O. Alpha-methyl-L-tryptophan PET detects epileptogenic cortex in children with intractable epilepsy. Neurology. 2003;60(6):960–8. doi: 10.1212/01.wnl.0000049468.05050.f2. 25. [DOI] [PubMed] [Google Scholar]
- 17.Juhász C, Chugani DC, Muzik O, et al. Relationship between EEG and positron emission tomography abnormalities in clinical epilepsy. J ClinNeurophysiol. 2000;17(1):29–42. doi: 10.1097/00004691-200001000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Asano E, Chugani DC, Juhász C, Muzik O, Chugani HT. Surgical treatment of West syndrome. Brain Dev. 2001;23(7):668–76. doi: 10.1016/s0387-7604(01)00305-9. [DOI] [PubMed] [Google Scholar]
- 19.Koman LA, Smith BP, Shilt JS. Cerebral palsy. Lancet. 2004;363(9421):1619–31. doi: 10.1016/S0140-6736(04)16207-7. [DOI] [PubMed] [Google Scholar]
- 20.Bejar RF, Vaucher YE, Benirschke K, et al. Postnatal white matter necrosis in preterm infants. J Perinatol. 1992;12(1):3–8. [PubMed] [Google Scholar]
- 21.Cioni G, Di Paco MC, Bertuccelli B, et al. MRI findings and sensorimotor development in infants with bilateral spastic cerebral palsy. Brain Dev. 1997;19(4):245–53. doi: 10.1016/s0387-7604(97)00569-x. [DOI] [PubMed] [Google Scholar]
- 22.Hashimoto K, Hasegawa H, Kida Y, et al. Correlation between neuroimaging and neurological outcome in periventricular leukomalacia: diagnostic criteria. Pediatr Int. 2001;43(3):240–5. doi: 10.1046/j.1442-200x.2001.01374.x. [DOI] [PubMed] [Google Scholar]
- 23.Kułak W, Sobaniec W, Kubas B, et al. Spastic cerebral palsy: clinical magnetic resonance imaging correlation of 129 children. J Child Neurol. 2007;22(1):8–14. doi: 10.1177/0883073807299953. [DOI] [PubMed] [Google Scholar]
- 24.Lee ZI, Byun WM, Jang SH, et al. Diffusion tensor magnetic resonance imaging of microstructural abnormalities in children with brain injury. Am J Phys Med Rehabil. 2003;82:556–559. doi: 10.1097/01.PHM.0000073830.15643.6A. [DOI] [PubMed] [Google Scholar]
- 25.Son SM, Ahn YH, Sakong J, et al. Diffusion tensor imaging demonstrates focal lesions of the corticospinal tract in hemiparetic patients with cerebral palsy. Neurosci Lett. 2007;420:34–38. doi: 10.1016/j.neulet.2007.04.054. [DOI] [PubMed] [Google Scholar]
- 26.Korzeniewski SJ, Birbeck G, DeLano MC, et al. A systematic review of neuroimaging for cerebral palsy. J Child Neurol. 2008;23:216–227. doi: 10.1177/0883073807307983. [DOI] [PubMed] [Google Scholar]
- 27.Volpe JJ, Herscovitch P, Perlman JM, et al. Positron emission tomography in the newborn: extensive impairment of regional cerebral blood flow with intraventricular hemorrhage and hemorrhagic intracerebral involvement. Pediatrics. 1983;72(5):589–601. [PubMed] [Google Scholar]
- 28.Volpe JJ, Herscovitch P, Perlman JM, et al. Positron emission tomography in the asphyxiated term newborn: parasagittal impairment of cerebral blood flow. Ann Neurol. 1985;17(3):287–96. doi: 10.1002/ana.410170312. [DOI] [PubMed] [Google Scholar]
- 29.Altman DI, Volpe JJ. Positron emission tomography in newborn infants. ClinPerinatol. 1991;18(3):549–62. Review. [PubMed] [Google Scholar]
- 30.Kerrigan JF, Chugani HT, Phelps ME. Regional cerebral glucose metabolism in clinical subtypes of cerebral palsy. Pediatr Neurol. 1991;7(6):415–25. doi: 10.1016/0887-8994(91)90024-f. [DOI] [PubMed] [Google Scholar]
- 31.Inder TE, Huppi PS, Warfield S, et al. Periventricular white matter injury in the premature infant is followed by reduced cerebral cortical gray matter volume at term. Ann Neurol. 1999;46(5):755–60. doi: 10.1002/1531-8249(199911)46:5<755::aid-ana11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 32.Batista CE, Chugani HT, Juhász C, et al. Transient hypermetabolism of the basal ganglia following perinatal hypoxia. Pediatr Neurol. 2007;36(5):330–3. doi: 10.1016/j.pediatrneurol.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 33.Hoon AH, Jr, Lawrie WT, Jr, Melhem ER, et al. Diffusion tensor imaging of periventricular leukomalacia shows affected sensory cortex white matter pathways. Neurology. 2002;59(5):752–6. doi: 10.1212/wnl.59.5.752. 10. [DOI] [PubMed] [Google Scholar]
- 34.Lee JD, Park HJ, Park ES, et al. Assessment of regional GABA(A) receptor binding using 18F-fluoroflumazenil positron emission tomography in spastic type cerebral palsy. Neuroimage. 2007;34(1):19–25. doi: 10.1016/j.neuroimage.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Chugani DC, Muzik O, Juhász C, et al. Postnatal maturation of human GABAA receptors measured with positron emission tomography. Ann Neurol. 2001;49(5):618–26. [PubMed] [Google Scholar]
- 36.Bell MJ, Hallenbeck JM. Effects of intrauterine inflammation on developing rat brain. J Neurosci Res. 2002;70:570–579. doi: 10.1002/jnr.10423. [DOI] [PubMed] [Google Scholar]
- 37.Cai Z, Pan ZL, Pang Y, et al. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res. 2000;47:64–72. doi: 10.1203/00006450-200001000-00013. [DOI] [PubMed] [Google Scholar]
- 38.Saadani-Makki F, Kannan S, Lu X, et al. Intrauterine administration of endotoxin leads to motor deficits in a rabbit model: a link between prenatal infection and cerebral palsy. Am J Obstet Gynecol. 2008;199:651–657. doi: 10.1016/j.ajog.2008.06.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Baud O, Vartanian T, et al. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci U S A. 2005;102:9936–9941. doi: 10.1073/pnas.0502552102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 41.Vilhardt F. Microglia: phagocyte and glia cell. Int J Biochem Cell Biol. 2005;37:17–21. doi: 10.1016/j.biocel.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 42.Nimmerjahn A, Kirchhoff F, Helmchen F. Restingmicroglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 43.Billiards SS, Haynes RL, Folkerth RD, et al. Development of microglia in the cerebral white matter of the human fetus and infant. J Comp Neurol. 2006;497:199–208. doi: 10.1002/cne.20991. [DOI] [PubMed] [Google Scholar]
- 44.Lawson LJ, Perry VH, Dri P, et al. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 45.Haynes RL, Folkerth RD, Keefe RJ, et al. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. 2003;62:441–450. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- 46.Espey MG, Chernyshev ON, Reinhard JF, Jr, et al. Activated human microglia produce the excitotoxin quinolinic acid. Neuroreport. 1997;8:431–434. doi: 10.1097/00001756-199701200-00011. [DOI] [PubMed] [Google Scholar]
- 47.Dommergues MA, Plaisant F, Verney C, et al. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience. 2003;121:619–628. doi: 10.1016/s0306-4522(03)00558-x. [DOI] [PubMed] [Google Scholar]
- 48.Fern R, Moller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20:34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167–1171. doi: 10.1038/nature04301. [DOI] [PubMed] [Google Scholar]
- 50.Dammann O, Leviton A. Infection remote from the brain, neonatal white matter damage, and cerebral palsy in the preterm infant. Semin Pediatr Neurol. 1998;5:190–201. doi: 10.1016/s1071-9091(98)80034-x. [DOI] [PubMed] [Google Scholar]
- 51.Nelson KB, Dambrosia JM, Grether JK, et al. Neonatal cytokines and coagulation factors in children with cerebral palsy. Ann Neurol. 1998;44:665–675. doi: 10.1002/ana.410440413. [DOI] [PubMed] [Google Scholar]
- 52.Yoon BH, Romero R, Park JS, et al. Fetal exposure to an intra-amniotic inflammation and the development of cerebral palsy at the age of three years. Am J Obstet Gynecol. 2000;182:675–681. doi: 10.1067/mob.2000.104207. [DOI] [PubMed] [Google Scholar]
- 53.Hailer NP, Vogt C, Korf HW, et al. Interleukin-1beta exacerbates and interleukin-1 receptor antagonist attenuates neuronal injury and microglial activation after excitotoxic damage in organotypic hippocampal slice cultures. Eur J Neurosci. 2005;21:2347–2360. doi: 10.1111/j.1460-9568.2005.04067.x. [DOI] [PubMed] [Google Scholar]
- 54.Lehnardt S, Lachance C, Patrizi S, et al. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garnier M, Boujrad N, Ogwuegbu SO, et al. The polypeptide diazepam-binding inhibitor and a higher affinity mitochondrial peripheral-type benzodiazepine receptor sustain constitutive steroidogenesis in the R2C Leydig tumor cell line. J Biol Chem. 1994;269:22105–22112. [PubMed] [Google Scholar]
- 56.Banati RB. Visualising microglial activation in vivo. Glia. 2002;40:206–217. doi: 10.1002/glia.10144. [DOI] [PubMed] [Google Scholar]
- 57.Gerhard A, Banati RB, Goerres GB, et al. [11C](R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology. 2003;61:686–689. doi: 10.1212/01.wnl.0000078192.95645.e6. [DOI] [PubMed] [Google Scholar]
- 58.Gerhard A, Pavese N, Hotton G, et al. In vivo imaging of microglial activation with [11C]®-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis. 2006;21:404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 59.Papadopoulos V, Baraldi M, Guilarte TR, et al. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 60.Kannan S, Saadani-Makki F, Muzik O, et al. Microglial activation in perinatal rabbit brain induced by intrauterine inflammation: detection with 11C-(R)-PK11195 and small-animal PET. J Nucl Med. 2007;48:946–954. doi: 10.2967/jnumed.106.038539. [DOI] [PubMed] [Google Scholar]
- 61.Brenner DJ, Hall EJ. Computed tomography–an increasing source of radiation exposure. N Engl J Med. 2007;357:2277–2284. doi: 10.1056/NEJMra072149. [DOI] [PubMed] [Google Scholar]